
Abstract
Background
The dramatic increase of antimicrobial resistance in the healthcare realm has become inexorably linked to the abuse of antibiotics over the years. Therefore, this study seeks to identify potential postbiotic metabolites derived from lactic acid bacteria such as Lactiplantibacillus plantarum that could exhibit antimicrobial properties against multi-drug resistant pathogens.
Results
In the present work, the genome sequence of Lactiplantibacillus plantarum PA21 consisting of three contigs was assembled to a size of 3,218,706 bp. Phylogenomic analysis and average nucleotide identity (ANI) revealed L. plantarum PA21 is closely related to genomes isolated from diverse niches such as dairy products, food, and animals. Genome mining through the BAGEL4 and antiSMASH database revealed four bacteriocins in a single cluster and four regions of biosynthetic gene clusters responsible for the production of bioactive compounds. The potential probiotic genes indirectly responsible for postbiotic metabolites production were also identified. Additionally, in vitro studies showed that the L. plantarum PA21 cell-free supernatant exhibited antimicrobial activity against all nine methicillin-resistant Staphylococcus aureus (MRSA) and three out of 13 Klebsiella pneumoniae clinical isolates tested.
Conclusion
Results in this study demonstrates that L. plantarum PA21 postbiotic metabolites is a prolific source of antimicrobials against multi-drug resistant pathogens with potential antimicrobial properties.
Similar content being viewed by others


Background
Antimicrobial resistance (AMR) is becoming more prevalent and continues to intimidate the ability to treat bacterial infections effectively. The reckless use of antibiotics has led to this very day where the potency of customary antimicrobials is diminishing due to the emergence of multi-drug resistant (MDR) bacteria. Methicillin resistant Staphylococcus aureus (MRSA) and Klebsiella pneumoniae are among clinically important MDR bacteria that are not only confined to healthcare settings but are also increasingly observed in the community [1, 2]. The World Health Organization (WHO) [3] attests that if this predicament is left unaddressed, AMR could lead to the resurgence of a pre-antibiotic era, where even the most basic infections can become fatal. Therefore, the emphasis on the discovery of new antimicrobial pipelines to tackle AMR and prevent a global health crisis is mandatory.
Lactic acid bacteria (LAB), in particular Lactiplantibacillus plantarum (previously known as Lactobacillus plantarum) [4], has been a subject of tremendous research lately due to its potential reservoir of novel antimicrobial compounds. This is largely due to its distinct properties which are attributed to a combination of factors such as the production of organic acids and bioactive compounds such as exopolysaccharides and bacteriocins [5]. Moreover, recent studies have highlighted the potential of L. plantarum as a candidate for the development of novel postbiotics, referring to bioactive compounds that are generated through the metabolic fermentation activity of LAB [6, 7]. The concept of postbiotics is particularly tempting because it single-handedly challenges the orthodox approach of purifying single compounds as analeptics. Postbiotics presents the possibility of harnessing the synergistic effects of multiple bioactive compounds which may offer a better therapeutic potential than any individual compound. Unlike previous reports on prebiotics and probiotics which are typically associated with live bacteria, postbiotics also offer a rather intriguing and safer alternative to combat AMR especially in immunocompromised individuals [6, 8].
Bioprospecting for postbiotic metabolites through the identification of biosynthetic gene clusters (BGCs) is one strategy to tap into the vast diversity of bacterial bioactive compounds. BGCs are modular gene units that work together to produce specific metabolites [9]. Advancements in genomics have facilitated a deeper understanding of how bacteria can be further exploited as a potential solution to AMR. Genome mining is such a tool that utilises bioinformatics to analyse bacterial genomes for the presence of BGCs and pathways that are involved in the production of bioactive compounds [10]. The search for antimicrobial compounds from L. plantarum PA21, in parallel with the advancements in genomics envisages a promising and swift avenue for the exploration of novel compounds in fighting AMR associated infections.
In this study, the phylogenetic relationship, and species classification of L. plantarum PA21 against other L. plantarum genomes was inferred. In addition, the whole-genome sequence of L. plantarum PA21 was mined to its potential antimicrobial compound while the probiotic features contributing to the antimicrobial activity was identified using the genome information. Subsequently, the bacterial inhibitory action of the antimicrobial substance from L. plantarum PA21 against MRSA and K. pneumoniae were investigated. The foundation laid on its genomic basis is expected to contribute to the overarching goals of developing targeted natural antimicrobial agents.
Results
Genome assembly and gene prediction
The gene prediction and annotation of L. plantarum PA21 were made using PATRIC (Supplementary Figure S1). The genome characteristics of L. plantarum PA21 are summarised in Table 1. The genome assembled into three contigs with N50 of 1,157,435. The genome size of 3,218,706 bp upon annotation was predicted to contain 3,118 protein coding sequences (CDS), 69 tRNAs, 14 rRNAs and 63 repeat regions. No plasmids were detected in the sequenced genome.
Distribution of L. plantarum in environmental habitats
L. plantarum was detected in a variety of environments ranging from gastrointestinal tracts of animals to freshwater and marine ecosystem (Fig. 1). Notably, high prevalence was detected in wastewater (15.90%) followed by pig gut (9.60%) and activated sludge (6.40%). The mean relative abundance throughout the environments was very low. The mean relative abundance of human, animal and insect gut metagenome displayed noticeable man relative abundance in general with high variability among samples with high standard deviation.

The prevalence and mean relative abundance of L. plantarum across different environments
Average nucleotide identity (ANI)
To investigate the genomic linkage and species boundaries, average nucleotide identity (ANI) analysis was undertaken on each Lactiplantibacillus genome assembly available from the NCBI database alongside L. plantarum PA21. The analysis of L. plantarum PA21 with other selected Lactiplantibacillus genomes revealed the formation of several clades through the heatmap clustering analysis of the ANI (Fig. 2). Based on the clustering, three distinct clades were identified. L. plantarum PA21 was found to be located in Clade A, specifically in a sub cluster consisting of other L. plantarum genomes originating from different isolation sources (Supplementary table S1). Notably, there are no patterns of clusters consists of similar isolation sources except for the first subcluster in Clade A which consist of genomes isolated from food-based sample namely, L. plantarum GR0128, L. plantarum subsp plantarum GR1184, L. plantarum subsp plantarum GR1186 and L. plantarum subsp plantarum GR1187.

