
Abstract
Barley (Hordeum vulgare) is one of the most popular cereal crops globally. Although it is a diploid species, (2n = 2x = 14) the study of its genome organization is necessary in the framework of plant breeding since barley is often used in crosses with other cereals like wheat to provide them with advantageous characters. We already have an extensive knowledge on different stages of the meiosis, the cell division to generate the gametes in species with sexual reproduction, such as the formation of the synaptonemal complex, recombination, and chromosome segregation. But meiosis really starts with the identification of homologous chromosomes and pairing initiation, and it is still unclear how chromosomes exactly choose a partner to appropriately pair for additional recombination and segregation. In this work we present an exhaustive molecular analysis of both telomeres and subtelomeres of barley chromosome arms 2H-L, 3H-L and 5H-L. As expected, the analysis of multiple features, including transposable elements, repeats, GC content, predicted CpG islands, recombination hotspots, G4 quadruplexes, genes and targeted sequence motifs for key DNA-binding proteins, revealed a high degree of variability both in telomeres and subtelomeres. The molecular basis for the specificity of homologous recognition and pairing occurring in the early chromosomal interactions at the start of meiosis in barley may be provided by these polymorphisms. A more relevant role of telomeres and most distal part of subtelomeres is suggested.
Similar content being viewed by others

Core ideas
-
Barley is a useful species for wheat breeding
-
Unzip the molecular bases for homologous chromosome recognition in meiosis is essential for plant breeding
-
Telomeres and subtelomeres contribute to chromosome recognition and pairing during meiosis in barley
-
A molecular analysis of barley subtelomeres revealed a high polymorphism for all the features analyzed
Introduction
Barley (Hordeum vulgare) is one of the first domesticated plants and is also the fourth most popular cereal crop globally [83]. Barley has three primary applications: malt production, human consumption, and animal feed [59]. In a breeding context, barley is frequently employed in crosses with other cereal crops, such as wheat, to provide them with advantageous characteristics. For instance, the incorporation of both wild and cultivated barley species (Hordeum chilense and H. vulgare) into bread wheat were established several years ago [34, 62]. These methods have not only been used to transfer desirable agronomic traits into wheat but also to investigate chromosome dynamics [17, 65, 82]. Thus, breeders can develop interspecific genetic crosses to obtain new genetic combinations to be used as new crops or enlarge the genetic basis of current crops. Unfortunately, hybridization between wheat and a related species such as barley produces only a low level of chromosome pairing and recombination having undesirable implications in the transfer to wheat of alien genes controlling important agronomical traits. Therefore, understanding the genome organization and how chromosomes interact at the beginning of meiosis in both species is key for genetics and plant breeding purposes.
Chromosome recognition and pairing must occur at the beginning of meiosis. Information about other meiotic processes, such as the formation of the synaptonemal complex, recombination and chromosome segregation is available [13, 41, 43], but how chromosomes specifically identify a partner to properly pair for further recombination and segregation remains to be elucidated. Because chromosome recognition and pairing are highly dynamic processes that only occur between specific chromosome regions and may not be synchronized from one nucleus to the next, these initial chromosome interaction studies are still challenging [112]. In higher eukaryotes, telomere-mediated reorganization at the beginning of meiosis seems to be a widely conserved first step in the homology search process of homologous chromosome recognition and pairing [14, 91, 112]. A DNA conformational change has been described in wheat and barley chromosomes when telomeres (and subtelomeres) correctly associated at the onset of meiosis, which is triggered along the chromosomes and is directly correlated with homologous recognition and pairing [16, 79].
Telomeres, stretches of repetitive sequences found at the ends of chromosomes, exhibit a high degree of conservation across eukaryotic organisms, also serve the essential function of preventing various chromosome-related issues, including end-to-end fusion, recombination, and the degradation of chromosome ends [49, 55,56,57, 60]. Moreover, these structures may also play a role in regulatory processes and meiosis [11, 12, 76, 97,98,99]. At the beginning of meiosis, a structure (known as “bouquet”) formed by the association and clustering of telomeres at the inner nuclear envelope in many species including wheat and barley, facilitates the initial interactions between homologues for recognition and pairing to enable subsequent recombination [8, 69, 72, 91, 113].
Additional chromosomal regions, particularly subtelomeres located adjacent to telomeres, are also considered when determining pairing specificity as chromosomes come into close proximity within the bouquet structure. This is because the telomeric DNA sequence itself is highly conserved [16]. Although subtelomeres are an attractive target for research, their polymorphism presents a technical obstacle [1]. Subtelomeres play a role in telomere maintenance through processes such as recombination or epigenetic modification. They are gene-rich areas, but they are less conserved than telomeres and often contain recombination hotspots [86, 21, 47, 51]. Furthermore, it's worth noting that the nucleotide sequences of telomeres and subtelomeres in many sequenced genomes have not been comprehensively characterized, despite their significant functional relevance [45, 57, 63], making the evaluation of their putative conserved roles more difficult.
