
Abstract
Background
Intramuscular fat (IMF) is an important factor in meat quality, and triglyceride (TG) and Phospholipids (PLIP), as the main components of IMF, are of great significance to the improvement of meat quality.
Results
In this study, we used 30 RNA sequences generated from the transcriptome of chicken breast muscle tissues at different developmental stages to construct a gene expression matrix to map RNA sequence reads to the chicken genome and identify the transcript of origin. We used weighted gene co-expression network analysis (WGCNA) and identified 27 co-expression modules, 10 of which were related to TG and PLIP. We identified 150 highly-connected hub genes related to TG and PLIP, respectively, which were found to be mainly enriched in the adipocytokine signaling pathway, MAPK signaling pathway, mTOR signaling pathway, FoxO signaling pathway, and TGF-beta signaling pathway. Additionally, using the BioMart database, we identified 134 and 145 candidate genes related to fat development in the TG-related module and PLIP-related module, respectively. Among them, RPS6KB1, BRCA1, CDK1, RPS3, PPARGC1A, ACSL1, NDUFAB1, NDUFA9, ATP5B and PRKAG2 were identified as candidate genes related to fat development and highly-connected hub genes in the module, suggesting that these ten genes may be important candidate genes affecting IMF deposition.
Conclusions
RPS6KB1, BRCA1, CDK1, RPS3, PPARGC1A, ACSL1, NDUFAB1, NDUFA9, ATP5B and PRKAG2 may be important candidate genes affecting IMF deposition. The purpose of this study was to identify the co-expressed gene modules related to chicken IMF deposition using WGCNA and determine key genes related to IMF deposition, so as to lay a foundation for further research on the molecular regulation mechanism underlying chicken fat deposition.
Similar content being viewed by others


Introduction
With the continuous development of the broiler industry, the consumption of chicken as the main source of animal protein in China is increasing every year [1]. However, high-density breeding and high-intensity growth selection lead to continuous decline in the quality of chicken, due to changes in lipids and aroma compounds in chicken intramuscular fat (IMF), IMF are important precursors of flavor directly affecting the tenderness, flavor, color and juiciness of chicken meat, and thus are closely related to meat quality [2]. Therefore, to improve meat quality it is extremely important to elucidate the molecular mechanism underlying IMF deposition.
The mechanism of IMF deposition is a very complex, dynamic process consisting of a series of steps, such as adipocyte differentiation, lipid synthesis, transport and decomposition [3], which is affected by age, nutrition, environment and other factors [4]. It involves many metabolic pathways and related genes, such as PPARG, GPAT1, FABP4 and FABP5 [5], as well as signal pathways, such as IGF/IGF-1, MAPK, TGF-β /BMPs, Wnt/β-catenin [6], and gene expression varies at different stages. The number and volume of adipocytes are closely related to the content of IMF. Fat in other animals such as pigs is mainly synthesized in adipose tissue, while fat in poultry such as chicken is mainly synthesized in the liver [7]. Most chicken fatty acids are first synthesized in the liver and then transported by low-density lipoproteins or chylomicrons for storage in adipose tissue as triglycerides (TG), while exogenous fat is absorbed through the small intestine and transported to various target tissues [8], such as fat and muscle tissues, in the form of chylous particles. These fats are eventually hydrolyzed into fatty acids by lipoproteases and then oxidized to provide energy or deposit [9] in the target tissue.
The main components of IMF are TG, phospholipids (PLIP) and cholesterol [10]. TG are composed of glycerol and fatty acids, and as the main component of IMF play a crucial role in muscle lipid metabolism [11]. PLIP are a major component of cellular membranes and IMFs, which are hydrolyzed to produce a mixture containing fatty acids and phosphoric acid [12], and due to their rich fatty acid content PL are the main flavor compound, which is closely related to meat quality. Therefore, it is critical to study the role of TG and PLIP in the process of IMF deposition.
With the development of second-generation sequencing, transcriptome sequencing has been widely used in the study of gene expression and regulation at the genome-wide level [13]. There are a growing number of studies on chicken IMF deposition using transcriptome sequencing technology. Numerous genes involved in IMF and abdominal fat deposition in broilers have been identified through transcriptome data differential expression analysis and weighted gene co-expression network analysis (WGCNA) [14]. To date, many genes have been confirmed to be related to chicken IMF deposition. Differential expression analysis and WGCNA are currently the main methods available for analyzing transcriptome data [15]. Differential expression analysis is mainly used to analyze the differences in expression between samples from two groups and WGCNA is a method to construct a gene co-expression network based on gene expression data [16], which mainly analyzes the relationship between the co-expression of genes. To analyze the relationship between the co-expression of genes, genes are separated into multiple modules, and then key genes related to phenotypes are identified through correlation analysis between modules and sample phenotypes [17]. Therefore, this study mainly constructed a co-expression network based on the transcriptome data of pectoral muscle tissues at six different developmental stages using the WGCNA method to study the regulation of IMF deposition and identified key genes related to the regulation of IF deposition. In addition, this study provides a more comprehensive theoretical basis for the molecular mechanism underlying IMF deposition.