The heatmap depicts the average nucleotide identity (ANI) whole-genome comparison of other 75 Lactiplantibacillus plantarum genomes corresponding to its isolation sources
Phylogenomic analysis
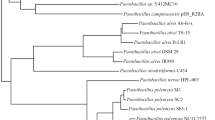
To delve deeper into the evolutionary relationship of L. plantarum PA21 with other Lactiplantibacillus genomes, phylogenomic analysis was performed (Fig. 3). The phylogenetic tree grouped the genomes of L. plantarum into clusters based on taxonomic classification. The tree displayed the formation of three main large clades (clades 1–3). Interestingly, the taxonomic placement in the tree was not consistent with ANI’s genome placement.

Phylogenomic maximum likelihood tree of 75 other Lactiplantibacillus genomes with L. plantarum PA21 and the outgroup, Bacillus cereus (accession: GCF_021655335.1). The branch lengths have been ignored and circled blue icon indicated bootstrap support for the depicted nodes in display. Binary data representing distinct shapes are used to denote the isolation source of the genome
Pangenome analysis
Pangenome analysis of the genome (n = 16) clustered together in clade 1 resulted in 4559 gene cluster of which 785 gene clusters (17.21%) are singletons (only present in one genome), 2418 gene clusters (53%) are core gene cluster (exist in all 16 genomes) and 1356 gene clusters (29.74%) are accessory gene clusters (exist in 2–15 genomes) (Fig. 4). Out of 2418 core gene clusters, 2132 gene clusters are single copy core genes. Based on ANI clustering as shown, the 16 genomes can be separated into two cluster (cluster A – dark brown and cluster B – light brown). Through functional enrichment analysis between these clusters (Supplementary Table S4-S6), several genes were found to be significantly enriched in either cluster. Thorough inspection of the genes suggests that some of the genes involved in metabolism such as Keratan sulphate degradation, Nitrate assimilation, Dissimilatory nitrate reduction and Entner-Doudoroff pathway. As vividly shown in Fig. 2, the habitat distribution of the Clade 1 does not suggest the functional enrichment of these set of genes are due to habitat exclusivity. Analysis of the singletons found in L. plantarum PA21 revealed about 44 gene clusters solely present in L. plantarum PA21 where only 24 gene clusters are annotated while the other 20 gene clusters failed to be annotated by any of gene functional database employed in this study (Supplementary Table S3).

Pangenome plot of 16 genomes of L. plantarum generated through Anvio. Dark brown and light brown layer represent genomes from two clusters demarcated based on the ANI clustering (top right corner). Presence/absence of gene clusters represent by the presence/absence of dark/light brown box. The genomes are clustered based on the presence/absence of the gene clusters
Bacteriocin and secondary metabolite clusters gene prediction
The genome information on L. plantarum PA21 was further investigated through bioinformatic analysis to gain genetic insights on their potential secondary metabolites. The antiSMASH and BAGEL4 server were used to screen for candidate BGCs. The screening revealed several BGCs that could encode antimicrobial compounds (Table 2). The L. plantarum PA21 was detected harbouring a single bacteriocin gene operon by BAGEL4 (Fig. 5) consisting of four bacteriocin structural genes (plnJK, plnN, plnA and plnEF). Further, two genes, HlyD and LanT were detected downstream of plantaricin E/F. Whereas, the antiSMASH database revealed four active metabolite regions.

Bacteriocin gene cluster organisation and its localization on genome map
Probiotic genes and virulence-associated genes
The likelihood of probiotic-associated genes in L. plantarum PA21 responsible for adherence, immunomodulatory activity, stress, bile, and acid tolerance was analysed (Table 3). The genome screening revealed a palette of probiotic genes with similarity hits of more than 70%. These included luxS, msa, lamA, srtA, clpC and dltB genes. Also, as anticipated, no virulence genes were detected to be present in the genome by the VFDB database.
Antimicrobial activity of L. plantarum PA21
The antimicrobial activity of L. plantarum PA21 CFS was tested against nine MRSA and 13 K. pneumoniae clinical isolates. The inhibition zone and antimicrobial activity for all the tested strains were tabulated in Table 4. The highest antimicrobial activity was observed against K. pneumoniae 7839 (1373.8 mm2/mL). Yet, the CFS was only able to inhibit three out of 13 K. pneumoniae isolates. High antimicrobial activity was also observed against MRSA 10 (1202.4 mm2/mL). Overall, the effect of CFS was greater on MRSA isolates than on K. pneumoniae isolates. Following this, CFS-susceptible strains, MRSA 10, MRSA 14, MRSA 20, K. pneumoniae 6163, K. pneumoniae 7839 and K. pneumoniae 0926 were arbitrarily selected for subsequent tests.
Effect of pH, enzyme and temperature on L. plantarum PA21 CFS
The antimicrobial activity of L. plantarum PA21 CFS upon various pH, enzymes and heat treatments was analysed against MRSA 10 (Table 5). L. plantarum PA21 CFS completely lost its antimicrobial activity from pH 5 to pH 10, however, antimicrobial activity remained between pH 2 to pH 4 compared to untreated CFS. Interestingly, the antimicrobial activity of CFS was lost when treated with proteinase K and trypsin. However, the CFS of L. plantarum PA21 remained active at all tested temperatures with a slight decrease in its inhibitory effects at 121 °C.
