
Abstract
Background
Diabetic foot ulcer (DFU) is one of the most common and severe complications of diabetes, with vascular changes, neuropathy, and infections being the primary pathological mechanisms. Glutamine (Gln) metabolism has been found to play a crucial role in diabetes complications. This study aims to identify and validate potential Gln metabolism biomarkers associated with DFU through bioinformatics and machine learning analysis.
Methods
We downloaded two microarray datasets related to DFU patients from the Gene Expression Omnibus (GEO) database, namely GSE134431, GSE68183, and GSE80178. From the GSE134431 dataset, we obtained differentially expressed Gln-metabolism related genes (deGlnMRGs) between DFU and normal controls. We analyzed the correlation between deGlnMRGs and immune cell infiltration status. We also explored the relationship between GlnMRGs molecular clusters and immune cell infiltration status. Notably, WGCNA to identify differentially expressed genes (DEGs) within specific clusters. Additionally, we conducted GSVA to annotate enriched genes. Subsequently, we constructed and screened the best machine learning model. Finally, we validated the predictions' accuracy using a nomogram, calibration curves, decision curve analysis (DCA), and the GSE134431, GSE68183, and GSE80178 dataset.
Results
In both the DFU and normal control groups, we confirmed the presence of deGlnMRGs and an activated immune response. From the GSE134431 dataset, we obtained 20 deGlnMRGs, including CTPS1, NAGS, SLC7A11, GGT1, GCLM, RIMKLA, ARG2, ASL, ASNS, ASNSD1, PPAT, GLS2, GLUD1, MECP2, ASS1, PRODH, CTPS2, ALDH5A1, DGLUCY, and SLC25A12. Furthermore, two clusters were identified in DFU. Immune infiltration analysis indicated the presence of immune heterogeneity in these two clusters. Additionally, we established a Support Vector Machine (SVM) model based on 5 genes (R3HCC1, ZNF562, MFN1, DRAM1, and PTGDS), which exhibited excellent performance on the external validation datasetGSE134431, GSE68183, and GSE80178 (AUC = 0.929).
Conclusion
This study has identified five Gln metabolism genes associated with DFU, revealing potential novel biomarkers and therapeutic targets for DFU. Additionally, the infiltration of immune-inflammatory cells plays a crucial role in the progression of DFU.
Similar content being viewed by others

Introduction
Diabetic foot ulcer (DFU) is a severe complication of diabetes. Non-healing and persistent inflammation are hallmarks of chronic diabetic ulcers and significant factors contributing to the difficulty of DFU healing. Among approximately 150 million diabetes patients worldwide, 15% to 20% develop foot ulcers, and 40% to 80% of these patients experience ulceration combined with diabetic foot infections [1]. Non-healing DFU generate a significant socioeconomic burden, estimated to cost $4 billion annually, with the cost of each amputation surgery potentially exceeding $53,500 [2]. In the clinical management of DFU, various approaches are used, including intravenous administration of antibiotics and local treatments such as silver ion dressings, insulin topical application, and anti-inflammatory gels. However, long-term use of these treatments can lead to drug resistance and even delay wound healing [3, 4]. Therefore, researching the molecular mechanisms of DFU and developing new therapies that can suppress chronic wound inflammation are crucial for improving treatment outcomes and prognosis for patients with DFU.
Wound healing is a highly complex and regulated biological process. It has been reported that immune-inflammatory regulation plays a crucial role in several stages of chronic wound healing. Neutrophils are the first inflammatory white blood cells to migrate to the wound site, where they eliminate invading pathogens through various mechanisms and initiate subsequent stages of inflammation and non-inflammatory responses [5]. Macrophages play a role in hemostasis, and pro-inflammatory M1 macrophages infiltrate after injury to clear bacteria, dead cells, and debris from the wound site [6]. During the phase of wound healing characterized by proliferation and repair, M2 polarization stimulates the movement and growth of fibroblasts, keratinocytes, and endothelial cells to facilitate the restoration of the dermal and epidermal layers as well as the vascular system. Recent research has highlighted the detrimental effects of lacking lymphocytes in diabetic mice, further deteriorating wound healing. This suggests that when there is a deficiency in innate immune regulatory function, an imbalance in M1 polarization, inadequate angiogenesis, and reduced wound healing can be exacerbated [7].
Glutamine (Gln) is the predominant amino acid found in the blood, and cultured cancer cells efficiently take it up. Researchers have extensively investigated glutamine's functions, including its contribution to aerobic glycolysis, support of the TCA cycle, and provision of reduced carboxylation for lipid synthesis [8]. Furthermore, the metabolism of glutamine is pivotal in enhancing cell growth and viability through its ability to mitigate oxidative stress and preserve the integrity of mitochondrial membranes [9]. Gln serves as an energy source for both tumor cells and immune cells [10]. It is worth noting that M2 macrophages and naive macrophages have different utilization rates of Gln, which affects their inflammatory phenotype. Reduced metabolism of Gln favors pro-inflammatory M1 macrophages [11]. Studies have shown that manipulating glutamine metabolism presents a promising strategy to transition tumor-associated macrophages from an M2 phenotype to an M1 phenotype, thereby bolstering anti-tumor inflammatory immune responses. Moreover, the metabolism of glutamine also exerts an impact on Th1 cell differentiation and the activation of effector T cells, underscoring its potential to reshape the tumor microenvironment and enhance the efficacy of immunotherapy [12]. Although the combination of targeting glutamine along with immunotherapy shows significant promise in oncology, our comprehension of the precise role of glutamine metabolism in immunogenicity and immunotherapy remains constrained. Considering the existing research void, our study seeks to thoroughly examine the interplay between glutamine metabolism and immunotherapy within the framework of DFU. Nonetheless, the exact mechanisms underlying the pathogenesis of DFU remain elusive.