Materials and methods
Experimental material
The Jinxing yellow (JXY) chickens obtained from the Institute of Animal Sciences of Chinese Academy of Agricultural Sciences (Beijing, China) of different ages used in this study were fed the same diet and raised under the same conditions at the Changping Experimental Base of the Institute of Animal Sciences of Chinese Academy of Agricultural Sciences (Beijing, China). The chickens were stunned by electric shock and killed by cervical dislocation at different developmental stages. Samples were collected at six different developmental stages: D1 (the first day after birth), D7, D35, D63, D91, and D119. Body weight, breast muscle weight, and abdominal fat weight were recorded at slaughter, and tissue samples of breast muscle were collected, quickly frozen in liquid nitrogen, and stored at -80 °C. The specific sample information is shown in Table 1.
Collection of phenotypic data related to intramuscular fat
In order to obtain phenotypic traits related to IMF, the contents of TG and PLIP were determined in pectoral muscle samples at six different developmental stages: D1 (the first day after birth), D7, D35, D63, D91, and D119. Briefly, samples of 2.0 g of breast muscle tissue of JXY chicken were homogenized and analyzed to determine the content of TG and PL in breast muscle tissue [18], using the appropriate detection kits (Nanjing Jiancheng Bioengineering Research Institute of China and Beijing Leichuang Biotechnology Co., Ltd. of China).
RNA library construction and transcriptome sequencing analysis
Transcriptome sequencing was performed by Beijing Guoke Biological Technology Co., Ltd. (Beijing, China). Total RNA was extracted using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA). To ensure the accuracy of the data, before RNA sequencing (RNA-seq) we determined the purity (OD260/280), concentration, and nucleic acid absorption peak of the RNA using a Nanodrop spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA), and the RNA integrity using an Agilent 2100 Bioanalyzer system (Agilent Technologies Inc., Santa Clara, CA, USA). After determining that the sample met the RNA quality criteria (based on purity and RNA integrity index), the library was constructed, and the library quantity and quality were determined using Qubit 2.0 for preliminary quantification. In addition, before proceeding to the subsequent experiment, the insert size of the library was detected using the Agilent 2100 Bioanalyzer system and determined to meet the expectations and library inspection was completed by accurately quantifying the effective concentration of the library (library effective concentration > 2nM) by quantitative real-time polymerase chain reaction (qPCR). After determining that the quantity and quality requirements of the library were met, different libraries were pooled according to the target amount of data of the sequencing machine, and sequenced using the Illumina Novaseq platform (Illumina Inc., San Diego, CA, USA). High-quality sequencing sequencing data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-N and low quality reads from raw data. In addition, Q20, Q30, GC-content and sequence duplication level of the clean data were determined. All the downstream analyses were based on high-quality clean data [19]. Raw sequences were transformed into clean reads after data processing, and the clean reads were then mapped to the chicken reference genome sequence. Only reads with a perfect match or one mismatch were further analyzed and annotated based on the reference genome [20].
Construction of the weighted co-expression network
Using the Illumina sequencing platform to obtain transcriptome data, 21,987 genes were identified from the breast muscle samples of JXY chicken at six different developmental stages, namely D1 (the first day after birth), D7, D35, D63, D91, and D119. The data were used to construct the co-expression network by WGCNA using the WGCNA package in the R software. Abnormal samples in WGCNA will affect the analysis of module results, so before WGCNA, abnormal outlier samples were removed, 30 samples were clustered, and no outlier samples were found. The 21,987 genes identified were used to build a co-expression module, by first calculating the correlation coefficient of genes. In order to determine whether genes have similar expression patterns, it is necessary to set an appropriate soft threshold [21], so that the connection between genes in the network obeys the scale-free network. When the scale-free fitting index is 0.9, the soft threshold is 4(Fig. 2B), indicating good network connectivity. Therefore, 4 is chosen as the most appropriate soft threshold for building a co-expression module. Then, a co-expression network [22] was built through an appropriate soft threshold, and genes were classified according to their expression pattern. Genes with similar expression pattern were classified into one module, and different colors represent different modules. To associate modules with traits, principal component analysis [23] was performed on all genes within the modules. The traits related to IMF, mainly included TG and PLIP. The correlation of modules with IMF-related traits was calculated and displayed as a heatmap. At p < 0.05, modules were considered to be significantly associated with phenotypic traits.
Screening of hub genes
In WGCNA, gene significance (GS) refers to the absolute value of the correlation between genes and phenotypic traits, and module membership (MM) to the correlation of the module eigengene and the gene expression profile. Genes that are highly correlated with a phenotypic trait are important genes in the modules associated with that trait [22]. Thus, we selected the gene with high GS and MM values as the hub gene in the module and set |GS|>0.2, and the gene at |MM|>0.8 is the hub gene in each corresponding module. We then used Cystoscope 3.9.0 to construct the gene co-expression network to identify the top 30 genes with the highest degree of connectivity in the module as true hub genes and visualize them [24].