Discussion
The emergence of AMR in bacteria has by far been the biggest health challenge in modern medicine. With conventional antibiotics beginning to lose their potency against such pathogenic bacteria, natural antimicrobial compounds as alternatives are being greatly sought-after. Data from the current work suggests that L. plantarum PA21 postbiotic metabolites could be capitalised on as natural antimicrobial agents. The L. plantarum PA21 genome was first sequenced to understand its genetic makeup, identification, and function. In a comprehensive outlook, the genome size of L. plantarum PA21 (3.2 Mb) was within the range of most L. plantarum species (3.0 to 3.3 Mb) [11, 12]. Interestingly, the L. plantarum PA21 genome was devoid of a plasmid. Though it is common for most L. plantarum to carry one or more plasmids [13], some strains do not [14]. The absence of plasmid may have advantageous implications since it reduces the risk for horizontal gene transfer and the spread of antibiotic resistance genes to other bacteria. Also, this characteristic enables L. plantarum PA21 to be exploited for genetic manipulation studies.
Comparative genomic analysis employing phylogenomic tree inference and ANI provided valuable insights on the taxonomic status of the L. plantarum PA21 genome. The genome of interest, L. plantarum PA21 previously isolated from a tropical plant, was clustered together with other genomes of L. plantarum isolated from different isolation sources such as dairy, animal, and food. The analyses from this study were consistent with the findings from Liu et al. (2022) [15] whereby a wide genome association study of L. plantarum revealed that plant derived L. plantarum was dispersed relatively in the phylogenetic tree although the plant-based genome sample was small (n = 4) while ANI clustering corresponded with the three clades formation observation in a bigger sample size (n = 455). The clustering in this study, which is an uneven distribution of niche specific genomes in the phylogenetic tree, indicated a close evolutionary relationship between L. plantarum PA21 and other L. plantarum strains despite being isolated at different geographic locations and niches. Similar finding was also reported in the works of Carpi et al. (2022) [16] where each clade consisted of strain originates from multiple niches. Discrepancy between ANI and phylogenomic using single copy core genes can be attributed to the methodological differences. ANI measures overall genome similarity while the phylogenomic approach utilized the variation in the set of conserved genes. This study utilized pangenome analysis through Anvio of which each protein sequences from each selected genomes were blasted against one another and clustered into “gene cluster”. Large portion of the gene clusters were attributed to singletons (17%) suggesting the genomes of L. plantarum are quite adaptive with capacity to respond and evolve in response to diverse environmental conditions as depicted in the niche distribution of the L. plantarum. There are no patterns of gene cluster that are specific to niches as depicted in the works of Leulier et al. (2016) [17] suggesting L. plantarum have nomadic lifestyle.
There is ever-growing evidence that bacteriocins are among the bioactive compounds produced by L. plantarum, responsible for the inhospitable environment towards MDR bacteria [18, 19]. Thereby, the success of postbiotic metabolites in inhibiting MDR pathogens is potentially exemplified by the presence of bacteriocins. Genomic analysis of L. plantarum PA21 revealed the presence of four bacteriocins, akin to plantaricins (pln) in succession to genes encoding immunity proteins - common in most L. plantarum. The structural genes of plnE and plnF as well as plnJ and plnK placed adjacently to each other in the same operon operate based on a two-peptide bacteriocin system - plnEF and plnJK. In a classical study by Anderssen et al. (1998) [20], the complementary peptides identified from L. plantarum C11 were 1000 times more active against antagonistic strains when combined. The activation of plnEF and plnJK must first be preceded by plnA that interacts with the histidine protein kinase of a three-component regulatory system in the regulation of bacteriocin production. Nonetheless, with plnA acting as an ancillary peptide, little has been known about plnN which is purported to be a double glycine leader bacteriocin-like peptide [21]. Although the function of plnN was not explored further, its localization within the bacteriocin operon indicates the likelihood that it may be involved in bacteriocin regulation. Besides, the complete genome analysis also revealed the presence of BGCs such as terpenes, T3PKS, RiPP-like and cyclic-lactone inducers which are required for the synthesis of antimicrobial compounds. These BGCs were responsible for antagonising the growth of indicator strains, as documented in earlier investigations [22, 23]. So, the results of genome mining to some extent show that L. plantarum PA21 might have the potential to produce antimicrobial compounds.
Postbiotic metabolites are often the products of live bacterial metabolic processes, ergo the association of postbiotic metabolites to probiotic genes may likely be relevant [24]. Hence, a genome-based approach was adopted to evaluate the probiotic potential of L. plantarum PA21. Tolerance to stress, bile and acid is an important characteristic of any probiotic bacteria to weather through various unfavourable conditions. The physiological stress related genes in the L. plantarum PA21 genome included luxS and clpC. In L. plantarum, luxS gene has been an integral part in the biosynthesis of autoinducer-2 (AI-2) signalling molecules that mediates interspecies communication. Whilst commonly associated with stress tolerance, studies have also shown the substantial role of luxS in adhesion, acid and bile stress [25]. Most notably, is the ability to regulate bacteriocin production. The deletion of luxS altered the metabolic pathways thus affecting bacteriocin production in L. plantarum KLDS.0391 [26]. The clpC gene on the other hand is a member of regulatory ATPases implicated in stress tolerance and response. The response is effectuated through the ability to restore protein function as well as target misfolded proteins for degradation [27]. It was reported that gene encoding ClpC protein was also vital in bile and acid tolerance [28].
Gene coding for protein DltB which modulates the immune response through D-alanylation of lipoteichoic acid was discerned in the L. plantarum PA21 genome. The lipoteichoic acid is a component of the bacterial cell wall that has been associated with pro-inflammatory interaction through Toll-like receptor 2 (TLR2) [29]. Therefore, the dltB gene is an important entity in the immunomodulatory effects in L. plantarum PA21. In addition, the L. plantarum PA21 probiotic characteristic was strengthened with the presence of adhesion genes coding for Msa, LamA and SrtA proteins. The adhesive property in probiotic strains is of paramount importance, by virtue of its ability to mediate adhesion to cells and prevent adhesion and invasion of pathogens [30]. Overall, the probiotic-associated genes in L. plantarum PA21 were mostly pleiotropic genes in which one gene could have multiple functions and tolerance against different stressors and may not just be restricted to a single function. Furthermore, these genes are likely to have an indirect impact on the postbiotic metabolite production by affecting its overall growth and metabolism.
