
Abstract
Background
Haloxylon ammodendron holds significance as an ecological plant, showcasing remarkable adaptability to desert conditions, halophytic environments, and sand fixation. With its potential for carbon sequestration, it emerges as a promising candidate for environmental sustainability. Furthermore, it serves as a valuable C4 plant model, offering insights into the genetic foundations of extreme drought tolerance. Despite the availability of plastid and nuclear genomes, the absence of a mitochondrial genome (mitogenome or mtDNA) hinders a comprehensive understanding of its its mtDNA structure, organization, and phylogenetic implications.
Results
In the present study, the mitochondrial genome of H. ammodendron was assembled and annotated, resulting in a multi-chromosomal configuration with two circular chromosomes. The mtDNA measured 210,149 bp in length and contained 31 protein-coding genes, 18 tRNA and three rRNA. Our analysis identified a total of 66 simple sequence repeats along with 27 tandem repeats, 312 forward repeats, and 303 palindromic repeats were found. Notably, 17 sequence fragments displayed homology between the mtDNA and chloroplast genome (cpDNA), spanning 5233 bp, accounting for 2.49% of the total mitogenome size. Additionally, we predicted 337 RNA editing sites, all of the C-to-U conversion type. Phylogenetic inference confidently placed H. ammodendron in the Amaranthacea family and its close relative, Suaeda glacum.
Conclusions
H. ammodendron mtDNA showed a multi-chromosomal structure with two fully circularized molecules. This newly characterized mtDNA represents a valuable resource for gaining insights into the basis of mtDNA structure variation within Caryophyllales and the evolution of land plants, contributing to their identification, and classification.
Similar content being viewed by others


Background
H. ammodendron is a small C4 perennial tree belonging to the Amaranthaceae family, formerly known as Chenopodiaceae [1]. The genus Haloxylon encompasses approximately 11 species, distributed from Iran to Mongolia and Northwestern China [2]. Referred to as the King of psammophytic woody plants, H. ammodendron demonstrates remarkable adaptability to severe environmental conditions, including desert conditions and sandstorms [3]. Widely found in the deserts of northwest China, it plays a crucial role in environmental protection through wind control, carbon sequestration, and sand fixation [4]. However, due to increasing overexploitation and the dramatic climate change occurring in central Asia, Haloxylon plants face a threat [5].
The declining numbers of these plants and the subsequent ecological impacts have sparked significant interest in genetic research on Haloxylon resources. Long et al. [6] provided the first transcriptome resource of H. ammodendron to investigate genes that confer drought stress resistance to the plant. For a thorough understanding of the resilience of H. ammodendron to drought stress, Gao et al. [7] conducted a complete transcriptome analysis under osmotic conditions, paired with evaluations of physiological factors such as proline, betaine, soluble sugar levels, and peroxidase activity. Using cpDNA genes (trnS-trnG and trnV) and ITS (Internal transcribed spacer) (ITS1-ITS4) sequences, Chen at al [8]. assessed 420 individuals from 36 populations of Haloxylon for their genetic diversity and historical demography. Suo et al. [9] utilized SSR marker-derived DNA markers to distinguish H. ammodendron and Haloxylon persicum species. Later, the cpDNA of H. ammodendron and H. persicum were assembled using short-read sequencing data. Recently, a high-quality chromosome-level assembly of H. ammodendron was released [10], paving the way for an in-depth investigation of genes of interest concerning its high tolerance to drought and salinity. Despite the availability of these genomic resources, the mitochondrial genome (mitogenome or mtDNA) of H. ammodendron is lacking, hampering mtDNA-based structural and functional evolutionary studies.
The mitogenome, often referred to as the cell’s powerhouse, plays a crucial role in cellular energy production [11]. Housed in the mitochondria, this distinct genome is responsible for producing essential proteins for oxidative phosphorylation, leading to the creation of adenosine triphosphate (ATP)– the primary energy currency of the cell [12, 13]. High variations in mtDNA size coupled with structural complexity, are the major factors slowing the pace of fully and accurately assembled mitogenome projects. For example, mitogenome sizes vary over 40-fold in the single genus Silene, from 253 kbp in Silene latifolia to more than 11 Mbp in Silene conica [14]. Variation in repetitive sequences, the incorporation of foreign sequences, and gain or loss of non-coding regions are among the key factors contributing to genome size variability [15, 16].
Furthermore, while a single circular mitogenome is common in angiosperms, various configurations may occur, including linear, fragmented, loop-like, branched, and multi-chromosomal structures [11]. The dynamic nature of mtDNA presents challenges in fully assembling the genomes of certain species compared to cpDNA assembly.
However, advancements in long-read sequencing technologies have made the assembly of plant mitochondrial genomes more achievable, facilitating comparative studies of plant mitogenome structure and evolutionary implications [17, 18]. In the present study, short and long-read sequencing data were used to generate the first mtDNA of H. ammodendron. The characterization of repeats content, sequence collinearity, DNA migration and RNA editing sites were explored.