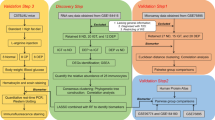
With the development of bioinformatics and machine learning [13, 14], several previous studies have utilized the GEO database to analyze relevant targets of DFU [15, 16]. Hence, we propose that Gln-metabolism related genes (GlnMRGs) play a significant role in DFU development. In this study, unsupervised clustering analysis was used to identify two distinct clusters based on the expression matrix of GlnMRGs. Machine learning models were then constructed using the key modules of DFU and the two clusters of WGCNA [17, 18], and the key models were determined based on the diagnostic sensitivity of the models [19]. This study aims to clarify the potential of GlnMRGs in enhancing immune diagnostics and therapeutic approaches for the management of DFU [20, 21]. The flowchart of this study is illustrated in Fig. 1.

Flow chart of this study
Materials and methods
Raw data processing
The analysis utilized the Gene Expression Omnibus (GEO) datasets GSE134431, GSE80178, and GSE68183. GSE134431 was used as the training set, while GSE134431, GSE80178 and GSE68183 served as the validation set. GSE68183 contains samples from 3 DF patients and 3 normal skin samples. GSE80178 contains samples from 9 DF patients and 3 normal skin samples [22]. GSE134431 contains ulcer samples from 13 DF patients and skin samples from 8 DF patients [23]. The dataset of GlnMRGs is obtained from MSigDB. The information for all GEO datasets is presented in Table 1.
Data filtering and processing
Precise mRNA data was acquired by applying Perl-based matching and sorting to the transcript data. Subsequently, for GSE134431, data normalization was performed.
The raw data from the gene expression matrix of the GEO dataset was processed and normalized using the R package “limma” (version 3.52.4). The SVA package was used to correct for batch effects between GSE134431, GSE68183, and GSE80178.
Cluster analysis of DFU patients
We determined the ideal cluster count by assessing a combination of criteria, including the cumulative distribution function curve, consensus clustering score, and consistency matrix. For this study, we established a maximum cluster count of k = 9.
Immune cell infiltration
Immune cell composition in DFU tissue was analyzed using CIBERSORT. We used the limma package to construct barplots and corplots to display the results of immune cells. CIBERSORT was used to obtain the deconvolution p-values for each sample through Monte Carlo sampling. A transcriptional feature matrix of 22 immune cells was used for computational simulation. The sum of the percentages of the 22 immune cells in each sample is set to 1. Here, we set the number of computational simulations to 1,000, and samples with a p-value < 0.05 were identified as precise immune cell subtypes.
Enrichment analysis
We utilized Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) to investigate biological functions and pathways. We employed R to assess how the differentially expressed GlnMRGs influence biological processes (BP), molecular functions (MF), and cellular components (CC), with this analysis performed using the Gene Set Variation Analysis (GSVA) method. GSVA scores were calculated using the R package “limma” (version 3.52.4), and |t value of GSVA scores| > 2 were considered to be significantly altered.
Co-expression gene identification
The Weighted Gene Co-Expression Network Analysis (WGCNA) algorithm is used to classify genes and identify relationships between modules and features.. A co-expression network was constructed using the top 25% variable genes from dataset GSE134431. The dynamic tree-cutting method with a threshold of 0.25 was employed to merge modules. Finally, we mapped the modules with the strongest correlation from the two classification methods.
Building prediction models based on multiple machine learning methods
The identification of cluster-specific GlnMRGs involves the use of WGCNA and the cross-analysis of DEGs within gene clusters. Vnnmap is used for visualizing overlapping genes. We used the “caret” R package to build machine learning models based on two different GlnMRG clusters, including Generalized Linear Model (GLM), Extreme Gradient Boosting (XGB), Support Vector Machine (SVM), and Random Forest (RF). GLM establishes the mathematical expectation of the response variable through a link function and can predict the relationship between linear combinations of variables [24]. XGB can be understood as a parallel prediction model with multiple trees. It iterates continuously, generating a new tree in each iteration, thereby ensuring that the predicted values remain close to the true values [25]. SVM is a type of generalized linear classifier that performs binary data classification through supervised learning. It is unparalleled in addressing small sample sizes, non-linearity, and high-dimensional pattern recognition [26]. In addition, RF is an ensemble machine learning method that uses a variety of independent decision trees to predict classification or regression [27].
Using different clusters as response variables, DEGs specific to selected clusters are chosen as explanatory variables. The DFU samples are randomly divided into training and validation sets in a 7:3 ratio. The “caret” R package automatically adjusts the parameters of these machine learning models through grid search, using default parameters for grid search. Subsequently, evaluation is performed using 5-fold cross-validation. Then, the “DALEX” package (version 2.4.2) is used to interpret four machine learning models and visualize the residual distribution and feature importance. The “pROC” package (version 1.18.0) is used to visualize the area under the receiver operating characteristic curve (AUC). Once the best machine learning model is determined, the top 5 key variables are available for predicting gene correlations in DFU.
Construction and independent validation analysis of column line chart model
Using the R package “rms” (version 6.3.0), a column line chart model was constructed, with corresponding scores for each predictive variable. The “Total Score” is the sum of the scores for the predictive variables. Additionally, calibration curve and decision curve analysis (DCA) were used to estimate the predictive ability of the column line chart model. The external datasets GSE134431, GSE68183, and GSE80178 were used to validate the model’s ability to differentiate between DFU and normal controls. Furthermore, the R package “pROC” was used to visualize the receiver operating characteristic (ROC) curve.
Drug-gene interactions
In the realm of disease diagnosis, the progress in bioinformatics has underscored the growing significance of creating biological models and pinpointing efficacious biomarkers. Nonetheless, the utilization of these biomarkers in clinical contexts remains of paramount importance. Predicting drug responses using informative markers is imperative for the future prevention and treatment of DFU. The utilization of the DGIdb database helps predict drug-gene interactions for cross-genetic and generated central genes in the RF model, enabling accurate drug prediction and providing references for therapeutic interventions.