Functional enrichment analysis of modules and selection of candidate genes
Gene ontology (GO; http://www.geneontology.org/) is widely used in bioinformatics research, and the gene product aspects are divided into three categories: biological process (BP), cellular component (CC), and molecular function (MF). The Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/) pathway enrichment analysis can identify the metabolic and signal transduction pathways in which the proteins encoded by regulated genes are involved. In order to investigate the key modules of the hub genes, the KOBAS (version 3.0) software (http://kobas.cbi.pku.edu.cn/kobas3/?t=1) was used to identify enriched KEGG pathways and GO terms associated with significant phenotypic traits (P < 0.05 was considered significant). The genes in the module were annotated using the BioMart database (http://www.biomarbiomart.org/) [25] with the reference genome GRCg6a. We annotated the results based on their function and selected genes associated with fat development. Based on the above data, candidate genes affecting fat deposition and development were identified.
Quantitative real-time PCR (qPCR)
In order to verify the accuracy of RNA-seq analyses, three genes were selected for qPCR verification, primers, based on the sequence of each gene, were designed by the NCBI/Primer-Blast software, and sequence alignment was performed by BLASTN. Total RNA was extracted from each pectoral muscle tissue using TRIzol reagent (Invitrogen) and reverse transcribed using (2.0 µg) of total RNA each sample using the reverse transcription kit FastQuant RT Kit (Tiangen, Beijing, China) to obtain the cDNA. The qPCR reaction was performed in final volume of 10 µL, using 5 µL of 2 × iQTM SYBR Green Supermix, 0.5 µL of upstream primer, 0.5 µL of downstream primer, and 3 µL of cDNA. The samples were subjected to 40 cycles of amplification (95 °C 3 min, 95 °C 3 s, 60 °C 34 s) on a QuantStudio 7 Flex system (Applied Biosystems, Foster City, CA, USA). Amplification of each sample was performed in triplicate. ACTB was used as the internal reference gene, and the collected data were analyzed by the 2−ΔΔCT method.
Statistical analyses
The significance of differences between means was assessed using the IBM SPSS Statistics (version 26) software (IBM Corporation, Armonk, NY, USA). A P-value of less than 0.05 was considered statistically significant. Data are presented as the mean ± standard error of the mean.
Results
Phenotypic data associated with intramuscular fat
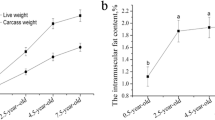
We measured the contents of TG and PLIP in the breast muscle tissue of JXY chickens at different developmental stages (Table 2). With increasing age, the contents of TG and PLIP increased, and the PLIP and TG content at ages 35 days and above was significantly higher than that at 1 and 7 days old. Also, there was no significant difference between the contents of TG at 1 and 7 days of age.
Quantitative real-time PCR (qPCR)
The fluorescence qPCR analysis of three selected genes showed that their expression trend was consistent with the transcriptome sequencing results. The qPCR analysis results were significantly correlated with those of the transcriptome sequencing analysis results, indicating that the transcriptome sequencing data were reliable as Fig. 1.

Correlation analysis of qPCR and RNA-seq data, the x-axis is the transcriptome expression value of the gene, and the y-axis is the qPCR value of the gene. * Indicates P < 0.05; ** indicates P < 0.01
Construction of co-expression modules
The gene expression matrix was constructed using the RNA sequences from chicken breast muscle tissues at different ages. Analysis of the 30 samples using the flashClust toolkit (R language), revealed that there were no outlier samples, so all samples were retained. The clustering diagram of the 30 samples is shown in Fig. 2A. Based on the scale-free network, the most suitable soft threshold was chosen to construct the co-expression network. When the scale-free fitting index is 0.9, the soft threshold is 4 (Fig. 2B), so 4 was chosen as the most suitable soft threshold to construct the co-expression module. The genes were divided into different modules, and the gene clustering tree was drawn. The mergeCutHeight for modules generation was set to 0.2, merge modules with a height lower than 0.2 and the minimum number of genes in a module was set to 100 to obtain 27 co-expression modules (Fig. 2C), and the number of genes in these modules ranged from 106 to 9765 (Table 3).