To substantiate the results from genome analysis, the antimicrobial potential of CFS was tested on MRSA and K. pneumoniae clinical isolates. To achieve a state of parity, chloramphenicol, a broad-spectrum antibiotic, was used as the positive control. The results clearly indicated that CFS of L. plantarum PA21 was more selective against Gram-positive bacteria than Gram-negative bacteria. This was also evident in several other studies where the CFS from Lactiplantibacillus sp. showed greater inhibition against Gram-positive bacteria such as MRSA than Gram-negative bacteria such as K. pneumoniae, Pseudomonas aeruginosa and Escherichia coli [31, 32]. One possible justification to account for the occurrence of such narrow inhibition spectrum by L. plantarum PA21 might be attributable to the differences in the MDR isolates’ resistance profiles and/or the composition of its CFS. Such a narrow spectrum might also be linked to the predicted bacteriocins in the L. plantarum PA21 genome as they are generally more potent against Gram-positive pathogens than Gram-negative pathogens [33]. This stems from the relevance of a thick peptidoglycan layer in Gram-positive bacteria that is usually porous in contrast to the outer lipid membrane-protected peptidoglycan layer in Gram-negative bacteria that serves as a formidable barrier. Bacteriocins therefore easily surmount this circumstance by disrupting the cell membrane and forming pores in Gram-positive bacteria [34].
Following that, the effects of heat, pH, and enzymes on bacteriocin activity of L. plantarum PA21 was analysed against MRSA 10. The antimicrobial activity of L. plantarum PA21 CFS was relatively unaffected across a wide range of temperature. Interestingly, when CFS was treated with proteolytic enzymes, the antimicrobial activity was completely lost, indicating the presence of proteinaceous substances in the CFS. In contrast, no antimicrobial activity by L. plantarum PA21 CFS was observed from pH 5 onwards. Thus, it is speculated that the acidity of CFS besides bacteriocin is partly responsible for the inhibitory effect as antimicrobial activity drastically declined in parallel to the increase in pH. These results corroborated with previous findings on L. plantarum NTU102 CFS that similarly exhibited a narrow pH range (pH 1 to pH 4) with unrecorded activity at neutral and alkaline pH while antimicrobial activity when treated with proteolytic enzymes such as pepsin, proteinase K and trypsin, significantly reduced [35]. Similarly, when Qian et al. (2020) [36] neutralized the CFS of L. plantarum strains, a significant reduction in antimicrobial activity was observed against the tested pathogens. Nevertheless, the primary antimicrobial effect exerted by the L. plantarum PA21 CFS on the clinical isolates may likely be due to the production of a range of antimicrobials such as organic acids which reduces the overall pH of the environment in tandem with other bioactive compounds such as bacteriocins. Therefore, all findings support that L. plantarum PA21 postbiotic metabolites could contribute to the antimicrobial potential against MDR pathogens and are worth bioprospecting.
Conclusions
An insight to the genome of L. plantarum PA21 revealed an assortment of bioactive compounds and probiotic features that could contribute to the antimicrobial properties against MDR pathogens. Whilst this study did not confirm the exact postbiotic metabolites involved, it did partially substantiate the presence of what would have been an array of metabolites that contributed to the antimicrobial activity of K. pneumoniae and MRSA clinical isolates. Ultimately, the insights gained on L. plantarum PA21 gene signatures and its regulatory mechanisms may be of assistance as potential antimicrobial agents in overcoming the AMR tide. Further research is necessary to fully characterise and elucidate the mechanisms underlying its various bioactivities against MDR pathogens and assess the safety and efficacy of L. plantarum PA21 in vitro for application as a postbiotic both as a standalone therapy or in combination with other treatments.
Materials and methods
Strains and growth conditions
The L. plantarum PA21 strain previously isolated from a tropical plant Pandanus amaryllifolius [37] has been deposited in the Microbial Culture Collection Unit (UNiCC) UPM (accession no: UPMC267) and was used as the test strain for postbiotic metabolite production while the MDR strains of K. pneumoniae and MRSA provided by Hospital Pengajar Universiti Putra Malaysia (HPUPM) were used as the indicator strains in this study. L. plantarum PA21 were grown in de Man, Rogosa and Sharpe (MRS) broth (Merck, Germany) at 37 °C for 24 h aerobically. The K. pneumoniae and MRSA strains were grown overnight in Luria Bertani (LB) broth and Brain Heart Infusion (BHI) broth, respectively at 37 °C for 24 h.
Genomic DNA isolation and whole genome sequencing (WGS)
L. plantarum PA21 was cultivated overnight in MRS broth at 37 °C and overnight culture was centrifuged the next day at 12,000 rpm for five minutes to obtain bacterial cells. The genomic DNA of L. plantarum PA21 was isolated using the PrimeWay Genomic DNA extraction kit (1st BASE, Malaysia). DNA quality was checked using a spectrophotometer (260/230 > 1.8 and 260/280 > 2.0; Implen, Germany) and 1% agarose electrophoresis gel. The whole genome sequencing of L. plantarum PA21 was carried out using the MinION MK1C (Oxford Nanopore Technologies, UK) with a R9.4.1 flow cell at Nanyang Technological University, Singapore. Prior to sequencing, the DNA was barcoded along with other microbial DNA through the PCR barcoding process using Rapid PCR Barcoding Kit’s protocol (SQK-RPB004). Base calling was done using guppy v3.2.2 (https://timkahlke.github.io/LongRead_tutorials/BS_G.html) to produce fastq files using the base calling model of dna_r9.4.1_450bps_hac.cfg.