Results
Genome assembly and annotation
In this study, we utilized Illumina short-reads and PacBio HiFi long-reads to assemble the mtDNA of H. ammodendron. Importantly, the data was not generated within this study; instead, we sourced the Illumina short-reads and PacBio long-reads from a previously conducted study on the nuclear genome of the species. Specifically, we accessed 49.4 Gbp of clean short reads (Table S1) from the NCBI SRA database under accession number SRR17127859. The assembly was facilitated using the GetOrganelle software, version 1.7.5 [19]. In parallel, the PacBio dataset comprised 22.6 Gbp of long-reads, also retrieved from NCBI SRA under the accession SRR17129371. When visualizing the short-read assembly graph using Bandage version 0.8.1 [20], an intricate and circular multi-branch mtDNA structure was assembled encompassing nodes or contigs (Fig. 1). These nodes were delineated by overlapping regions depicted by black lines on the assembly graph. Overlapping regions were resolved by aligning associated branch nodes with the PacBio HiFi dataset. Due to the presence of repeats, recombinant sequences could occur, leading to different configurations. Therefore, four potential genomic paths were inferred. Paths 1–1 and 1–2 were considered major configurations, while paths 2 − 1 and 2–2 represented minor configurations (Fig. 1, Table S2, Table S3). The major configuration was obtained by extending 2000 bp on both ends based on the repetitive sequence. As for the minor configuration, the 2000 bp extension was not supported by the long-reads data (Fig. 1, Table S2, Table S3). Consequently, two fully circular sequences representing the mtDNA of H. ammodendron, were obtained (Fig. 1). The two circularized contigs, namely Chromosome 1 and Chromosome 2, exhibited similar GC content (44%) spanning 121,403 bp and 88,746 bp respectively (Table 1).

Diagram of mitogenome recombination. The major configuration of Haloxylon ammodendron mitogenome is two independent circular mtDNA, with three pairs of repeats that can mediate gene recombination: HRS1, HRS2 and HRS3. According to the supported long-reads, the frequencies of mitogenome recombination mediated by HRS1, HRS2 and HRS3 were 44.2%, 4.92%, and 0.21%, respectively
The annotation of the H. ammodendron mtDNA is summarized in Table 2. A total of 31 protein-coding genes, 18 tRNA and three rRNA were predicted. Among the protein-coding genes, 24 core genes typically found in angiosperms [21], and seven variably present genes were identified. The core genes include five ATP synthase genes (atp1, atp4, atp6, atp8, and atp9), nine NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9), four cytochrome c biogenesis genes (ccmB, ccmC, ccmFC, and ccmFN), three cytochrome c oxidase genes (cox1, cox2, and cox3), one membrane transport protein gene (mttB), one mature enzyme gene (matR), and one ubiquinol-cytochrome c reductase gene (cob). The non-core genes include one ribosomal large subunit gene (rpl5), five ribosomal small subunit genes (rps3, rps4, rps7, rps12, rps13), and one succinate dehydrogenase gene (sdh4). Regarding tRNA, three were in duplicate copy (trnC-GCA, trnG-GCC, trnP-UGG), while one was in triplicate copy (trnfM-CAU). Among the three rRNA, rrn26 was present in duplicate copy.
Gene transfer
Putative gene transfer between organelles was unveiled through alignment of the newly assembled cpDNA (Fig. 2A) with the mtDNA of H. ammodendron. Homologous sequence fragments between the organelles are illustrated in Fig. 2B. According to the sequence similarity analysis, 17 DNA fragments were identified as homologous between the mtDNA and cpDNA, with a total length of 5233 bp, accounting for 2.49% of the mtDNA (Table S4). Among them, the longest fragment measured 1158 bp in length. By annotating these homologous sequences, eight complete genes were found on the 17 homologous fragments, including one protein-coding gene (petG) and seven tRNA genes (trnD-GUC, trnH-GUG, trnM-CAU, trnN-GUU, trnP-UGG, trnS-GCU, trnW-CCA). Additionally, we investigated the mtDNA trace in the nuclear genome (Table S5). The longest alignment, with a length of 15 Kbp, was identified on chromosome 1 (Table S5).

H. ammodendron plastome overview and sequence similarity. (A) Depiction of the H. ammodendron plastome, providing a visual representation of its organization and key features. (B) Illustration highlighting sequence similarity between the mitochondrial and chloroplast organelles. Yellow arcs denote regions of the mitochondrial genome, while the purple arc signifies the chloroplast genome. The interconnecting green lines emphasize homologous segments shared between the two genomes
Repeat sequences analysis
Microsatellites were screened for each set of chromosomes in the H. ammondendron mtDNA (Fig. 3A). For chromosome 1, a total of 42 simple sequence repeats (SSRs) were identified, comprising eight monomeric SSRs, eight dimeric SSRs, four trimeric SSRs, 17 tetrameric SSRs, four tetrameric SSRs, and one hexameric SSRs (Fig. 3A). Among the SSRs, the repeat motifs (A)10, (GAAA)3 and (CT)5 were found to be prevalent (Table S6). Seventeen tandem repeat sequences were identified, exhibiting a matching identity greater than 70% and lengths ranging from 14 to 31 bp (Fig. 3B, Table S7). Dispersed repeat sequences in chromosome 1 were also examined, detecting 467 repeat sequence pairs with a length equal to or greater than 30 bp. Among these, 234 pairs were palindromic repeats, 233 were forward repeats, and one was found to be a reverse repeat type. The longest observed palindromic repeat sequence spanned 123 bp, while the longest forward repeat sequence was 175 bp in length (Table S8).