Sample clustering. There are no abnormal samples in the clustering tree

R2 = 0.9, β = 5; when R2 = 0.9, the optimal soft threshold is 5

Gene clustering tree. The upper part of the figure is the gene hierarchy clustering tree, and the lower part is the gene module. Each color represents a module
Module-phenotype correlation analysis
Phenotypic traits associated with IMF include TG and PLIP contents. A module eigengene (ME) refers to the level of gene expression in the module, and the phenotype-module-related heat map is shown in Fig. 3. A total of 10 modules were found to be significantly associated with the IMF trait of the pectoral muscle (P < 0.05). Eight modules, namely the brown, pink, turquoise, green, black, salmon, light green and grey60, were significantly correlated with TG content (P < 0.05), among which the salmon, black, pink, green, and turquoise modules were significantly negatively correlated with TG content (r=-0.5, p = 0.005; r=-0.51, p = 0.04;r=-0.46,p = 0.01;-0.71,p = 1e-5;r=-0.64,p = 2e-04), and the brown, lightgreen, and grey60 modules were significantly positively correlated with TG content (r = 0.71, P = 1e-0.5; r = 0.37, p = 0.04;r = 0.45, p = 0.01;), with the brown module showing the strongest correlation with TG content. Seven modules, namely the brown, pink, lightcyan, turquoise, green, black and cyan modules, were significantly correlated with PLIP content (P < 0.05), among which the pink, lightcyan, cyan, green, black, and turquoise modules were significantly negatively correlated with PLIP content (r=-0.63, p = 2e-04; r=-0.39,p = 0.03;r=-0.4,p = 0.03, r=-0.51, p = 0.04;r=-0.68,p = 3e-05;r=-0.79,p = 2e-07), and brown was significantly positively correlated with PLIP content (r = 0.84, p = 9e-09), showing the strongest correlation.

Correlation heat map between phenotype and module, The X-axis represents phenotypic information, and the Y-axis represents modules. The darker the color, the higher the correlation. Red represents positive correlation, blue represents negative correlation, and P < 0.05 indicates a significant correlation
Selection of candidate genes in relevant modules
Database After annotating the genes in the relevant modules using the BioMart database, we selted genes associated with fat deposition based on their functional annotation. There were 134 candidate genes associated with fat development in TG-related modules (Table 4), and 145 candidate genes associated with fat development in PLIP-related modules (Table 5). According to the above results, this experiment identified candidate genes that affect fat deposition and development.
Hub genes in fat deposition-related modules
A module that is significantly associated with the IMF-related phenotype means that the genes in the module are involved and play a key role in IMF formation. Therefore, for follow-up analysis, we focused on the genes in the module. To identify the hub genes in the module, the scatter plot of GS and MM correlations in the module was drawn (Fig. 4a, b), and the genes with |GS|>0.2 and |MM|>0.8 from each of the 5 modules associated with TG and PLIP were selected as the hub genes. We found 865, 390, 1539, 612, and 369 hub genes significantly associated with TG in the brown, pink, turquoise, green and black modules, respectively. We also found 872, 387, 1662, 684 and 473 hub genes significantly associated with PLIP in the brown, pink, turquoise, green and black modules, respectively. Overlapping hub genes identified by association with TG and hub gene identified by association with PLIP revealed that most of the genes overlapped (Fig. 5). Then, after constructing the gene co-expression network using Cystoscope3.9.0, the top thirty genes with the highest degree of connectivity in each module were selected as the true hub genes. Ultimately, 150 hub genes associated with TG and 150 hub genes associated with PLIP were identified, and the identified hub genes were visualized with Cytoscape 3.9.0 (Fig. 6a, b).

Scatter plot of the GS and MM. (a) Scatter plot of the GS and MM correlation in TG related modules. (b) Scatter plot of the GS and MM correlation in PLIP related modules. The ordinate axis represents the correlation between gene and phenotype, the abscissa axis represents the correlation between gene and module, and different colors represent different modules. The gene in the upper right corner of each figure is highly phenotype-related and is the hub gene in the module

Overlap of hub genes in the TG and PLIP-related modules, overlapping hub genes are important genes

Visualization of the top 30 genes in specific modules: black, brown, turquoise, green, pink. (a) Top 30 hub genes with the highest degree of connectivity in TG-related modules. (b) Top 30 hub genes with the highest degree of connectivity in PLIP-related modules. The thicker the line, the higher the correlation, and more lines indicate that the gene is more important
Functional enrichment analysis of TG and PLIP-related hub genes
The KEGG enrichment analysis of hub genes in the modules was mainly focused on important signaling pathways, such as the adipocytokine signaling pathway, MAPK signaling pathway, mTOR signaling pathway, FoxO signaling pathway, ErbB signaling pathway, TGF-beta signaling pathway, etc. GO term annotations were used to select the top ten enriched most significant terms in BP, CC, and MF (Fig. 7a, b). These GO terms were mainly annotated to translation (GO:0006412), mitochondrial respiratory chain complex I (GO:0005747) and structural constituent of ribosome (GO:0003735). The BPs annotated to these GO terms are all related to the formation of lipids. Among the 150 TG-related candidate genes nine hub genes, namely RPS6KB1, BRCA1, CDK1, RPS3, PPARGC1A, ACSL1, NDUFAB1, NDUFA9 and ATP5B, are also hub genes in the TG-related module, and among the 150 PLIP-related candidate genes, NDUFAB1, NDUFA9, ATP5B, PRKAG2, ACSL1, BRCA1, CDK1 and RPS3 are the hub gene in the PLIP-related module. Therefore, RPS6KB1, BRCA1, CDK1, RPS3, PPARGC1A, ACSL1, NDUFAB1, NDUFA9, ATP5B and PRKAG2 may be the key genes affecting IMF deposition(Fig. 8). These genes are mainly enriched in Adipocytokine signaling pathway, Insulin signaling pathway, FoxO signaling pathway.