Genome assembly and annotation
Demultiplexed fastq files were assembled using Flye (https://github.com/fenderglass/Flye), a long-read assembler to produce contigs. Further correction of the draft sequences from Flye was done using Medaka (https://github.com/nanoporetech/medaka). The genome was annotated in Pathosystems Resource Integration Center (PATRIC) [38].
Phylogenomic analysis and average nucleotide identity (ANI)
Phylogenomic analysis was done through GToTree pipeline [39]. In brief, representative genomes from the genus of Lactiplantibacillus and several complete genomes of L. plantarum were selected and downloaded using GToTree by listing the Refseq’s accession ID. The genome of L. plantarum PA21 and the outgroup, Bacillus cereus (accession: GCF_021655335.1) were included in the analysis by using the locally available fasta files. The ORF of these genomes were predicted using Prodigal v2.6.3 [40] and HMMsearch v3.3.2 [41] was used to search for an in-house set of HMM of Firmicutes. The outputs were then trimmed using trimAl v1.4.rev15 [42] and concatenated before parsing to Iqtree2 together with the partition file (http://www.iqtree.org) using ultrafast bootstrap approximation for phylogenetic estimation of maximum likelihood tree [43]. Taxonkit [44] was used to provide genus and species name of the input genome based on RefSeq accession ID. Tree was visualised using Interactive Tree of Life (itol) web server (https://itol.embl.de/) with branch length ignored to further visualise the clade separation.
Similar sets of genomes were subjected to ANI analysis using fastANI [45]. The heatmap and clustering were done using ANIclustermap (https://github.com/moshi4/ANIclustermap). Protologger version 1.0 was used mainly to infer environmental distribution utilizing the built-in Integrated Microbial NGS Platform (IMNGS) database of 1000 amplicon sequencing data [46, 47]. The output was parsed and visualized using matplotlib.
Pangenome analysis
Pangenome analysis was done using the program Anvi’o version 8 [48]. In brief, the genomes from Clade 1 were used as the input genomes. The fasta files were first formatted using anvi-reformat-fasta program before being converted into individual contigs database using anvi-gen-contigs-database. The contigs database were annotated with functional annotation from Ncbi-cogs, Pfam, Kofam and CAZyme using anvi-run-ncbi-cogs, anvi-run-pfams, anvi-run-kegg-kofams and anvi-run-cazymes, respectively. The individual contigs database were then combined into a genome storage and were used to create a pangenome using anvi-gen-genome-storage and anvi-pan-genome. The pangenome was analyzed using anvi-display-pan and was summarized using anvi-summarize.
Genome mining
The secondary metabolite prediction tools, BAGEL 4 [49] and antiSMASH [50] were mined to determine putative bacteriocin clusters and genes related to biosynthesis of antimicrobial proteins. The linear genome of L. plantarum PA21 mapped to putative bacteriocin clusters was constructed using ProKsee [51]. All tools were used at default settings. The image of gene operon was generated using DNA Features Viewer (https://github.com/Edinburgh-Genome-Foundry/DnaFeaturesViewer) based on BAGEL4 output.
Determination of probiotic and virulence genes
The annotated genome was screened for similar probiotic genes through the NCBI database. The selection of probiotic related genes was based on a previous report that defined the genetic determinants involved in probiotic Lactobacillus sp [30]. BLASTP was used for the identification of genes with similar amino acid sequences using a default configuration. Then, the annotated genes of L. plantarum PA21 were analysed through the virulence factors database (VFDB) [52] using Abricate (https://github.com/tseemann/abricate).
Antimicrobial activity
The antimicrobial activity by L. plantarum PA21 CFS was evaluated on clinical isolates through the agar well diffusion method described by Tagg and Mcgiven [53]. The overnight L. plantarum PA21 broth culture was centrifuged (4000 rpm, 10 min, 4 °C) to obtain cell-free supernatant (CFS). Briefly, 100 uL of CFS was pipetted into the wells (8–9 mm) of Mueller-Hinton (MH) agar seeded with indicator strains adjusted to an optical density (OD) of 0.08–0.1 at 595 nm. MRS broth only served as the negative control while antibiotic disk; chloramphenicol (30 ug) served as the positive control. The inhibition zones were measured after incubation at 37 °C for 24 h and the antimicrobial activity (AU) were reported as a unit area of the inhibition zone per unit volume sample loaded into well (mm2/mL) [54].
Effect of pH, enzyme and temperature on L. plantarum PA21 CFS
To determine the effect of pH, the CFS of L. plantarum PA21 was adjusted to pH values ranging from 2.0 to 10.0 using 1 M HCL and 1 M NaOH. The proteinaceous nature of antimicrobial substances in CFS was determined by incubating the CFS with 1 mg/mL proteinase K and 1 mg/mL trypsin (Sigma-Aldrich, USA) at 37 °C for two hours. The effect of temperature was investigated by incubating the CFS at 30, 50, 70, 100 and 121 °C for 15 min. Antimicrobial activity was then determined through the agar well diffusion method. Untreated CFS was used as a control.
Data analysis
The results in this study were presented as means ± standard deviation (n = 3). All statistical analysis was performed using GraphPad Prism 9 version 9.0 for Windows.
Data availability
The L. plantarum PA21 whole genome sequence reported in this study has been deposited at NCBI under Bioproject with the accession number PRJNA987396.
Abbreviations
- CFS:
-
Cell-free supernatant
- MDR:
-
Multi-drug resistant bacteria
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- ANI:
-
Average nucleotide identity
- BGC:
-
Biosynthetic gene cluster
References
Tsouklidis N, Kumar R, Heindl SE, Soni R, Khan S. Understanding the fight against resistance: hospital-acquired methicillin-resistant Staphylococcus Aureus vs. community-acquired methicillin-resistant Staphylococcus aureus. Cureus. 2020;12(6). https://doi.org/10.7759/cureus.8867.