Ethics approval and consent to participation
This manuscript does not pertain to a clinical trial; therefore, ethical approval and consent for participation are not relevant.
Results
Expression of GlnMRGs in DFU patients
We identified 20 differentially expressed GlnMRGs (deGlnMRGs). Among them, CTPS1, NAGS, SLC7A11, GGT1, GCLM, RIMKLA, ARG2, ASL, ASNS, ASNSD1, and PPAT showed higher expression levels in DFU testicular tissue. In contrast, the expression level of GLS2, GLUD1, MECP2, ASS1, PRODH, CTPS2, ALDH5A1, DGLUCY, and SLC25A12 in DFU testicular tissue was significantly lower than in normal controls (Fig. 2A, B). The chromosomal positions of the GlnMRGs were calculated and visualized in the form of circles (Fig. 2C). Subsequently, we performed correlation analysis on these genes (Fig. 2D, E). Most of these genes showed positive correlations with each other.

Identification of deGlnMRGs in DFU (A) The expression levels of GlnMRGs (B) Heatmap of deGlnMRGs (C) The location of GlnMRGs on chromosomes (D) Gene relationship network diagram of deGlnMRGs (E) Correlation analysis of deGlnMRGs. Red and green colors represent positive and negative correlations, respectively. The correlation coefficient was expressed as the area of the pie chart
Immune infiltration analysis
The immune environment plays a critical role in the occurrence and progression of DFU. The distribution of immune cells in various samples is shown in Fig. 3A. The immune cell differences between the DFU and control groups are depicted in Fig. 3B. Immune cell populations Mast cells activated and Neutrophils are higher in DFU compared to the control group, while population NK cells activated and T cells CD8 are lower in DFU compared to the control group. The correlation analysis between immune cells and GlnMRGs is shown in Fig. 3C.

Expression of Immune cells. A and B Expression of immune cells in different clusters. C Correlation between GlnMRGs and immune cells
Cluster analysis
When k is set to 2, the highest within-group correlation is observed, indicating that GlnMRGs can be used to divide DFU patients into two groups (Fig. 4A). Figure 4B demonstrates significant differences in principal component analysis (PCA) between these two clusters. We also examined the expression of GlnMRGs in different clusters based on this cluster analysis. The levels of CTPS1, SLC7A11, GLUL, GOT1, ASNS, PPAT, PRODH, CTPS2, and DGLUCY were found to have significant differences between the two groups (Fig. 4C, D). In addition, we also analyzed the results of immune cell infiltration based on the clusters (Fig. 4E, F).

Identification of GlnMRGs clusters in DFU (A) Consensus clustering matrix when k = 2. B PCA visualized the distribution of the two clusters. C Boxplots of GlnMRGs expressed between the two clusters. D Heatmap of the expression patterns of the GlnMRGs between the two clusters. E Relative abundance maps of 22 infiltrating immune cells between the two clusters. F Boxplots of immune infiltration differences between the two clusters
Functional enrichment analysis
Performing GSVA enrichment analysis using GlnMRGs. The pathway is mainly enriched in aminoacyl tRNA biosynthesis, steroid biosynthesis, and alpha linolenic acid metabolism (Fig. 5A). The GO analysis results include histone H4 K20 methylation, intestinal epithelial structure maintenance, and regulation of cell projection size (Fig. 5B).

Enrichment analysis for GlnMRGs. a KEGG. b GO
Gene module selection and construction of co-expression networks
We have built a co-expression network for both normal control and DFU patients using WGCNA, and identified key gene modules associated with DFU. We identified the co-expression gene modules under this condition (Fig. 6A). Following that, the dynamic cut algorithm yielded a total of 26 co-expression modules, each represented by distinct colors, and generated a TOM heat map (Fig. 6B,C,D). In addition, by applying the genes from these 26 modules, we analyzed the similarity and continuity of co-expression between module clinical features (normal control and DFU). We found that the red module had the strongest association with DFU, which included 222 hub genes (Fig. 6E). Furthermore, there is a positive correlation between the red module and the module-related genes (Fig. 6F).

Co-expression network of DEGs in DFU (A) Set soft threshold power (B) The cluster tree dendrogram of co-expression modules is shown in different colors (C) Cluster diagram of module eigengenes (D) TOM heatmap of 26 modules (E) Heatmap of correlation analysis of module eigengenes with clinical features. Rows and columns represent modules and clinical features, respectively (F) Scatter plot of the genetic significance of the blue module members with DFU
Furthermore, we analyzed key gene modules closely associated with Gln metabolism clusters using WGCNA. When the soft threshold parameter β is set to 12 and R^2 is 0.9, a scale-free network is constructed under this condition (Fig. 7A). Twenty-two modules were identified as important. The heatmap displays the TOM of all module-related genes (Fig. 7B,C,D). The examination of the association between modules and clinical characteristics (Cluster1 and Cluster2) underscored the importance of the pink module (Fig. 7E). Furthermore, the correlation analysis results indicate a significant positive correlation between the pink module and its corresponding HUB genes (Fig. 7F).

Co-expression network of DEGs between the two Gln clusters (A) Set soft threshold power (B) The cluster tree dendrogram of co-expression modules is shown in different colors (C) Cluster diagram of module eigengenes (D) TOM heatmap of 22 modules (E) Heatmap of correlation analysis of module eigengenes with clinical features. Rows and columns represent modules and clinical features, respectively (F) Scatter plot of the genetic significance of the turquoise module members with Cluster1
Construction of the model
Mapping the pink module genes of Gln metabolism clusters and the red module genes of DFU, a total of 63 cluster-specific GlnMRGs (Additional file 1: Appendix 1) were obtained (Fig. 8A,B). The residual distribution results of the four models indicate that SVM has the highest residual (Fig. 8B). Figure 8C ranks the top 10 significant feature variables for each model based on root mean square error. The ROC results of the four machine learning models indicate that the AUC value of SVM is 1.000 (Fig. 8D). Therefore, selecting SVM model (R3HCC1, ZNF562, MFN1, DRAM1, and PTGDS) (Fig. 8E) as the best model is because we believe it can best differentiate between different patient groups.