The majority of studies on subtelomeres have concentrated on the distal 500 Kb of each chromosome arm in the species under investigation, including humans, Arabidopsis, rice, and wheat [1, 40, 52, 53, 63]. However, the detailed molecular organization of subtelomeres is still unclear.
Several hypotheses regarding the functions of subtelomeres in chromosome dynamics and genome stability have been proposed due to their high polymorphism. For instance, in rice, subtelomeres may play a role in promoting recombination and the insertion of transposons [23]. In wheat and rye, recombining areas are frequently involved in chromosome recognition and pairing between homologues [101]. In fact, in all eukaryote kingdoms, including plants, recombination occurs significantly in the subtelomeric region [26, 47, 51, 89]. The presence of an extra pair of barley homologous chromosomes with terminal deletions in the wheat background has further highlighted the significance of subtelomeres in the processes of chromosome pairing and recognition. This is evident as chromosome recognition, pairing, and recombination fail when subtelomeres are absent [16].
Therefore, the subsequent crucial step involves conducting an in-depth molecular analysis of both telomeres and subtelomeres to gain a deeper understanding of their structural characteristics and any distinctive traits that may be associated with the initial stages of chromosome recognition and pairing. A comprehensive analysis has previously been carried out in wheat, revealing substantial polymorphism among homologous chromosomes across all the examined features [1].
In this study, we have identified and characterized the telomeric and subtelomeric sequences in barley chromosome arms where telomeric sequences were annotated (2H-L, 3H-L, and 5H-L), thereby enabling the precise delineation of the subtelomeric sequence's starting point. To the best of our knowledge, this represents the first molecular analysis of barley telomere sequences to date. Additionally, we have undertaken a detailed examination of subtelomeres within the 500 Kb distal subtelomeric regions and expanded this investigation to cover a broader chromosome region (5 Mb) in those chromosomes where telomeric sequences were identified. Our findings contribute to our understanding of the molecular structure of barley telomeres and subtelomeres and raise the possibility that the distinct patterns of various DNA repetitive sequences and DNA protein-binding sequences may play a role in chromosome specificity, which is required for homologous chromosome associations and recombination.
Materials and methods
Plant material and fluorescence in situ hybridization
Barley root tips from H. vulgare were used for in situ hybridization experiments. Three DNA probes were fluorescently labelled following standard procedures, the barley subtelomeric sequence HvT01 [10], the highly conserved telomeric sequence pAt74 originally isolated from A. thaliana [85], and the GAA satellite sequence isolated from barley [74] for identifying chromosomes. All the methods for preparing mitotic chromosomes spreads and in situ hybridization experiments have been described previously [77, 78].
DNA sequences
All the sequences analyzed in this study were obtained from NCBI (RefSeq: MorexV3 https://www.ncbi.nlm.nih.gov/assembly/GCF_904849725.1).
DNA sequence analysis and prediction tools
Telomere analysis was done utilizing NCBI sequences. Plots of this analysis were done with the informatic software GraphPad Prism 6.
Subtelomere analysis was done as follows. Prediction of coding genes and non-coding RNAs was done using EnsemblPlants (https://plants.ensembl.org/index.html). In EnsemblPlants, coding genes were detected by IPK database (Leibniz Institute of Plant Genetics and Crop Plant Research) and non-coding RNAs were detected by EoRNA database (Barley Expression Database that displays gene and transcript abundance using Barley Reference Transcript (BaRTv1.0) from The James Hutton Institute).
Low complexity domains (A, AG and G rich domains), simple repeats, DNA transposons, long terminal repeats (LTR), type I LINE transposons and type I SINE transposons were detected using RepeatMasker (Interspersed Repeat Masking Based on Protein Similarity, http://repeatmasker.org/cgi-bin/RepeatProteinMaskRequest) and Censor (https://www.girinst.org/censor/index.php). Parameters for the analysis with Censor were sets as follows: the sequence source used was Triticum, and this source was composed of wheat, barley and rye.
Emboss CpG plot (https://www.bioinformatics.nl/cgi-bin/emboss/cpgplot) was used for CG content calculation and CpG island prediction [84]. Parameters for the analysis were as follows: window Size (100), minimum length of a reported island (200), minimum observed/expected before a CpG island is reported (0.6), minimum average percentage of C plus G in a set of 10 windows that are required before a CpG island is reported (50).
Hot and cold recombination spots were predicted with iRSpot-EL (http://bliulab.net/iRSpot-EL/) [50]. Size of sliding window (in Kb) and step size parameters were set at 2 and 200, respectively.