Repeat patterns in the H. ammodendron mitochondrial genome. (A) Chart showing the count of Simple Sequence Repeats (SSRs) distributed in chromosome 1 and chromosome 2. (B) Visualization of the number of tandem and dispersed repeats found in both chromosomes, offering insight into the genome’s complexity and evolutionary history
In chromosome 2, 24 SSRs were detected (Fig. 3A). Among these SSRs, nine were monomeric, two were dimeric, three were trimeric, nine were tetrameric, and one was the pentameric type. No hexameric SSRs were found in this chromosome. The most prominent SSR motif was thymine (T)10, among others (Table S7). A total of 10 tandem repeat sequences with a matching identity greater than 78% and lengths ranging from 18 to 42 bp were identified (Fig. 3B, Table S8). Dispersed repeat sequences in chromosome 2 were also examined, detecting 148 pairs of repeat sequences with a length equal to or greater than 30 bp. Among these, 69 pairs were palindromic repeats, and 79 were forward repeats. No reverse or complement repeats were detected in both chromosomes. The longest observed palindromic repeat sequence spanned 56 bp, while the longest forward repeat sequence was 114 bp in length (Table S8).
RNA editing
RNA editing, a widespread phenomenon in land plants, entails modifications within the coding region of the transcript involving the addition, loss, or conversion of nucleotides [22]. The current study identified 337 RNA editing sites, all of which were cytidine to uridine (C to U) conversion types. The predicted RNA editing sites in various genes are shown in Fig. 4. The ccmB and ccmFN genes exhibited the highest number (25) of RNA editing sites. Conversely, no RNA editing sites were observed in the sdh4 gene. The amino acid transition exhibiting the highest abundance was from serine to leucine, comprising 19.58% (66 sites) of the observed transitions (Table S9).

RNA editing sites in H. ammodendron mitochondrial genes. This figure presents a comprehensive count of RNA editing sites discovered within 31 mitochondrial protein-coding genes of H. ammodendron, shedding light on post-transcriptional modifications and their potential implications
Codon usage
The codon usage pattern of the H. ammodendron mtDNA was presented in Fig. 5. Codons exhibiting a relatively synonymous codon usage (RSCU) value greater than one were regarded as being favored by the corresponding amino acids. Apart from the start codon AUG and the tryptophan codon UGG, both of which had an RSCU value of 1, there were widespread codon usage preferences in the H. ammodendron mitochondrial protein-coding genes (Fig. 5). For example, alanine (Ala) showed a strong preference for the codon GCU, with the highest RSCU value of 1.64. In addition, leucine (Leu) preferred the codon UUA, with an RSCU value of 1.61 (Table S10).

Codon usage preferences in H. ammodendron‘s mitochondrial genome. Graphical representation illustrating the Relative Synonymous Codon Usage (RSCU) values. This showcases the preferences of certain codons encoding the same amino acid, hinting at evolutionary pressures and translational optimization
Phylogenetic analysis and collinearity analysis
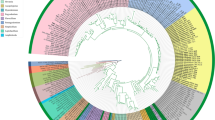
The phylogenetic tree based on mtDNA (Fig. 6) was constructed using a maximum likelihood approach, incorporating a diverse set of 30 species representing various orders (Table S11). The resulting tree topology aligns with the most recent classification the Angiosperm Phylogeny Group (APG) provided. H. ammodendron and the Amaranthaceae species Spinacia oleracea, Chenopodium quinoa, and Suaeda glauca were placed within the Caryophyllales order. Interestingly, H. ammodendron exhibited a close phylogenetic relationship with S. glauca, another member of the Amaranthaceae family, widely distributed in the arid areas of northwest China [23]. These results were robustly supported by Bayesian phylogenetic tree inference (Figure S1). To identify both conserved and unique mitochondrial regions among Caryophyllales species, we performed pairwise alignments using Spinacia oleracea (NC_035618.1), S. glauca (NC_060419.1), Alternanthera philoxeroides (MN166292.1), Beta macrocarpa (NC_015994.1), Silene latifolia (NC_014487.1), Mirabilis jalapa (NC_056991.1), and Fallopia aubertii (MW664926.1) mtDNA (Fig. 7). The alignment results were provided in Fig. 7 and Table S12. Although collinear blocks were noticeable within Caryoplyllales species, their length is relatively short, indicating a non-conservative mitogenome structure. It is worth pointing out that the homologous block arrangement is inconsistent between H. ammodendron and its congeners, implying an evolutionary mtDNA rearrangement occurrence. Additionally, some blank regions were observed in H. ammodendron, representing species-specific sequences lacking homology with other species.