Construction of SVM, RF, XGB, and GLM machine models. A Crossover genes of the Gln clusters module and the DFU module. B The cumulative residual distribution of the four models. C Residual Boxplots of the four machine learning models, where the red dots indicate the root mean square of the residuals (D) ROC analysis of four machine learning models with fivefold cross-validation in the test set. E The important features in SVM, RF, XGB, and GLM
Evaluating machine learning models
We constructed line plots to evaluate the predictive performance of the SVM model (Fig. 9A). The calibration curve and DCA were used to evaluate the predictive accuracy of the line plot model. The calibration curve shows the minimum error between the actual DFU cluster risk and the predicted risk (Fig. 9B). Furthermore, DCA demonstrates that the line plot has high accuracy and can provide reference for clinical decision-making (Fig. 9C). Subsequently, we used the validation dataset GSE134431, GSE80178, and GSE68183 (Fig. 9D) to validate the model, and the ROC result showed an AUC of 0.929, indicating perfect discrimination. The immune-related analysis of the model genes is presented in Fig. 9E, providing an explanation for the immune function of the model.

Validation of a 5-gene-based SVM model. A Construction of a nomogram to predict DFU risk based on a 5-gene SVM model (B, C) Calibration curves (D) ROC of the 5-gene-based SVM model (E) SVM model Model immune-related heatmap
Drug-gene interactions
The all interacting genes were used for drug prediction (Additional file 1: Appendix 2).
Discussions
DFU represent a complex condition that is unlikely to be effectively addressed using a solitary medication or other intervention measures. The focus of modern drug therapy strategies is to accelerate the healing of chronic wounds and prevent the recurrence of DFU [28]. Over the last three decades, our comprehension of the molecular underpinnings of DFU has notably expanded. This has underscored the potential of biomarkers for the development of diagnostic tools, risk assessment, clinical trials, targeted therapies, and the discovery of novel drug targets [29, 30]. However, the pathogenesis of DFU is still unclear. Therefore, we hypothesize that there is a intricate connection between GlnMRGs and the occurrence of DFU. In our study, we explored our hypothesis through bioinformatics methods to aid in understanding DFU [31]. In this study, we have explored the potential of GlnMRGs as a biomarker in DFU, aiming to contribute to the diagnosis, detection, and treatment of DFU.
Gln metabolism, as an interesting regulatory node, is receiving increasing attention, and it varies in different clinical settings. Gln is among the most plentiful non-essential amino acids found in the body's circulation (amino acids synthesized by the body and not required in the diet), playing a role in nearly all biosynthetic pathways within rapidly dividing cells [32]. Furthermore, it acts as a source of nitrogen for the production of purines and pyrimidines and serves as a precursor for the synthesis of proteins and glutathione. Gln metabolism initiates with the enzyme glutaminase, which deaminates it to yield glutamate. Gln serves as a vital intermediate metabolite with diverse biosynthetic roles within cells [33]. Multiple studies have linked disrupted Gln metabolism to cancer development, and medications aimed at targeting Gln metabolism have received approval for treating various malignant tumors. As cancer advances from premalignant lesions to clinically detectable tumors and subsequently evolves into metastatic malignancies, metabolic requirements and characteristics may undergo alterations. Recent studies have emphasized the role of GlnMRGs in various non-tumor age-related diseases. For example, Wu et al. established metabolic characteristic models for Alzheimer's disease [12] and osteoporosis [34] related to glutamine metabolism. The physiological role of glutamine metabolism in the progression of DFU is not yet clear, and this could be an interesting research direction.
This study utilized unsupervised clustering analysis and further employed the expression profile of GlnMRGs to elucidate the diverse patterns of Gln regulation within GlnMRGs, thereby identifying two distinct clusters of GlnMRGs. In addition, machine learning models were constructed based on disease and key genes identified by WGCNA in the two distinct GlnMRGs clusters, which is an innovative approach in this study. With the progress and development of research, machine learning models are increasingly used for predicting DFU [35]. Compared to traditional univariate analysis, machine learning often employs a multivariate analysis approach that takes into account the relationships between variables. Therefore, machine learning models are more accurate and the results are more reliable. The “caret” R package that we used is a comprehensive machine learning toolkit designed to address prediction problems. Its key feature is the ability to quickly prepare all the necessary components, including data preprocessing, model training, and the entire process of model prediction [36]. We compared the prediction performance of four machine learning models: XGB, SVM, GLM, and RF. The SVM-based prediction model we constructed had very high predictive effectiveness in the test set (AUC = 0.833). In an externally validated dataset, the SVM model based on 5 genes accurately predicted DFU with an AUC of 0.929, demonstrating its good diagnostic value for DFU. Additionally, we constructed a column line chart model for diagnosing DFU using five genes: R3HCC1, ZNF562, MFN1, DRAM1, and PTGDS. Our results showed that the model had good predictive performance, indicating its potential for clinical application. In conclusion, the SVM model based on five genes for distinguishing DFU subtypes was satisfactory.