Caneiras C, Lito L, Melo-Cristino J, Duarte A. Community-and hospital-acquired Klebsiella pneumoniae urinary tract infections in Portugal: virulence and antibiotic resistance. Microorganisms. 2019;7(5):138. https://doi.org/10.3390/microorganisms7050138.
World Health Organization. Global priority list of antibiotic-resistance bacteria to guide research, discovery, and development of new antibiotics, Geneva: World Health Organization. https://policycommons.net/artifacts/1818147/global-priority-list-of-antibiotic-resistant-bacteria-to-guide-research-discovery-and-development/2555608/. 2017. Accessed 5 July 2023.
Zheng J, Wittouck S, Salvetti E, Franz CM, Harris HM, Mattarelli P, O’toole PW, Pot B, Vandamme P, Walter J, Watanabe K, Wuyts S, Felis GE, Ganzle MG, Lebeer S. A taxonomic note on the genus Lactobacillus: description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int J Syst Evol Microbiol. 2020;70(4):2782–858. https://doi.org/10.1099/ijsem.0.004107.
Dawwam GE, Saber II, Yassin MH, Ibrahim HF. Analysis of different bioactive compounds conferring antimicrobial activity from Lactobacillus plantarum and Lactobacillus acidophilus with gas chromatography-mass spectrometry (GC-MS). Egypt Acad J Biol Sci. 2022;14(1):1–10. https://doi.org/10.21608/EAJBSG.2022.213620.
Chuah LO, Foo HL, Loh TC, Mohammed Alitheen NB, Yeap SK, Abdul Mutalib NE, Rahim RA, Yusoff K. Postbiotic metabolites produced by Lactobacillus plantarum strains exert selective cytotoxicity effects on cancer cells. BMC Complement Altern Med. 2019;19:1–12. https://doi.org/10.1186/s12906-019-2528-2.
Rather IA, Choi SB, Kamli MR, Hakeem KR, Sabir JS, Park YH, Hor YY. Potential adjuvant therapeutic effect of Lactobacillus plantarum probio-88 postbiotics against SARS-COV-2. Vaccines. 2021;9(10):1067. https://doi.org/10.3390/vaccines9101067.
Hossain MI, Mizan MFR, Roy PK, Nahar S, Toushik SH, Ashrafudoulla M, Jahid IK, Lee J, Ha SD. Listeria monocytogenes biofilm inhibition on food contact surfaces by application of postbiotics from Lactobacillus curvatus B. 67 and Lactobacillus plantarum M. 2. Food Res Int. 2021;148:110595. https://doi.org/10.1016/j.foodres.2021.110595.
Medema MH, Kottmann R, Yilmaz P, Cummings M, Biggins JB, Blin K, Zhang C. Minimum information about a biosynthetic gene cluster. Nat Chem Biol. 2015;11(9):625–31. https://doi.org/10.1038/nchembio.1890.
Albarano L, Esposito R, Ruocco N, Costantini M. Genome mining as new challenge in natural products discovery. Mar Drugs. 2020;18(4):199. https://doi.org/10.3390/md18040199.
Kleerebezem M, Boekhorst J, Van Kranenburg R, Molenaar D, Kuipers OP, Leer R, Tarchini R, Peters SA, Sandbrick HM, Fiers MWE, Stiekema W, Lankhorst RMK, Bron PA, Hoffer SM, Groot MNN, Kerkhoven R, de Vries M, Ursing B, de Vos WM, Siezen RJ. Complete genome sequence of Lactobacillus plantarum WCFS1. PNAS. 2003;100(4):1990–5. https://doi.org/10.1073/pnas.033770410.
Nikodinoska I, Makkonen J, Blande D, Moran C. Whole genome sequence data of lactiplantibacillus plantarum IMI 507027. Data Br. 2022;42:108025. https://doi.org/10.1016/j.dib.2022.108025.
McLeod A, Fagerlund A, Rud I, Axelsson L. Large plasmid complement resolved: complete genome sequencing of Lactobacillus plantarum MF1298, a candidate probiotic strain associated with unfavorable effect. Microorganisms. 2019;7(8):262. https://doi.org/10.3390/microorganisms7080262.
Axelsson L, Rud I, Naterstad K, Blom H, Renckens B, Boekhorst J, Kleerebezem M, Hijum SV, Siezen RJ. Genome sequence of the naturally plasmid-free Lactobacillus plantarum strain NC8 (CCUG 61730). J Bacteriol. 2012;194(9). https://doi.org/10.1128/JB.00141-12.
Li K, Wang S, Liu W, Kwok LY, Bilige M, Zhang W. Comparative genomic analysis of 455 lactiplantibacillus plantarum isolates: Habitat-specific genomes shaped by frequent recombination. Food Microbiol. 2022;104:103989. https://doi.org/10.1016/j.fm.2022.103989.
Carpi FM, Coman MM, Silvi S, Picciolini M, Verdenelli MC, Napolioni V. Comprehensive pan-genome analysis of lactiplantibacillus plantarum complete genomes. J Appl Microbiol. 2022;132(1):592–604. https://doi.org/10.1111/jam.15199.
Martino ME, Bayjanov JR, Caffrey BE, Wels M, Joncour P, Hughes S, Gillet B, Kleerebezem M, Hijum SAFT, Leulier F. Nomadic lifestyle of Lactobacillus plantarum revealed by comparative genomics of 54 strains isolated from different habitats. Environ Microbiol. 2016;18(12):4974–89. https://doi.org/10.1111/1462-2920.13455.
Hassan MU, Nayab H, Rehman TU, Williamson MP, Haq KU, Shafi N, Shafique F. Characterisation of bacteriocins produced by Lactobacillus spp. isolated from the traditional Pakistani yoghurt and their antimicrobial activity against common foodborne pathogens. BioMed Res Int. 2020;2020. https://doi.org/10.1155/2020/8281623.