Phylogenetic placement of H. ammodendron within Caryophyllales. A Maximum Likelihood phylogenetic tree that delineates the evolutionary relationship of H. ammodendron with other species in the Caryophyllales order. Node labels display support values, with maximum-likelihood bootstrap values offering a measure of the tree’s reliability

Comparative collinearity in caryophyllales mitogenomes. A landscape view showcasing the genome collinearity between H. ammodendron and its Caryophyllales counterparts. The diagram helps in understanding genomic rearrangements, evolutionary events, and conservation levels among these species
Discussion
In land plants, mtDNA has undergone extensive genomic reorganization and gene arrangements, resulting in rapid structural evolution. Studying plant mtDNA offers a path into their structural organization, genetic diversity, and evolutionary dynamics, which have implications for understanding their functional role in biological processes such as plant growth, energy production, and respiration.
In the present study, we took advantage of both short- and long-read sequencing data to assemble the first mtDNA of H. ammodendron. The assembly mitogenome length was 210,149 bp, which is about 50% shorter than S. glauca (NC_060419.1, assembly size: 474,330 bp), and P. aculeate (NC_067638.1, assembly size: 515,187 bp) but in similar range with M. jalapa (NC_056991.1, assembly size: 267,334 bp), all belonging to the same order: Caryophyllales. The observed genome size variation could be imputable to several factors, including repeat elements density, intragenic regions gain or loss, and the introgression of foreign DNA [24,25,26].
Repeats elements such as microsatellites, tandem and dispersed repeats are prone to recombination leading to isomeric sequence sets in the mitogenome [14, 27, 28]. Therefore, the repeats dynamism could impact DNA maintenance and evolution of the plant mtDNA [16]. Unusually sized repeats within plant mtDNA exert significant influence, marked by their excessive size relative to coding capacity, a low mutation rate in genes, and a substantial rearrangement rate [16]. Besides, ubiquitous short tandem repeats play pivotal roles in mtDNA evolution, contributing to tandem duplications and structural variations [29]. It is worth to mention that homologous recombination, facilitated by rich repeated sequences in plant mtDNA, emerges as a major evolutionary driver [30]. Additionally, repeat-mediated recombination stands out as a highly active and substantial contributor to structural rearrangements in mtDNA [31].
One of the complexities of the mtDNA is the variability in molecular conformation. Several configurations have been documented, including circular, linear, sigma-like, fragmented, and multi-chromosomal structures [30]. Here we reported a multi-chromosomal mitogenome architecture that consists of two circular molecules. Similar findings were reported in other Caryophyllales species, such as Fagopyrum esculentum (10 circular chromosomes) [32], and Fallopia multiflora (two circular chromosomes) [33]. In the same plant order, a multi-chromosomal architecture with more than 50 chromosomes was also found for one of the symbolic plants, Silene conica, well known for its massive mitogenome spanning 11 Mb [14]. However, the close relatives S. glauca (NC_060419.1) [18] and Chenopodium quinoa (NC_041093.1) [17] present a single molecule in a fully circularized conformation based on long reads data-based assembly. The origin and maintenance of the multi-ring structure in plant mtDNA are areas of ongoing research [34,35,36,37,38,39]. It is believed that mechanisms such as recombination and rearrangement events, possibly mediated by repeated sequences [40,41,42], could play a role in generating and maintaining this phenomenon. The balance between the recombination and replication of mitochondrial rings was proposed as a key aspect that could influence the occurrence of mtDNA multi-chromosome structure [43, 44]. Besides, gain or loss of chromosome sets is suggested to be one of the key biological events leading to the diversity of mtDNA structures across different organisms [35], providing insights into the mechanisms driving the mitochondrial evolution.
DNA transfer among chloroplasts and mitochondria allows genetic exchange between cellular compartments and can serve as a driver for expansion or contraction of mitochondrial genomes in plants. Our study detected sequence transfer between the cpDNA and the mtDNA, indicating possible horizontal or intracellular gene transfer [45]. Sequences exchange between mtDNA and cpDNA was also noted for other Caryophyllales members including Mammillaria huitzilopochtli [46].
RNA editing is a post-transcriptional mechanism in higher plant organelles, contributing to improved protein folding [47]. In angiosperm mitochondria, extensive cytidine to uridine RNA editing occurred, playing a crucial role in transcript expression [48]. Interestingly, all protein-coding genes analyzed from H. ammomendron mtDNA showed C- to-U conversion, which may affect the start or end positions of the coding genes. It is worth noting that proteins translated from edited RNAs could play a critical function in an environment change context, resulting in plant acclimatization and survival capabilities [49]. Indeed, H. ammodendron is well known for its remarkable tolerance to saline and dry environments [50, 51].
The maximum likelihood phylogenetic inference resulted in a tree with strong support values, placing H. ammodendron in the close relative taxon S. glauca. Our tree topology is in perfect agreement with the recent results based on the Angiosperm Phylogeny Group (APG IV) classification [52], ITS [53], plastid markers [54], the recent nuclear whole genome sequence data [10], and the Bayesian phylogenetic tree indicating the reliability of the mitogenome-based tree.