Using the SVM algorithm, we identified 5 central GlnMRGs (R3HCC1, ZNF562, MFN1, DRAM1, and PTGDS) and validated their diagnostic ability using an external dataset, indicating their potential impact on the pathogenesis of DFU. However, the pathways by which these genes might be linked to the control of particular transcription factors related to Gln management have yet to be elucidated. The R3HCC1 gene encodes a protein consisting of the R3H domain and a coiled-coil region known as coiled-coil containing 1. This protein is thought to exhibit nucleic acid-binding capabilities. Specifically, the R3H domain has the ability to bind to single-stranded DNA and RNA in a sequence-specific manner [37]. As an illustration, the R3H domain modulates the interaction between Rbs1 (a poly[A] mRNA binding protein) and RNA polymerase III [38]. However, it is unfortunate that research on R3HCC1 in DFU remains lacking. Mitochondria are highly dynamic organelles that engage in ongoing fusion and fission processes to preserve their appropriate morphology, a prerequisite for their regular functioning [39]. Mitochondrial fusion entails the combination of two mitochondria and is facilitated by the mitochondrial fusion protein (MFN)-1. MFN1 is situated on the outer mitochondrial membrane and participates in the initial phases of membrane fusion [40, 41]. Research has demonstrated that mice with a deficiency of MFN1 in their skeletal muscles display mitochondrial dysfunction and experience significant muscle atrophy [42]. Simultaneously, mitochondrial dynamics also govern cellular apoptosis [43]. Overexpression of MFN1 promotes mitochondrial fusion and maintains mitochondrial function, while knocking down MFN1 accelerates cellular apoptosis [44]. DRAM1 is a transmembrane protein characterized by six transmembrane domains and is predominantly situated within lysosomes [45, 46]. Being a target gene of p53, DRAM1 assumes a critical role in p53-dependent autophagy and cellular apoptosis [47]. Prostaglandins (PG) are a class of biologically active endogenous metabolites derived from arachidonic acid through the cyclooxygenase (COX) pathway. PTGDS, also known as PGD2, plays a crucial role in vascular function [48]. Research indicates that in diabetic wounds, there is a decrease in COX-1 expression, while COX-2 levels significantly increase [49]. It's important to highlight that a marked enhancement in wound healing is distinguished by a substantial elevation in the synthesis of PGE2/PGD2, which exhibits co-localization with the induced COX-1 within the newly formed tissue surrounding the wound [50].
According to research, DFU wounds exhibit characteristics such as high glucose levels, the accumulation of advanced glycation end-products, hypoxia, and ischemia. There is also a significant presence of mast cells activated, neutrophils, NK cells activated, and T cells CD8 in the local wound microenvironment. Under the complex interplay of numerous cytokines, this ultimately results in an unfavorable immune microenvironment that hinders the rapid repair of DFU wounds [51, 52]. Recently, the immune-inflammatory microenvironment has been identified as one of the reasons for the failure of DFU treatment [53]. The pathological features of an abnormal immune microenvironment in DFU primarily involve changes in the types and functions of immune cells, persistent presence of pro-inflammatory factors leading to sustained tissue damage, and local abnormal cell proliferation. This is responsible for the occurrence of chronic inflammation or acute exacerbations of chronic inflammation at DFU sites, which are fundamentally different from pure acute inflammation. Neutrophils are part of the innate immune system and undergo apoptosis after performing their functions at the wound site. Macrophages eventually phagocytose apoptotic neutrophils, providing a strong signal for the resolution of inflammation [54]. Neutrophils also play a crucial role in vascular formation and maturation. However, in the disrupted immune microenvironment of DFU, coupled with improper extracellular matrix regulation and the pro-inflammatory environment induced by high glucose and hypoxia at the wound site, neutrophils are continuously recruited. Excessive neutrophils hinder vascular formation and maturation, leading to altered vascular permeability and impaired function. This compromised vascular function fails to provide effective microcirculation, weakening wound contraction [55]. The abnormal immune-inflammatory microenvironment leads to impaired NK cell and T cell function, hinders granulation tissue formation, exacerbates inflammation, prevents keratinocytes from migrating and properly epithelializing the wound, delays tissue maturation, and results in the development of chronic non-healing wounds. To successfully treat DFU immune inflammation and restore a normal biological microenvironment at the wound site, both basic and clinical research on DFU must explore unique biomarkers from the perspective of immune-inflammatory interactions.
Research on biomarkers in the context of DFU is still relatively limited. Recently, bioinformatics analysis has become a valuable tool for exploring the intricate and complex connections between cell apoptosis and DFU [56, 57]. A comprehensive study has indicated and identified potential biomarkers associated with DFU through transcriptomics and proteomics bioinformatic models, and a study has shown that CXCL11, DDX60, IFI44, and IFI44L are key hub genes with the potential to serve as molecular targets for immunotherapy in DFU [58]. However, there are currently very few predictive model studies on Gln and DFU. This study, based on the research of Gln mechanisms, provides a reference for identifying effective immunotherapies in the treatment of DFU. Firstly, we collected comprehensive data on GlnMRGs from continuously updated GEO databases to expand upon early research. Secondly, GSE134431 was used as the primary dataset for analysis, and it was combined with GSE80178 and GSE68183 to validate the general pattern in the model. GO and KEGG analyses, as well as GSVA analysis, added credibility to this study. Lastly, there are currently almost no predictive models for GlnMRGs that can provide specific recommendations for future immunoinflammatory research or treatments based on Gln metabolism interference in DFU. This study constructed a diagnostic model for Glu and DFU through machine learning methods, and also combined with immune cell infiltration, enriching our understanding of their immune regulatory interactions. These findings, elucidated through computer algorithms, not only describe the general paradigm of the intricate interactions between Gln, DFU, and the immune system but also pave the way for integrating detailed molecular insights with clinical relevance on a new scientific trajectory. Furthermore, the continuous development of artificial intelligence offers valuable insights for healthcare practitioners and has the potential to shape a more nuanced understanding of DFU and treatment strategies in the future. This can foster rich clinical insights and guide exploration and treatments in the intertwined fields of immunotherapy and foot pathophysiology.