Zhu X, Zhao Y, Sun Y, Gu Q. Purification and characterisation of plantaricin ZJ008, a novel bacteriocin against Staphylococcus spp. from Lactobacillus plantarum ZJ008. Food Chem. 2014;165:216–23. https://doi.org/10.1016/j.foodchem.2014.05.034.
Anderssen EL, Diep DB, Nes IF, Eijsink VG, Nissen-Meyer J. Antagonistic activity of Lactobacillus plantarum C11: two new two-peptide bacteriocins, plantaricins EF and JK, and the induction factor plantaricin A. Appl Environ Microbiol. 1998;64(6):2269–72. https://doi.org/10.1128/AEM.64.6.2269-2272.1998.
Diep DB, Håvarstein LS, Nes IF. Characterization of the locus responsible for the bacteriocin production in Lactobacillus plantarum C11. J Bacteriol. 1996;178(15):4472–83. https://doi.org/10.1128/jb.178.15.4472-4483.1996.
Tenorio-Salgado S, Castelán‐Sánchez HG, Dávila‐Ramos S, Huerta‐Saquero A, Rodríguez‐Morales S, Merino‐Pérez E, de la Fuente LFR, Solis-Pereira SE, Perez-Rueda E, Lizama‐Uc G. Metagenomic analysis and antimicrobial activity of two fermented milk kefir samples. Microbiologyopen. 2021;10(2):e1183. https://doi.org/10.1002/mbo3.1183.
Hegemann JD, Van Der Donk WA. Investigation of substrate recognition and biosynthesis in class IV lanthipeptide systems. J Am Chem Soc. 2018;140(17):5743–54. https://doi.org/10.1021/jacs.8b01323.
Salminen S, Collado MC, Endo A, Hill C, Lebeer S, Quigley EM, Sanders ME, Shamir R, Swann JR, Szajewska H, Vinderola G. The International Scientific Association of Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of postbiotics. Nat Rev Gastroenterol Hepatol. 2021;18(9):649–67. https://doi.org/10.1038/s41575-021-00440-6.
Jia FF, Zheng HQ, Sun SR, Pang XH, Liang Y, Shang JC, Zhu ZT, Meng XC. Role of luxS in stress tolerance and adhesion ability in Lactobacillus plantarum KLD. 0391. BioMed Res Int. 2018;2018. https://doi.org/10.1155/2018/4506829.
Jia FF, Pang XH, Zhu DQ, Zhu ZT, Sun SR, Meng XC. Role of the luxS gene in bacteriocin biosynthesis by Lactobacillus plantarum KLDS1. 0391: a proteomic analysis. Sci Rep. 2017;7(1):13871. https://doi.org/10.1038/s41598-017-13231-4.
Corcoran BM, Stanton C, Fitzgerald G, Ross RP. Life under stress: the probiotic stress response and how it may be manipulated. Curr Pharm Des. 2008;14(14):1382–99.
Goel A, Halami PM, Tamang JP. Genome analysis of Lactobacillus plantarum isolated from some Indian fermented foods for bacteriocin production and probiotic marker genes. Front Microbiol. 2020;40. https://doi.org/10.3389/fmicb.2020.00040.
Lebeer S, Vanderleyden J, De Keersmaecker SC. Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. Nat Rev Microbiol. 2010;8(3):171–84. https://doi.org/10.1038/nrmicro2297.
Lebeer S, Vanderleyden J, De Keersmaecker SC. Genes and molecules of lactobacilli supporting probiotic action. Microbiol Mol Biol Rev. 2008;72(4):728–64. https://doi.org/10.1128/mmbr.00017-08.
Bazireh H, Shariati P, Azimzadeh Jamalkandi S, Ahmadi A, Boroumand MA. Isolation of novel probiotic Lactobacillus and Enterococcus strains from human salivary and fecal sources. Front Microbiol. 2020;11:597946. https://doi.org/10.3389/fmicb.2020.597946.
Zavistanaviciute P, Lele V, Antanaitis R, Televičius M, Ruzauskas M, Zebeli Q, Bartkiene E. Separate and synergic effects of Lactobacillus uvarum LUHSS245 and arabinogalactan on the in vitro antimicrobial properties as well as on the fecal and metabolic profile of newborn calves. Animals. 2020;10(4):593. https://doi.org/10.3390/ani10040593.
Bédard F, Hammami R, Zirah S, Rebuffat S, Fliss I, Biron E. Synthesis, antimicrobial activity and conformational analysis of the class IIa bacteriocin pediocin PA-1 and analogs thereof. Sci Rep. 2018;8(1):9029. https://doi.org/10.1038/s41598-018-27225-3c.
Du H, Zhou L, Lu Z, Bie X, Zhao H, Niu YD, Lu F. Transcriptomic and proteomic profiling response of methicillin-resistant Staphylococcus aureus (MRSA) to a novel bacteriocin, plantaricin GZ1-27 and its inhibition of biofilm formation. App Microbiol Biotechnol. 2020;104:7957–70. https://doi.org/10.1007/s00253-020-10589-w.
Lin TH, Pan TM. Characterization of an antimicrobial substance produced by Lactobacillus plantarum NTU 102. J Microbiol Immunol Infect. 2019;52(3):409–17. https://doi.org/10.1016/j.jmii.2017.08.003.
Qian Z, Zhao D, Yin Y, Zhu H, Chen D. Antibacterial activity of Lactobacillus strains isolated from Mongolian yogurt against Gardnerella vaginalis. BioMed Res Int. 2020;2020. https://doi.org/10.1155/2020/3548618.
Jalilsood T, Baradaran A, Song AAL, Foo HL, Mustafa S, Saad WZ, Yusoff K, Rahim RA. Inhibition of pathogenic and spoilage bacteria by a novel biofilm-forming Lactobacillus isolate: a potential host for the expression of heterologous proteins. Microb Cell Factories. 2015;14:1–14. https://doi.org/10.1186/s12934-015-0283-8.