Conclusions
In the present study, the mtDNA of H. ammodendron was characterized. The mtDNA exhibited a multi-chromosomal conformation with two circular molecules with a total length of 210,149 bp. It contains 31 protein-coding genes, 18 tRNA and 3 rRNA. A set of 17 sequence fragments were found homologous between the mtDNA and cpDNA, spanning 5,233 bp, accounting for 2.49% of the mitogenome. RNA editing assessment revealed that the protein-coding genes were mainly subjected to cytidine-to-uridine conversion. The phylogenetic inference showed a close relationship with S. glauca. The mtDNA resource provided by this study laid a foundation for further investigations into the comparative evolution and functional role of the H. ammodendron mtDNA.
Methods
Assembly and annotation
Before the mtDNA assembly, short-reads data were trimmed using fastp v0.23.4 [55]. To assemble the H. ammodendron mitogenome, a hybrid approach encompassing both short and long-read sequencing data was implemented. Firstly, using the Illumina short-reads data, a draft mtDNA assembly was conducted with GetOrganelle v1.7.5 [19] with the following settings:-R 15 -k 21,45,65,85,105 -F embplant_mt. The resulting genome graph sketch was then visualized using Bandage v. 0.8.1 [20], manually inspected and curated for node junctions. Using minimap2.2.26 (r1175), we mapped PacBio HiFi data onto the initial assembly to address conflictual nodes and repetitive regions [56].. In cases where multiple alternative connections exist at branching nodes, preference was given to connections supported by long reads. The obtained assembly was validated by mapping both short and long reads to ensure the connectivity and consistency of the assembly.
The assembly was annotated with GeSeq online tool (https://chlorobox.mpimp-golm.mpg.de/geseq.html#) [57] with the following parameters: Arabidopsis thaliana (NC_037304), Nicotiana tabacum (NC_ 006581), Glycine max (NC_020455), and Suaeda glauca (NC_060419.1) were set as reference mitogenomes; BLAT search (protein search identity = 25, rRNA, tRNA, DNA search identity = 85) for coding protein genes, rRNA and tRNA prediction. Additionally, tRNAscan-SE v2.0.7 [58] was used for tRNA prediction with a cut-off score for reporting tRNAs = 15. The annotated mtDNA was visualized using Apollo v2.7.0 [59] for manual correction. The genome map was rendered using the Organellar Genome DRAW (OGDRAW) tool [60].
Repeat sequence analysis
The identification of repetitive sequences, including microsatellite sequence repeats (SSRs), tandem repeats, and dispersed repeats, was performed using MISA (https://webblast.ipk-gatersleben.de/misa/) [61], TRF (https://tandem.bu.edu/trf/trf.unix.help.html) [62], and the REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) [63] web servers respectively.
RNA editing and codon usage bias analyses
The protein-coding sequences of the mitochondrial genome of H. ammodendron were extracted using the PhyloSuite toolbox [64]. Prediction of RNA editing sites was performed using the online tool PREPACT3 (http://www.prepact.de/) [65], while MEGA v7.0 software [66] was employed to calculate the relative synonymous codon usage (RSCU).
Collinearity analysis
Using the BLASTN v2.10.1 + tool, we conducted pairwise alignments to assess sequence similarity between H. ammodendron and its related species (Spinacia oleracea, Suaeda glauca, Alternanthera philoxeroides, Beta macrocarpa, Silene latifolia, Mirabilis jalapa, Fallopia aubertii) [67]. Homologous sequences exceeding 500 bp were retained as conserved collinear blocks for constructing the multiple synteny plot with the help of MCscanX implemented in TBTools v1.098746 [68].
Gene transfer analysis
To detect the putative gene transfer between cpDNA and mtDNA, we performed a de novo assembly and annotation of the cpDNA of our sample using GetOrganelle v1.7.5 [19] and GeSeq web server [57], respectively. Then, an alignment of organelles was performed using BLAST v2.10.1+ [67] with the following settings: E-value ≤ 1E-10, matching rate ≥ 70%, and matching length ≥ 40 bp. Utilizing the genome of H. ammodendron, we applied a similar approach by aligning the mtDNA onto the nuclear genome to investigate the sequence transfer between the mtDNA and the nuclear genome.
Phylogenetic analysis
To infer the mtDNA-based phylogenetic tree, a set of 15 conserved genes (atp1, atp4, atp6, atp8, ccmB, ccmC, ccmFC, ccmFN, cox2, matR, nad1, nad2, nad3, nad5, and nad6) from 30 species belonging to four orders (Caryophyllales, Ericales, Santalales, and Zygopgyllales) (Table S10) were extracted from each mtDNA and aligned using MAFFT v7.505 [69]. Poorly aligned sequence regions were trimmed with trimAl v 1.4.1 [70]. The resulting data matrix was then concatenated with PhyloSuite [64] (Supplementary file 1), and the tree was constructed using IQ-TREE v2.0.3 following the Maximum Likelihood approach with the GTR + I + G model [64]. To assess the tree topology, a Bayesian method was performed using MrBayes v3.2.6 tool [71] with the following settings: number of substitution types: 6; Model: 4by4; rates variation across sites: invgamma; number of generations: 10,000, sample frequency: 10; Burnin: 250. The tree was rendered with the Interactive Tree of Life tool (iTOL) v5.0 [72].