Translation
Although this study provides a theoretical foundation and research concept, our model still has some limitations. Firstly, the data used in the study is sourced from the GEO database, which poses challenges in determining the quality and reliability of the statistical data. To improve the quality and reliability of the statistical data, we selected GSE134431 as the primary dataset and utilized GSE13443, GSE80178, and GSE68183 for model validation, as these datasets have clear grouping. Secondly, a key challenge is the lack of fundamental understanding of the mRNA-level system related to Gln metabolism and DFU. Therefore, there is a lack of in-depth knowledge about its underlying mechanisms, and future research needs to design foundational experiments for further validation. Lastly, we can further investigate the selection of parameter values in our model, which can be done in future studies in conjunction with basic research to determine the optimal screening parameters for the model.
Conclusions
The development and progression of DFU result from intricate interactions among diverse targets, signaling pathways, and mechanisms, with regulatory processes that are synergistic and bidirectional. Gln metabolism impacts the synthesis of R3HCC1, ZNF562, MFN1, DRAM1, and PTGDS, and based on this, a diagnostic model was constructed. Future improvements involve increasing the quantity of data sources and conducting more scientific and clinical research to determine whether effective treatments can reduce the immunoinflammatory manifestations in DFU patients by modulating the targets and pathways of Gln metabolism. In conclusion, our research results provide broad potential biomarkers for DFU treatment strategies.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Liu Y, Yu M, LaMantia JN, et al. Associations between specialty care and improved outcomes among patients with diabetic foot ulcers. PLoS One. 2023;18(12):e0294813. https://doi.org/10.1371/journal.pone.0294813. Published 2023 Dec 19.
Sen CK. Human Wounds and its Burden: Updated 2022 Compendium of Estimates [published online ahead of print, 2023 Sep 27]. Adv Wound Care (New Rochelle). 2023. https://doi.org/10.1089/wound.2023.0150.
McIllhatton A, Lanting S, Lambkin D, Leigh L, Casey S, Chuter V. Reliability of recommended non-invasive chairside screening tests for diabetes-related peripheral neuropathy: a systematic review with meta-analyses. BMJ Open Diabetes Res Care. 2021;9(2):e002528. https://doi.org/10.1136/bmjdrc-2021-002528.
Orlando G, Prior Y, Reeves ND, Vileikyte L. Patient and Provider Perspective of Smart Wearable Technology in Diabetic Foot Ulcer Prevention: A Systematic Review. Medicina (Kaunas). 2021;57(12):1359. https://doi.org/10.3390/medicina57121359. Published 2021 Dec 13.
Wen Q, Liu D, Wang X, et al. A systematic review of ozone therapy for treating chronically refractory wounds and ulcers. Int Wound J. 2022;19(4):853–70. https://doi.org/10.1111/iwj.13687.
Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–55. https://doi.org/10.1038/nature12034.
Bechara N, Gunton JE, Flood V, Hng TM, McGloin C. Associations between Nutrients and Foot Ulceration in Diabetes: A Systematic Review. Nutrients. 2021;13(8):2576. https://doi.org/10.3390/nu13082576. Published 2021 Jul 27.
Li H, Wu Z, Zhang Y, Lu X, Miao L. Glutamine metabolism genes prognostic signature for stomach adenocarcinoma and immune infiltration: potential biomarkers for predicting overall survival. Front Oncol. 2023;13:1201297. https://doi.org/10.3389/fonc.2023.1201297. Published 2023 Jun 12.
Nandigama R, Upcin B, Aktas BH, Ergün S, Henke E. Restriction of drug transport by the tumor environment. Histochem Cell Biol. 2018;150(6):631–48. https://doi.org/10.1007/s00418-018-1744-z.
Yang WH, Qiu Y, Stamatatos O, Janowitz T, Lukey MJ. Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy. Trends Cancer. 2021;7(8):790–804. https://doi.org/10.1016/j.trecan.2021.04.003.
Oh MH, Sun IH, Zhao L, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest. 2020;130(7):3865–84. https://doi.org/10.1172/JCI131859.
Wu Z, Liu P, Huang B, et al. A novel Alzheimer’s disease prognostic signature: identification and analysis of glutamine metabolism genes in immunogenicity and immunotherapy efficacy. Sci Rep. 2023;13(1):6895. https://doi.org/10.1038/s41598-023-33277-x. Published 2023 Apr 27.
Wang W, Zhang L, Sun J, Zhao Q, Shuai J. Predicting the potential human lncRNA-miRNA interactions based on graph convolution network with conditional random field. Brief Bioinform. 2022;23(6):bbac463. https://doi.org/10.1093/bib/bbac463.
Zhang L, Yang P, Feng H, Zhao Q, Liu H. Using Network Distance Analysis to Predict lncRNA-miRNA Interactions. Interdiscip Sci. 2021;13(3):535–45. https://doi.org/10.1007/s12539-021-00458-z.
Huang Y, Wang D, Zhang W, et al. Identification of hub genes and pathways associated with cellular senescence in diabetic foot ulcers via comprehensive transcriptome analysis. J Cell Mol Med. https://doi.org/10.1111/jcmm.18043. Published online November 20, 2023.
Wang X, Jiang G, Zong J, et al. Revealing the novel ferroptosis-related therapeutic targets for diabetic foot ulcer based on the machine learning. Front Genet. 2022;13:944425. https://doi.org/10.3389/fgene.2022.944425. Published 2022 Sep 26.
Sun F, Sun J, Zhao Q. A deep learning method for predicting metabolite-disease associations via graph neural network. Brief Bioinform. 2022;23(4):bbac66. https://doi.org/10.1093/bib/bbac266.
Gao H, Sun J, Wang Y, et al. Predicting metabolite-disease associations based on auto-encoder and non-negative matrix factorization. Brief Bioinform. 2023;24(5):bbad259. https://doi.org/10.1093/bib/bbad259.