Pathway enrichment analysis (a) and GO term annotation (b) of genes in the modules. BP, biological processes; CC, cellular components; MF, molecular functions

According to the Wayne diagram of correlation analysis, overlapping genes are important candidate genes. Intersection of hub gene and candidate gene in TG and PLIP related module;
Discussion
At present, there are much research on intramuscular fat in poultry at home and abroad, and important progress has been made. For example, the main signaling pathways and genes involved in the regulation of intramuscular fat during development were explored; The molecular mechanism of fat differential deposition regulation was revealed by studying the difference of fat in different parts, and the biochemical mechanism of IMF influence on meat flavor was also analyzed. In general, the existing research mainly focus on the screening of candidate genes and pathways of IMF, and the deep-seated molecular mechanisms of its directed deposition regulation remain to be studied.
IMF is a lipid mixture composed of TG, PLIP and TCHO closely related to meat quality [26], of which TG and PLIP are the most abundant. TG, as the main component of IMF, is closely related to fat metabolism and is a representative indicator of IMF content [27]. Therefore, increasing the content of TG is an important way to regulate IMF content, while the synthesis of TG and PLIP has a crucial role in fat deposition in muscles. It is well established that IMF deposition is a complex physiological process mediated through various mechanisms involving multiple metabolic and signaling pathways regulated by proteins encoded by many functional candidate genes Several studies have shown that SLC16A7 is involved in TG deposition and mainly plays a role in muscle cells [28].The developmental stage is the major muscle and IMF formation period where myocytes and adipocytes interact when they are in close proximity in muscle tissue [29]. Thus, the identification of co-expressed genes in developing muscle tissue is important to understand the muscle formation process and the regulation of meat quality, which can reveal the molecular regulatory relationships between them. In this study, samples of JXY chicken breast muscle were collected from seven different developmental stages, namely D1 (first day after birth), D7, D35, D63, D91 and D119, and subjected to transcriptome sequencing and WGCNA to identify genes associated with IMF deposition at different developmental stages.
WGCNA can make full use of phenotypic information, convert the association between many genes and phenotypes into the relationship between gene set and phenotype, and cluster genes with similar expression patterns into one module, therefore we use WGCNA to construct a gene co-expression network [30]. at different developmental stages and identify key genes affecting IMF deposition according to TG and PLIP content phenotypes. WGCNA was used to identified 27 co-expression modules and found 8 and 7 modules significantly related to TG and PLIP, respectively. Additionally, it was also used to analyze the hub genes in the TG and PLIP significantly related modules and found that most of the genes affecting TG and PLIP overlapped. Since TG is composed of glycerol and fatty acids [31] and PLIP is composed of phosphate and fatty acids [32], we suggest that these genes affect the contents of TG and PLIP and thus IMF deposition mainly by affecting fatty acid content. Fatty acids are the basic components of cells [33], and their content and type are important factors affecting the nutritional value and taste of chicken meat as well as important precursors of volatile compounds [34], which play a key role in various biological processes, such as energy storage, metabolic regulation, and information transfer in animal bodies. Genetics and diet affect the composition of fatty acids, different fatty acids have different melting points, so for pigs and ruminants, fatty acids affect the hardness of adipose tissue [35].
Studies have shown that the FABP gene family is an important candidate gene family in the regulation of the IMF content, meat tenderness and flavor, and is related to lipid metabolism pathways, such as those involved in fat decomposition and production [36]. As a marker of adipocyte differentiation, FABP4 can regulate the expression of PPARG and play an important role in fatty acid transport and metabolism [37].
Enrichment analysis of genes in TG- and PL-related modules found 39 significant signaling pathways, including some important signaling pathways, such as MAPK signaling pathway, mTOR signaling pathway, and FoxO signaling pathway. Some studies have shown that the MAPK signaling pathway can affect the production of fat and regulate the lipid metabolism through the PPAR signaling pathway [38]. Also, as a target gene of microRNA miR-29a, CTRP6 can promote the proliferation and differentiation of intramuscular and subcutaneous adipocytes through the MAPK signaling pathway [39].
In this study, candidate genes related to fat deposition in TG- and PL-related modules were identified. Specifically, 134 genes related to fat deposition in TG-related modules and 145 genes related to fat deposition in PLIP-related modules were identified. Among the top 30 selected hub genes with the highest connectivity in TG- and PL-related modules, RPS6KB1, BRCA1, CDK1, RPS3, PPARGC1A, ACSL1, NDUFAB1, NDUFA9 and ATP5B genes were found to be both candidate genes related to adipogenesis and highly linked hub genes in the TG-related module, while NDUFAB1, NDUFA9, ATP5B, PRKAG2, ACSL1, BRCA1, CDK1 and RPS3 were found to be both adipogenesis-related and highly linked hub genes in the PLIP-related module. Therefore, RPS6KB1, BRCA1, CDK1, RPS3, PPARGC1A, ACSL1, NDUFAB1, NDUFA9, ATP5B and PRKAG2 may be key genes affecting IMF deposition in chickens.