Data availability
The data underpinning the conclusions drawn in this study are accessible in both the main manuscript and its supplementary information appendices. The PacBio Hi-Fi and Illumina sequencing reads are retrievable via the Sequence Read Archive accession numbers SRR17129371 and SRR17127859, respectively. Furthermore, the assembled sequences corresponding to chromosome 1 and chromosome 2 of the mitogenome have been submitted to the National Center for Biotechnology Information and are retrievable under the GenBank accession numbers OR296702 and OR296703, respectively.
References
Su P, Cheng G, Yan Q, Liu X. Photosynthetic regulation of C4 desert plant Haloxylon ammodendron under drought stress. Plant Growth Regul. 2007;51:139–47.
Bunge. (Chenopodiaceae). In: Flora of China Editorial Committee, ed. Flora of China. Vol. 5. St. Louis: Science Press, Beijing/Missouri Botanic Garden Press, 395–396. CABI Compendium. 2004;CABI Compe.
Yang WB, Feng W, Jia ZQ, Zhu YJ, Guo JY. Soil water threshold for the growth of Haloxylon Ammodendron in the Ulan Buh desert in arid northwest China. South Afr J Bot. 2014;92:53–8.
Ma Q, Wang X, Chen F, Wei L, Zhang D, Jin H. Carbon sequestration of sand-fixing plantation of Haloxylon Ammodendron in Shiyang River Basin: Storage, rate and potential. Glob Ecol Conserv. 2021;28:e01607.
Li J, Chang H, Liu T, Zhang C. The potential geographical distribution of Haloxylon across Central Asia under climate change in the 21st century. Agric for Meteorol. 2019;275:243–54.
Long Y, Zhang J, Tian X, Wu S, Zhang Q, Zhang J, et al. De novo assembly of the desert tree Haloxylon Ammodendron (C. A. Mey.) Based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genomics. 2014;15:1–11.
Gao HJ, Lü XP, Zhang L, Qiao Y, Zhao Q, Wang YP et al. Transcriptomic profiling and physiological analysis of Haloxylon Ammodendron in response to osmotic stress. Int J Mol Sci. 2018;19.
Chen Y, Ma S, Zhang D, Wei B, Huang G, Zhang Y, et al. Diversification and historical demography of Haloxylon ammodendron in relation to Pleistocene climatic oscillations in northwestern China. PeerJ. 2022;10:1–27.
Suo Z, Jia Z, Lu Q, Pan B, Jin X, Xu G, et al. Distinguishing Haloxylon persicum and H. ammodendron (Haloxylon Bunge, Amaranthaceae) using DNA marker. AASRI Procedia. 2012;1:305–10.
Wang M, Zhang L, Tong S, Jiang D, Fu Z. Chromosome-level genome assembly of a xerophytic plant, Haloxylon Ammodendron. DNA Res. 2022;29:1–9.
Chen Z, Zhao N, Li S, Grover CE, Nie H, Wendel JF, et al. Plant mitochondrial genome evolution and cytoplasmic male sterility. CRC Crit Rev Plant Sci. 2017;36:55–69.
Gray MW, Burger G, Lang BF. Mitochondrial evolution. Sci (80-). 1999;283:1476–81.
Gray MW. Mitochondrial evolution. Cold Spring Harb Perspect Biol. 2012;4.
Sloan DB, Alverson AJ, Štorchová H, Palmer JD, Taylor DR. Extensive loss of translational genes in the structurally dynamic mitochondrial genome of the angiosperm Silene latifolia. BMC Evol Biol. 2010;10.
Smith DR, Keeling PJ. Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc Natl Acad Sci. 2015;112:10177–84.
Wynn EL, Christensen AC. Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3 genes. Genomes Genet. 2019;9:549–59.
Maughan PJ, Chaney L, Lightfoot DJ, Cox BJ, Tester M, Jellen EN, et al. Mitochondrial and chloroplast genomes provide insights into the evolutionary origins of quinoa (Chenopodium quinoa Willd). Sci Rep. 2019;9:1–11.
Cheng Y, He X, Priyadarshani SVGN, Wang Y, Ye L, Shi C, et al. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda Glauca. BMC Genomics. 2021;22:1–15.
Jin J-J, Yu W-B, Yang J-B, Song Y, DePamphilis CW, Yi T-S, et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020;21:241.
Wick RR, Schultz MB, Zobel J, Holt KE, Bandage. Interactive visualization of de novo genome assemblies. Bioinformatics. 2015;31:3350–2.
Adams KL, Qiu Y, Stoutemyer M, Palmer JD. Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc Natl Acad Sci. 2002;99:9905–12.
Jobson RW, Qiu YL. Did RNA editing in plant organellar genomes originate under natural selection or through genetic drift? Biol Direct. 2008;3:1–22.
Zhang S, Ni X, Arif M, Yuan Z, Li L, Li C. Salinity influences cd accumulation and distribution characteristics in two contrasting halophytes, Suaeda Glauca and Limonium Aureum. Ecotoxicol Environ Saf. 2020;191 November 2019:110230.