Wu Z, Gao Y, Cao L, Peng Q, Yao X. Purine metabolism-related genes and immunization in thyroid eye disease were validated using bioinformatics and machine learning. Sci Rep. 2023;13(1):18391. https://doi.org/10.1038/s41598-023-45048-9. Published 2023 Oct 26.
Chen Z, Zhang L, Sun J, Meng R, Yin S, Zhao Q. DCAMCP: A deep learning model based on capsule network and attention mechanism for molecular carcinogenicity prediction. J Cell Mol Med. 2023;27(20):3117–26. https://doi.org/10.1111/jcmm.17889.
Hu H, Feng Z, Lin H, et al. Gene function and cell surface protein association analysis based on single-cell multiomics data. Comput Biol Med. 2023;157:106733. https://doi.org/10.1016/j.compbiomed.2023.106733.
Ramirez HA, Pastar I, Jozic I, et al. Staphylococcus aureus Triggers Induction of miR-15B-5P to Diminish DNA Repair and Deregulate Inflammatory Response in Diabetic Foot Ulcers. J Invest Dermatol. 2018;138(5):1187–96. https://doi.org/10.1016/j.jid.2017.11.038.
Sawaya AP, Stone RC, Brooks SR, et al. Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing. Nat Commun. 2020;11(1):4678. https://doi.org/10.1038/s41467-020-18276-0. Published 2020 Sep 16.
Takada Y, Miyagi R, Takahashi A, Endo T, Osada N. A Generalized Linear Model for Decomposing Cis-regulatory, Parent-of-Origin, and Maternal Effects on Allele-Specific Gene Expression. G3 (Bethesda). 2017;7(7):2227–34. https://doi.org/10.1534/g3.117.042895. Published 2017 Jul 5.
Babajide Mustapha I, Saeed F. Bioactive Molecule Prediction Using Extreme Gradient Boosting. Molecules. 2016;21(8):983. https://doi.org/10.3390/molecules21080983. Published 2016 Jul 28.
Krause L, McHardy AC, Nattkemper TW, Pühler A, Stoye J, Meyer F. GISMO–gene identification using a support vector machine for ORF classification. Nucleic Acids Res. 2007;35(2):540–9. https://doi.org/10.1093/nar/gkl1083.
Rigatti SJ. Random Forest. J Insur Med. 2017;47(1):31–9. https://doi.org/10.17849/insm-47-01-31-39.1.
Chen J, Qin S, Liu S, et al. Targeting matrix metalloproteases in diabetic wound healing [published correction appears in Front Immunol. 2023 Sep 11;14:1287048]. Front Immunol. 2023;14:1089001. https://doi.org/10.3389/fimmu.2023.1089001. Published 2023 Feb 17.
Meng R, Yin S, Sun J, Hu H, Zhao Q. scAAGA: Single cell data analysis framework using asymmetric autoencoder with gene attention. Comput Biol Med. 2023;165:107414. https://doi.org/10.1016/j.compbiomed.2023.107414.
Wang T, Sun J, Zhao Q. Investigating cardiotoxicity related with hERG channel blockers using molecular fingerprints and graph attention mechanism. Comput Biol Med. 2023;153:106464. https://doi.org/10.1016/j.compbiomed.2022.106464.
Zhao M, Yu WX, Liu SJ, et al. Identification and immuno-infiltration analysis of cuproptosis regulators in human spermatogenic dysfunction. Front Genet. 2023;14:1115669. https://doi.org/10.3389/fgene.2023.1115669. Published 2023 Mar 29.
DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29(3):313–24. https://doi.org/10.1038/onc.2009.358.
Natarajan SK, Venneti S. Glutamine Metabolism in Brain Tumors. Cancers (Basel). 2019;11(11):1628. https://doi.org/10.3390/cancers11111628. Published 2019 Oct 24.
Wang L, Deng C, Wu Z, Zhu K, Yang Z. Bioinformatics and machine learning were used to validate glutamine metabolism-related genes and immunotherapy in osteoporosis patients. J Orthop Surg Res. 2023;18(1):685. https://doi.org/10.1186/s13018-023-04152-2. Published 2023 Sep 14.
Lu Y, Liu X, Zhao J, et al. Single-cell profiling reveals transcriptomic signatures of vascular endothelial cells in non-healing diabetic foot ulcers. Front Endocrinol (Lausanne). 2023;14:1275612. https://doi.org/10.3389/fendo.2023.1275612. Published 2023 Dec 1.
Lee C, Kim H. Machine learning-based predictive modeling of depression in hypertensive populations. PLoS One. 2022;17(7):e0272330. https://doi.org/10.1371/journal.pone.0272330. Published 2022 Jul 29.
Grishin NV. The R3H motif: a domain that binds single-stranded nucleic acids. Trends Biochem Sci. 1998;23(9):329–30. https://doi.org/10.1016/s0968-0004(98)01258-4.
Cieśla M, Turowski TW, Nowotny M, Tollervey D, Boguta M. The expression of Rpb10, a small subunit common to RNA polymerases, is modulated by the R3H domain-containing Rbs1 protein and the Upf1 helicase. Nucleic Acids Res. 2020;48(21):12252–68. https://doi.org/10.1093/nar/gkaa1069.
Wai T, Langer T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab. 2016;27(2):105–17. https://doi.org/10.1016/j.tem.2015.12.001.
Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305(5685):858–62. https://doi.org/10.1126/science.1099793.
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160(2):189–200. https://doi.org/10.1083/jcb.200211046.
Chen H, Vermulst M, Wang YE, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141(2):280–9. https://doi.org/10.1016/j.cell.2010.02.026.
Pyakurel A, Savoia C, Hess D, Scorrano L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell. 2015;58(2):244–54. https://doi.org/10.1016/j.molcel.2015.02.021.