Most of the genes are related to fat deposition, some studies have found that the ELOVL6 gene is positively correlated with the proliferation of bovine precursor adipocytes, and the knockdown of ELOVL6 expression significantly downregulated the protein expression levels of CDK1 and PCNA, prolonging the cell cycle transition from G2 phase to M phase, thus inhibiting the proliferation of precursor adipocytes [40]. Another study found that stearic acid is transported to dairy cow mammary epithelial cells through FATP4, upregulating the expression of CDK1, and then activating the PI3K-mTOR-4EBP1/S6K and mTOR-SREBP-1 signaling axes to promote the synthesis of milk fat milk protein [41]. Research indicated a remarkable upregulation of BRCA1 in the LW pig, signifying its pivotal role in modulating fatty acid and lipid metabolism, alongside controlling obesity [42]. Likewise, PPARGC1A, acting as a transcription factor, exerts profound influence over the regulation of white adipocyte differentiation and the deposition of bovine intramuscular fat (IMF) [43]. Furthermore, research has shown PPARGC1A expedites the accumulation of IMF through its positive regulation of saturated and monounsaturated fatty acid metabolism [44].
Investigations have demonstrated that the overexpression of NDUFAB1 confers substantial protection against obesity and insulin resistance in murine models [45]. The porcine ATP5B gene exhibits extensive transcriptional expression across all tissues, with particular prominence in the delicate testis and adipose tissue, thereby implicating its involvement in skeletal muscle development and meat quality [46]. Moreover, PRKAG1, a distinguished member of the AMPK (AMP-activated protein kinase) family, substantially influences pork quality and backfat thickness [47]. Nevertheless, current literature remains scant in regard to the exploration of RPS6KB1, RPS3, CDK1, and NDUFA9 genes in adipose tissue. Thus, further investigations are imperative to unravel the intricate associations between the proteins, encoded by these genes, and their involvement in fat deposition.
Conclusion
Ten modules with significant correlation with IMF traits were identified by WGCNA. RPS6KB1, BRCA1, CDK1, RPS3, PPARGC1A, ACSL1, NDUFAB1, NDUFA9, ATP5B and PRKAG2 are not only candidate genes related to fat development but also hub genes in related modules, which may be important candidate genes regulating IMF deposition in chickens. Hub genes in WGCNA module mainly affect TG and PLIP contents by regulating fatty acid metabolism, thereby affecting IMF deposition. It is of great significance to improve meat quality and meat safety to meet people’s demand for high-quality ecological products and improving breeding technology.
Data Availability
The RNA sequencing raw data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA011927) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
References
Weng K, Huo W, Li Y, Zhang Y, Zhang Y, Chen G, Xu Q. Fiber characteristics and meat quality of different muscular tissues from slow- and fast-growing broilers. Poult Sci. 2022;101(1):101537.
Hou X, Zhang R, Yang M, Niu N, Wu J, Shu Z, Zhang P, Shi L, Zhao F, Wang L, et al. Metabolomics and lipidomics profiles related to intramuscular fat content and flavor precursors between Laiwu and Yorkshire pigs. Food Chem. 2023;404(Pt A):134699.
Fu RQ, Liu RR, Zhao GP, Zheng MQ, Chen JL, Wen J. Expression profiles of key transcription factors involved in lipid metabolism in Beijing-You chickens. Gene. 2014;537(1):120–5.
Zhang M, Li D, Zhai Y, Wang Z, Ma X, Zhang D, Li G, Han R, Jiang R, Li Z, et al. The Landscape of DNA methylation Associated with the Transcriptomic Network of Intramuscular adipocytes generates insight into intramuscular Fat Deposition in Chicken. Front cell Dev Biology. 2020;8:206.
Liu L, Liu X, Cui H, Liu R, Zhao G, Wen J. Transcriptional insights into key genes and pathways controlling muscle lipid metabolism in broiler chickens. BMC Genomics. 2019;20(1):863.
Yan J, Gan L, Qi R, Sun C. Adiponectin decreases lipids deposition by p38 MAPK/ATF2 signaling pathway in muscle of broilers. Mol Biol Rep. 2013;40(12):7017–25.
Lu Z, He XF, Ma BB, Zhang L, Li JL, Jiang Y, Zhou GH, Gao F. Increased fat synthesis and limited apolipoprotein B cause lipid accumulation in the liver of broiler chickens exposed to chronic heat stress. Poult Sci. 2019;98(9):3695–704.
Cui H, Zheng M, Zhao G, Liu R, Wen J. Identification of differentially expressed genes and pathways for intramuscular fat metabolism between breast and thigh tissues of chickens. BMC Genomics. 2018;19(1):55.
Nematbakhsh S, Pei Pei C, Selamat J, Nordin N, Idris LH, Abdull Razis AF. Molecular Regulation of Lipogenesis, Adipogenesis and Fat Deposition in Chicken. Genes 2021, 12(3).