Rice DW, Alverson AJ, Richardson AO, Young GJ, Sanchez-Puerta MV, Munzinger J, et al. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Sci (80-). 2013;342:1468–73.
Zhang S, Wang J, He W, Kan S, Liao X, Jordan DR, et al. Variation in mitogenome structural conformation in wild and cultivated lineages of sorghum corresponds with domestication history and plastome evolution. BMC Plant Biol. 2023;23:1–17.
Mower JP. Variation in protein gene and intron content among land plant mitogenomes. Mitochondrion. 2020;53:203–13.
Folkerts O, Hanson MR. Three copies of a single recombination repeat occur on the 443 kb mastercircle of the Petunia hybrida 3704 mitochondrial genome. Nucleic Acids Res. 1989;17:7345–57.
Siculella L, Damiano F, Cortese MR, Dassisti E, Rainaldi G, Gallerani R, et al. Gene content and organization of the oat mitochondrial genome. Theor Appl Genet. 2001;103:359–65.
Xia H, Zhao W, Shi Y, Wang XR, Wang B. Microhomologies are associated with tandem duplications and structural variation in plant mitochondrial genomes. Genome Biol Evol. 2020;12:1965–74.
Gualberto JM, Newton KJ. Plant mitochondrial genomes: Dynamics and mechanisms of Mutation. Annu Rev Plant Biol. 2017;68 February:225–52.
Zhong Y, Yu R, Chen J, Liu Y, Zhou R. Highly active repeat-mediated recombination in the mitogenome of the holoparasitic plant Aeginetia indica. Front Plant Sci. 2022;13:1–10.
Logacheva MD, Schelkunov MI, Fesenko AN, Kasianov AS, Penin AA. Mitochondrial genome of Fagopyrum esculentum and the genetic diversity of extranuclear genomes in buckwheat. Plants. 2020;9.
Kim CK, Kim YK. The multipartite mitochondrial genome of Fallopia multiflora (Caryophyllales: Polygonaceae). Mitochondrial DNA Part B Resour. 2018;3:155–6.
Sloan DB, Alverson AJ, Chuckalovcak JP, Wu M, McCauley DE, Palmer JD, et al. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012;10:e1001241.
Wu Z, Cuthbert JM, Taylor DR, Sloan DB. The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc Natl Acad Sci U S A. 2015;112:10185–91.
Bi C, Qu Y, Hou J, Wu K, Ye N, Yin T. Deciphering the multi-chromosomal mitochondrial genome of Populus simonii. Front Plant Sci. 2022;13:1–14.
Li X, Zhe M, Huang Y, Fan W, Yang J, Zhu A. The evolution of mitochondrial genomes between two Cymbidium Sister species: dozens of circular chromosomes and the maintenance and deterioration of genome synteny. Genes (Basel). 2023;14:864.
Yu R, Sun C, Zhong Y, Liu Y, Sanchez-Puerta MV, Mower JP, et al. The minicircular and extremely heteroplasmic mitogenome of the holoparasitic plant Rhopalocnemis phalloides. Curr Biol. 2022;32:470–479e5.
Szandar K, Krawczyk K, Myszczyński K, Ślipiko M, Sawicki J, Szczecińska M. Breaking the limits - multichromosomal structure of an early eudicot Pulsatilla patens mitogenome reveals extensive RNA-editing, longest repeats and chloroplast derived regions among sequenced land plant mitogenomes. BMC Plant Biol. 2022;22:109.
Yang H, Ni Y, Zhang X, Li J, Chen H, Liu C. The mitochondrial genomes of Panax notoginseng reveal recombination mediated by repeats associated with DNA replication. Int J Biol Macromol. 2023;:126359.
Jiang M, Ni Y, Li J, Liu C. Characterisation of the complete mitochondrial genome of Taraxacum mongolicum revealed five repeat-mediated recombinations. Plant Cell Rep. 2023;42:775–89.
Sloan DB. One ring to rule them all? Genome sequencing provides new insights into the master circle model of plant mitochondrial DNA structure. New Phytol. 2013;200:978–85.
Kozik A, Rowan BA, Lavelle D, Berke L, Schranz ME, Michelmore RW, et al. The alternative reality of plant mitochondrial DNA: one ring does not rule them all. PLOS Genet. 2019;15:e1008373.
Morley SA, Ahmad N, Nielsen BL. Plant Organelle Genome Replication Plants. 2019;8:358.
Archibald JM, Richards TA. Gene transfer: anything goes in plant mitochondria. BMC Biol. 2010;8:8–10.
Plancarte DC, Solórzano S. Structural and gene composition variation of the complete mitochondrial genome of Mammillaria huitzilopochtli (Cactaceae, Caryophyllales), revealed by de novo assembly. BMC Genomics. 2023;24:1–13.
Hao W, Liu G, Wang W, Shen W, Zhao Y, Sun J et al. RNA editing and its roles in Plant Organelles. Front Genet. 2021;12 September.
Knoop V. C-to-U and U-to-C: RNA editing in plant organelles and beyond. J Exp Bot. 2023;74:2273–94.