Yang X, Xue P, Chen H, et al. Denervation drives skeletal muscle atrophy and induces mitochondrial dysfunction, mitophagy and apoptosis via miR-142a-5p/MFN1 axis. Theranostics. 2020;10(3):1415–32. https://doi.org/10.7150/thno.40857. Published 2020 Jan 1.
Wei M, Zhu Z, Wu J, Wang Y, Geng J, Qin ZH. DRAM1 deficiency affects the organization and function of the Golgi apparatus. Cell Signal. 2019;63:109375. https://doi.org/10.1016/j.cellsig.2019.109375.
Kerley-Hamilton JS, Pike AM, Hutchinson JA, Freemantle SJ, Spinella MJ. The direct p53 target gene, FLJ11259/DRAM, is a member of a novel family of transmembrane proteins. Biochim Biophys Acta. 2007;1769(4):209–19. https://doi.org/10.1016/j.bbaexp.2007.02.002.
Crighton D, Wilkinson S, O’Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126(1):121–34. https://doi.org/10.1016/j.cell.2006.05.034.
Zou R, Zheng M, Tan M, Xu H, Luan N, Zhu L. Decreased PTGDS Expression Predicting Poor Survival of Endometrial Cancer by Integrating Weighted Gene Co-Expression Network Analysis and Immunohistochemical Validation. Cancer Manag Res. 2020;12:5057–75. https://doi.org/10.2147/CMAR.S255753. Published 2020 Jun 26.
Kämpfer H, Bräutigam L, Geisslinger G, Pfeilschifter J, Frank S. Cyclooxygenase-1-coupled prostaglandin biosynthesis constitutes an essential prerequisite for skin repair. J Invest Dermatol. 2003;120(5):880–90. https://doi.org/10.1046/j.1523-1747.2003.12140.x.
Kämpfer H, Schmidt R, Geisslinger G, Pfeilschifter J, Frank S. Wound inflammation in diabetic ob/ob mice: functional coupling of prostaglandin biosynthesis to cyclooxygenase-1 activity in diabetes-impaired wound healing. Diabetes. 2005;54(5):1543–51. https://doi.org/10.2337/diabetes.54.5.1543.
Lv D, Cao X, Zhong L, et al. Targeting phenylpyruvate restrains excessive NLRP3 inflammasome activation and pathological inflammation in diabetic wound healing. Cell Rep Med. 2023;4(8):101129. https://doi.org/10.1016/j.xcrm.2023.101129.
Song J, Zeng J, Zheng S, et al. Sanguisorba officinalis L. promotes diabetic wound healing in rats through inflammation response mediated by macrophage. Phytother Res. 2023;37(9):4265–81. https://doi.org/10.1002/ptr.7906.
Zhang Z, Zheng Y, Chen N, et al. San Huang Xiao Yan recipe modulates the HMGB1-mediated abnormal inflammatory microenvironment and ameliorates diabetic foot by activating the AMPK/Nrf2 signalling pathway. Phytomedicine. 2023;118:154931. https://doi.org/10.1016/j.phymed.2023.154931.
Wilgus TA, Roy S, McDaniel JC. Neutrophils and Wound Repair: Positive Actions and Negative Reactions. Adv Wound Care (New Rochelle). 2013;2(7):379–88. https://doi.org/10.1089/wound.2012.0383.
Wong SL, Demers M, Martinod K, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21(7):815–9. https://doi.org/10.1038/nm.3887.
Adikusuma W, Zakaria ZA, Irham LM, et al. Transcriptomics-driven drug repositioning for the treatment of diabetic foot ulcer. Sci Rep. 2023;13(1):10032. https://doi.org/10.1038/s41598-023-37120-1. Published 2023 Jun 20.
Tanwar AS, Shruptha P, Paul B, Murali TS, Brand A, Satyamoorthy K. How Can Omics Inform Diabetic Foot Ulcer Clinical Management? A Whole Genome Comparison of Four Clinical Strains of Staphylococcus aureus. OMICS. 2023;27(2):51–61. https://doi.org/10.1089/omi.2022.0184.
Rong Y, Yang H, Xu H, et al. Bioinformatic Analysis Reveals Hub Immune-Related Genes of Diabetic Foot Ulcers. Front Surg. 2022;9:878965. https://doi.org/10.3389/fsurg.2022.878965. Published 2022 Apr 5.
Acknowledgements
Not applicable.
Funding
The study was funded by the National Natural Science Foundation of China (82274528, 82305245); Construction Task Book for the Three-Year Action Plan for Accelerating the Inheritance and Innovative Development of Traditional Chinese Medicine in Shanghai (2021–2023) (ZY(2021–2023)-0211); Shanghai Municipal Health Commission Scientific Research Programme Mission Statement (202240228); Special Youth Project for Clinical Research of Shanghai Municipal Health Commission (20234Y0162); Clinical Research Talent Training Program of Shanghai University of Traditional Chinese Medicine Affiliated Hospital (2023LCRC06); Four Bright Foundations of Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine (SGKJ-202301); Shanghai Pudong New Area Health and Family Planning Commission’s Pudong Famous TCM Cultivation Program project (PWRZM2020-18).
Author information
Authors and Affiliations
Contributions
LGB and YX made equal contributions to this paper. SHS was responsible for writing the initial draft of the manuscript. SHS, FWJ and YX assisted in the implementation of this study. SHS contributed to the methodology, reviewed, and edited the manuscript. HRY and FWJ made significant contributions to the manuscript revision. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Appendix 1.
InterGenes. Appendix 2. Drug prediction.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shi, H., Yuan, X., Yang, X. et al. A novel diabetic foot ulcer diagnostic model: identification and analysis of genes related to glutamine metabolism and immune infiltration. BMC Genomics 25, 125 (2024). https://doi.org/10.1186/s12864-024-10038-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10038-2