Qiu F, Xie L, Ma JE, Luo W, Zhang L, Chao Z, Chen S, Nie Q, Lin Z, Zhang X. Lower expression of SLC27A1 enhances intramuscular Fat Deposition in Chicken via Down-regulated fatty acid oxidation mediated by CPT1A. Front Physiol. 2017;8:449.
Cui H, Liu L, Liu X, Wang Y, Luo N, Tan X, Zhu Y, Liu R, Zhao G, Wen J. A selected population study reveals the biochemical mechanism of intramuscular fat deposition in chicken meat. J Anim Sci Biotechnol. 2022;13(1):54.
Souillard C, Soula G. [Chicken erythrocyte nuclei: composition and analysis of phospholipids]. Biochimie. 1976;58(10):1263–71.
Li XJ, Yang H, Li GX, Zhang GH, Cheng J, Guan H, Yang GS. Transcriptome profile analysis of porcine adipose tissue by high-throughput sequencing. Anim Genet. 2012;43(2):144–52.
Xing S, Liu R, Zhao G, Liu L, Groenen MAM, Madsen O, Zheng M, Yang X, Crooijmans R, Wen J. RNA-Seq analysis reveals hub genes involved in Chicken Intramuscular Fat and Abdominal Fat Deposition during Development. Front Genet. 2020;11:1009.
Yuan H, Lu J. Consensus module analysis of abdominal fat deposition across multiple broiler lines. BMC Genomics. 2021;22(1):115.
Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Luo N, Shu J, Yuan X, Jin Y, Cui H, Zhao G, Wen J. Differential regulation of intramuscular fat and abdominal fat deposition in chickens. BMC Genomics. 2022;23(1):308.
Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE. 2012;7(2):e30619.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60.
Gao Z, Ding R, Zhai X, Wang Y, Chen Y, Yang CX, Du ZQ. Common Gene Modules Identified for Chicken Adiposity by Network Construction and Comparison. Front Genet. 2020;11:537.
Yip AM, Horvath S. Gene network interconnectedness and the generalized topological overlap measure. BMC Bioinformatics. 2007;8:22.
Li G, Zhao Y, Li Y, Chen Y, Jin W, Sun G, Han R, Tian Y, Li H, Kang X. Weighted gene coexpression network analysis identifies specific transcriptional modules and hub genes related to intramuscular fat traits in chicken breast muscle. J Cell Biochem. 2019;120(8):13625–39.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Haider S, Ballester B, Smedley D, Zhang J, Rice P, Kasprzyk A. BioMart Central Portal–unified access to biological data. Nucleic Acids Res 2009, 37(Web Server issue):W23–27.
Meng Q, Ma M, Zhang W, Bi Y, Cheng P, Yu X, Fu Y, Chao Y, Ji T, Li J, et al. The gut microbiota during the progression of Atherosclerosis in the perimenopausal period shows specific compositional changes and significant correlations with circulating lipid metabolites. Gut Microbes. 2021;13(1):1–27.
Liu L, Cui HX, Zheng MQ, Zhao GP, Wen J. Comparative analysis of differentially expressed genes related to triglyceride metabolism between intramuscular fat and abdominal fat in broilers. Br Poult Sci. 2018;59(5):514–20.
Wang Y, Liu L, Liu X, Tan X, Zhu Y, Luo N, Zhao G, Cui H, Wen J. SLC16A7 promotes triglyceride deposition by De Novo Lipogenesis in Chicken muscle tissue. Biology 2022, 11(11).
Yin Y, Chen F, Li J, Yang J, Li Q, Jin P. AURKA enhances autophagy of adipose derived stem cells to promote Diabetic Wound Repair via Targeting FOXO3a. J Invest Dermatol. 2020;140(8):1639–1649e1634.
Zhang J, Lan Q, Lin J. Identification of key gene modules for human osteosarcoma by co-expression analysis. World J Surg Oncol. 2018;16(1):89.
Lísa M, Netušilová K, Franěk L, Dvořáková H, Vrkoslav V, Holčapek M. Characterization of fatty acid and triacylglycerol composition in animal fats using silver-ion and non-aqueous reversed-phase high-performance liquid chromatography/mass spectrometry and gas chromatography/flame ionization detection. J Chromatogr A. 2011;1218(42):7499–510.
de Carvalho C, Caramujo MJ. The various roles of fatty acids. Molecules 2018, 23(10).
Das UN. Essential fatty acids in health and Disease. J Assoc Phys India. 1999;47(9):906–11.
Larick DK, Turner BE, Schoenherr WD, Coffey MT, Pilkington DH. Volatile compound content and fatty acid composition of pork as influenced by linoleic acid content of the diet. J Anim Sci. 1992;70(5):1397–403.
Wood JD, Enser M, Fisher AV, Nute GR, Sheard PR, Richardson RI, Hughes SI, Whittington FM. Fat deposition, fatty acid composition and meat quality: a review. Meat Sci. 2008;78(4):343–58.