Davis JJ, Wattam AR, Aziz RK, Brettin T, Butler R, Butler RM, Chlenski P, Conrad N, Dickerman A, Dietrich EM, Gabbard JL, Gerdes S, Guard A, Kenyon RW, Machi D, Mao C, Murphy-Olson D, Nguyen M, Nordberg EK, Olsen GJ, Olson RD, Overbeek JC, Overbeek R, Parrello B, Pusch GD, Shukla M, Thomas C, VanOeffelen M, Vonstein V, Warren AS, Xia F, Xie D, Yoo H, Stevens R. The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res. 2020;48(D1):D606–12. https://doi.org/10.1093/nar/gkz943.
Lee MD. GToTree: a user-friendly workflow for phylogenomics. Bioinform. 2019;35(20):4162–4. https://doi.org/10.1093/bioinformatics/btz188.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:1–11. https://doi.org/10.1186/1471-2105-11-119.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7(10):e1002195. https://doi.org/10.1371/journal.pcbi.1002195.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinform. 2019;25(15):1972–3. https://doi.org/10.1093/bioinformatics/btp348.
Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5(3):e9490. https://doi.org/10.1371/journal.pone.0009490.
Shen W, Re H. TaxonKit: a practical and efficient NCBI taxonomy toolkit. J Genet Genom. 2021;48(9):844–50. https://doi.org/10.1016/j.jgg.2021.03.006.
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9(1):5114. https://doi.org/10.1038/s41467-018-07641-9.
Hitch TC, Riedel T, Oren A, Overmann J, Lawley TD, Clavel T. Automated analysis of genomic sequences facilitates high-throughput and comprehensive description of bacteria. ISME Comm. 2021;1(1):16. https://doi.org/10.1038/s43705-021-00017-z.
Lagkouvardos I, Joseph D, Kapfhammer M, Giritli S, Horn M, Haller D, Clavel T. IMNGS: a comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci Rep. 2016;6(1):33721. https://doi.org/10.1038/srep33721.
Eren AM, Kiefl E, Shaiber A, Veseli I, Miller SE, Schechter MS, Fink I, Pan JN, Yousef M, Fogarty EC, Trigodet F, Watson AR, Esen OC, Moore RM, Clayssen Q, Lee MD, Kivenson V, Graham ED, Merrill BD, Karkman A, Blankenberg D, Eppley JM, Sjodin A, Scott JJ, Campos XV, Mckay LJ, McDaniel EA, Stevens SLR, Anderson RE, Fuessel J, Fernandez-Guerra A, Maignien L, Delmont TO, Willis AD. Community-led, integrated, reproducible multi-omics with anvi’o. Nat Microbiol. 2021;6(1):3–6. https://doi.org/10.1038/s41564-020-00834-3.
van Heel AJ, de Jong A, Song C, Viel JH, Kok J, Kuipers OP. BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018;46(W1):W278–81. https://doi.org/10.1093/nar/gky383.
Medema MH, Blin K, Cimermancic P, De Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39(suppl_2):W339–46. https://doi.org/10.1093/nar/gkr466.
Grant JR, Enns E, Marinier E, Mandal A, Herman EK, Chen CY, Graham M, Domselaar GV, Stothard P. Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023;gkad326. https://doi.org/10.1093/nar/gkad326.
Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, Jin Q. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33(suppl_1):D325–8. https://doi.org/10.1093/nar/gki008.
Tagg J, McGiven A. Assay system for bacteriocins. Appl Microbiol. 1971;21(5):943–943. https://doi.org/10.1128/am.21.5.943-943.1971.
Abbasiliasi S, Tan JS, Tengku Ibrahim TA, Ramanan RN, Vakhshiteh F, Mustafa S, Ariff AB. Isolation of Pediococcus acidilactici Kp10 with ability to secrete bacteriocin-like inhibitory substance from milk products for applications in food industry. BMC Microbiol. 2012;12:260. https://doi.org/10.1186/1471-2180-12-260.
Acknowledgements
The authors would like to thank Federico’s Lauro’s laboratory at Nanyang Technological University, Singapore for providing the facilities for genomic library preparation and sequencing.
Funding
This study was supported by the Ministry of Higher Education, Malaysia, through the Fundamental Research Grant Scheme (FRGS/1/2021/STG01/UPM/02/7; Project ID:19764). Sharleen Livina Isaac is the recipient of the Graduate Research Fellowship (GRF), UPM.
Author information
Authors and Affiliations
Contributions
Conceptualization, W.N.I.W.A.K.; Methodology, S.L.I.; Software, A.M.H. and A.Z.A.M.; Validation, W.N.I.W.A.K., A.M.H., A.A.-L.S. and R.A.R.; Formal Analysis, S.L.I., A.Z.A.M. F.S.R. and N.S.H.; Investigation, S.L.I., N.S.H. and A.Z.A.M.; Resources, W.N.I.W.A.K.; Data Curation, S.L.I.; Writing – Original Draft Preparation, S.L.I.; Writing – Review & Editing, W.N.I.W.A.K., A.M.H., A.A.-L.S. and R.A.R; Visualization, S.L.I.; Supervision, W.N.I.W.A.K.; Project Administration, W.N.I.W.A.K.; Funding Acquisition, W.N.I.W.A.K. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants performed by any of the authors.
Permission
We hereby confirm that the necessary permission to utilize L. plantarum PA21 strain isolated from Pandanus amaryllifolius in this study from Prof Raha Abdul Rahim has been obtained.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Isaac, S.L., Abdul Malek, A.Z., Hazif, N.S. et al. Genome mining of Lactiplantibacillus plantarum PA21: insights into its antimicrobial potential. BMC Genomics 25, 571 (2024). https://doi.org/10.1186/s12864-024-10451-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10451-7