Gommans WM, Mullen SP, Maas S. RNA editing: a driving force for adaptive evolution? BioEssays. 2009;31:1137–45.
Hu D, Lv G, Qie Y, Wang H, Yang F, Jiang L. Response of morphological characters and photosynthetic characteristics of Haloxylon ammodendron to water and salt stress. Sustain. 2021;13:1–12.
He XH, Si JH, Zhou DM, Wang CL, Zhao CY, Jia B, et al. Leaf chlorophyll parameters and photosynthetic characteristic variations with stand age in a typical desert species (Haloxylon Ammodendron). Front Plant Sci. 2022;13:1–13.
Chase MW, Christenhusz MJM, Fay MF, Byng JW, Judd WS, Soltis DE, et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc. 2016;181:1–20.
Pyankov VI, Artyusheva EG, Edwards GE, Black CC, Soltis PS. Phylogenetic analysis of tribe salsoleae (Chenopodiaceae) based on ribosomal its sequences: implications for the evolutIon of photosynthesis types. Am J Bot. 2001;88:1189–98.
Akhani H, Edwards G, Roalson EH. Diversification of the Old World Salsoleae s. l. (Chenopodiaceae): molecular phylogenetic analysis of Nuclear and Chloroplast Data sets and a revised classification. Int J Plant Sci. 2007;168:931–56.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100.
Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, et al. GeSeq - Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45:W6–11.
Lowe TM, Eddy SR. tRNAscan-SE: a program for Improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:0955–64.
Dunn NA, Unni DR, Diesh C, Munoz-Torres M, Harris NL, Yao E, et al. Apollo: democratizing genome annotation. PLoS Comput Biol. 2019;15:1–14.
Greiner S, Lehwark P, Bock R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019;47:W59–64.
Beier S, Thiel T, Münch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33:2583–5.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80.
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–42.
Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, et al. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 2020;20:348–55.
Lenz H, Rüdinger M, Volkmar U, Fischer S, Herres S, Grewe F, et al. Introducing the plant RNA editing prediction and analysis computer tool PREPACT and an update on RNA editing site nomenclature. Curr Genet. 2010;56:189–201.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative Toolkit developed for interactive analyses of big Biological Data. Mol Plant. 2020;13:1194–202.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Huelsenbeck JP, Ronquist F. MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5.
Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–6.
Acknowledgements
We sincerely thank the experimental personnel and bioinformatics analysts at Wuhan Benagen Technology Company Limited (www.benagen.com) and MitoRun research group participated in this project.
Funding
This study was supported by grants from the National Natural Science Foundation of China (82173066, 81974366, 82273208), the Natural Science Foundation of Guangdong Province (2020A1515011408), the Shenzhen Science and Technology Innovation Commission Project (JCYJ20200109120208018, JCYJ20190808155015181, JCYJ20220818101601004).
Author information
Authors and Affiliations
Contributions
L.Y., T.X. and H.X. designed the research. J.L., W.G., Z. Z. and Y.X. participated in the material preparation. L.Y., J.L. and W.G., performed the genome assembly, and evolutionary analysis. L.Y. and T.X. wrote the manuscript. H.X., Y.X. and T.X. revised the manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1: Supplementary Table 1.
Haloxylon ammodendron sequence reads archive information of short and long reads data. Supplementary Table 2. Using long-reads to validate potential recombinant DNA sequences based on BLASTn. Supplementary Table 3. The percentage of different genome configurations identified by long-reads in the Haloxylon ammodendron mitogenome. Supplementary Table 4. Results of sequence similarity analysis between chloroplast and mitochondrial genomes of Haloxylon ammondendron. Supplementary Table 5. Alignment of nuclear and mitochondrial genomes of Haloxylon ammodendron. Supplementary Table 6. Microsatellite repeats detected on Haloxylon ammodendron mitogenome. Supplementary Table 7. Tandem repeats results identified in Haloxylon ammodendron mitogenome. Supplementary Table 8. Dispersed repeats results detected in Haloxylon ammodendron mitogenome. Supplementary Table 9. RNA editing sites and conversion type within Haloxylon ammodendron mitochondrial genome. Supplementary Table 10. Relative synonymous codon usage of various amino acids in the Haloxylon ammodendron mitochondrial genome. Supplementary Table 11. List of species employed in the phylogenetic tree construction. Supplementary Table 12. Collinearity analysis results within the selected Caryophyllales species
Supplementary Material 2: Supplementary Figure 1.
Bayesian-based phylogenetic tree. The Bayesian tree inference was conducted using the MrBayes tool based on a conserved gene set. Node support values are highlighted in red. The tree was inferred using the 4by4 model. The tree was visualized using etetoolkit online viewer http://etetoolkit.org/treeview/
Supplementary Material 3: Supplementary file 1.
Alignment matrix used for the phylogenetic tree inference
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, L., Liu, J., Guo, W. et al. Insights into the multi-chromosomal mitochondrial genome structure of the xero-halophytic plant Haloxylon Ammodendron (C.A.Mey.) Bunge ex Fenzl. BMC Genomics 25, 123 (2024). https://doi.org/10.1186/s12864-024-10026-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10026-6