Chen XY, Niu JJ, Geng ZY. Gene expression and plasma lipid content in relation to intramuscular fat in Chinese indigenous Wuhua chicken. J Appl Poult Res. 2017;26(3):391–400.
Wang G, Kim WK, Cline MA, Gilbert ER. Factors affecting adipose tissue development in chickens: a review. Poult Sci. 2017;96(10):3687–99.
Guo L, Cui H, Zhao G, Liu R, Li Q, Zheng M, Guo Y, Wen J. Intramuscular preadipocytes impede differentiation and promote lipid deposition of muscle satellite cells in chickens. BMC Genomics. 2018;19(1):838.
Wu W, Zhang J, Zhao C, Sun Y, Pang W, Yang G. CTRP6 regulates porcine adipocyte proliferation and differentiation by the AdipoR1/MAPK signaling pathway. J Agric Food Chem. 2017;65(27):5512–22.
Junjvlieke Z, Mei CG, Khan R, Zhang WZ, Hong JY, Wang L, Li SJ, Zan LS. Transcriptional regulation of bovine elongation of very long chain fatty acids protein 6 in lipid metabolism and adipocyte proliferation. J Cell Biochem. 2019;120(8):13932–43.
Li F, Hu G, Long X, Cao Y, Li Q, Guo W, Wang J, Liu J, Fu S. Stearic acid activates the PI3K-mTOR-4EBP1/S6K and mTOR-SREBP-1 signaling axes through FATP4-CDK1 to promote milk synthesis in primary bovine mammary epithelial cells. J Agric Food Chem. 2022;70(13):4007–18.
Huang W, Zhang X, Li A, Xie L, Miao X. Genome-wide analysis of mRNAs and lncRNAs of intramuscular Fat related to lipid metabolism in two Pig breeds. Cell Physiol Biochem. 2018;50(6):2406–22.
Ramayo-Caldas Y, Fortes MR, Hudson NJ, Porto-Neto LR, Bolormaa S, Barendse W, Kelly M, Moore SS, Goddard ME, Lehnert SA, et al. A marker-derived gene network reveals the regulatory role of PPARGC1A, HNF4G, and FOXP3 in intramuscular fat deposition of beef cattle. J Anim Sci. 2014;92(7):2832–45.
Wheeler MA, Smith SD, Saito N, Foster HE Jr., Weiss RM. Effect of long-term oral L-arginine on the nitric oxide synthase pathway in the urine from patients with interstitial cystitis. J Urol. 1997;158(6):2045–50.
Zhang R, Hou T, Cheng H, Wang X. NDUFAB1 protects against obesity and insulin resistance by enhancing mitochondrial metabolism. FASEB J. 2019;33(12):13310–22.
Xu H, Xu Y, Liang X, Wang Y, Jin F, Liu D, Ma Y, Yuan H, Song X, Zeng W. Porcine skeletal muscle differentially expressed gene ATP5B: molecular characterization, expression patterns, and association analysis with meat quality traits. Mamm Genome. 2013;24(3–4):142–50.
Demeure O, Liaubet L, Riquet J, Milan D. Determination of PRKAG1 coding sequence and mapping of PRKAG1 and PRKAG2 relatively to porcine back fat thickness QTL. Anim Genet. 2004;35(2):123–5.
Acknowledgements
We thanks the Institute of Animal Science of CAAS,Chinese Academy of Agricultural Sciences for the support of this experiment.
Funding
The research was supported by Nanning Major Science and Technology Project in 2022 (20222025)(Guangxi, China),Modern Agricultural Industrial Technology System Guangxi Broiler Innovation Team Construction Special Fund (nycytxgxcxtd-04-20-4), China National Broiler Industry Technology System Construction Special Fund (CARS-41-Z16).
Author information
Authors and Affiliations
Contributions
JZ analyzed the data and draft the manuscript. YW prepared the tissue samples for sequencing. YS and MZ conducted data management. ZC and HC initiated this study, designed the experiments and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
.
This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the Ministry of Science and Technology (Beijing, China). The study was approved by the Research Ethics Committee of the Institute of Animal Sciences of the Chinese Academy of Agricultural Sciences (Beijing, China) (Protocol No. IAS2019-21). All methods were carried out in accordance with relevant guidelines and regulations. This study was carried out in compliance with the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).
Consent for publication
Not applicable.
Author details
1College of Animal Science and Technology, Qingdao Agricultural University, Qingdao 266109, China.
2State Key Laboratory of Animal Nutrition, Key Laboratory of Animal (Poultry) Genetics Breeding and Reproduction, Ministry of Agriculture, Institute of Animal Sciences, Chinese Academy of Agricultural Sciences, Beijing 100193, China.
3Guangxi Jingling Agriculture and animal Husbandry Group Co., LTD,Nanning 530049,China.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, J., Wang, Y., Su, Y. et al. RNA sequencing identifies key genes involved in intramuscular fat deposition in chickens at different developmental stages. BMC Genomics 25, 219 (2024). https://doi.org/10.1186/s12864-023-09819-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09819-y