Sequences associated with hot recombination spots [19] were identified (simple repeat: CCGCCGCCG, and sequences associated with transposable elements: CTCCCTCC, TTAGTCCCGGTT). These sequences were localized and displayed by means of MAST (MEME Suite 5.0.5) (https://meme-suite.org/meme/tools/mast) [7]. Parameters were set as follows: direct and reverse complement sequences were analyzed, and results combined, E-value ≤ 10 (MAST displays all sequences, exact or degenerate, matching query with E-values below the given specified threshold), the p-value of a hit must be less than 0.0001 to be shown in the output.
Distribution of predicted DNA-binding sites of putative barley proteins homologous to SMC1β cohesin (CCACCAGGTGGC), YY1 (GGGGGCAGTGG) and HMG proteins ([AT] n > 5) was obtained by means of MAST (MEME Suite 5.0.5) (https://meme-suite.org/meme/tools/mast) [7]. Parameters were set as follows: direct and reverse complement sequences were analyzed and results combined, E-value ≤ 10 (MAST displays all sequences, exact or degenerate, matching query with E-values below the given specified threshold); the p-value of a hit must be less than 0.0001 to be shown in the output.
In both regions (telomere and subtelomere), DNA analyser (G4 hunter) (https://bioinformatics.ibp.cz/#/) has been used for the study of G-quadruplexes. For this analysis we tried the standard threshold (1.2), but we finally used a more restrictive threshold (1.8).
Results
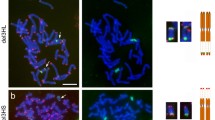
The sequences located at the ends of all barley (Hordeum vulgare) chromosomes were scrutinized using the data available from NCBI. Barley has 7 pairs of chromosomes (chromosomes 1H, 2H, 3H, 4H, 5H and 7H). In situ hybridization experiments were performed in somatic metaphase chromosome spreads to visualize the telomeric repeat and the subtelomeric region on barley. As expected, all barley chromosome ends contained the telomeric repeat and a high variability for the subtelomeric sequence was found among different barley chromosomes (Fig. 1). However, we found that only the assemblies of chromosomes 2H, 3H and 5H long arms contained plant terminal telomeric repeats (5’-TTTAGGG-3’). For this reason, we decided to restrict our study to the long arm ends of chromosomes 2H, 3H and 5H.

In situ hybridization (FISH) of barley chromosomes. a Telomeres in barley chromosomes (red). b Subtelomeres in barley chromosomes (green) and GAA (red). Chromosomes were counterstained with DAPI (blue). Scale bar represents 10 μm for both panels
We focused our study on the chromosome ends comprising the telomeric repeat sequences and the distal part of subtelomeres adjacent to the telomeric repeat sequences.We identified chromosome arms that included the telomeric repeat sequence (Table 1). We found differences on the length of the three telomeres analyzed, being 3H-L chromosome telomere the longest and 2H-L chromosome telomere the shortest.
By sequence analysis, we have detected nucleotide additions, deletions and substitutions within the consensus sequence that conforms this repetition unit along the telomeric sequence (Fig. 2). A small number of nucleotide substitutions have been identified in this study. We only detected a fraction of 0.16%, 0.014% and 0.13% of nucleotides substitutions in barley chromosome arms 2H-L, 3H-L and 5H-L, respectively (Fig. 2c). All of them were single base substitutions. We found that adenine was the base that suffered the most this type of mutation (50% of total substitutions). In contrast with nucleotide substitutions, a higher number of nucleotide additions and deletions were detected in this chromosome region. In detail, the percentage of additions was 1.73% on 2H-L chromosome arm, 0.81% on 3H-L chromosome arm and 1.2% on 5H-L chromosome arm. Besides, we annotated a 2.81%, 0.89% and 1.62% deletions on 2H-L, 3H-L and 5H-L chromosome arms, correspondingly (Table 2). We found additions and deletions within the whole telomeric region, reporting a higher concentration of both types of mutation on the two ends of the telomere (Fig. 2a, b). It is worthy to say that 2H-L chromosome arm contained the highest accumulation of mutations (substitutions, additions and deletions), 4.71%, in contrast with 3H-L and 5H-L chromosome arms, which only have 1.72 and 3.03%, respectively (Fig. 2d).

DNA variations for the repeat plant telomeric sequence (TTTAGGG) including nucleotides additions, deletions and substitutions along the telomeric sequence identified in chromosome arms 2H-L, 3H-L and 5H-L. a Additions (b) Deletions (c) Substitutions (d) All types of base mutations
In details, numerous deletions of one of the Ts in TTT were found on the three chromosome arms that were examined, a lower number of G deletion in GGG was observed. Many additions of G were also observed in GGG. On chromosome arm 2H-L, 114 deletions were detected, of which 82% were T deletions that variated the sequence from TTTAGGG to TTAGGG. Also, we found 62 additions, 87% of these mutations were G additions, transforming the canonical repeat sequence to TTTAGGGG. 131 deletions and 115 additions were located on chromosome arm 3H-L. T deletions correspond to 88% and G additions to 76.52%. On chromosome arm 5H-L, we found 96 deletions of whom 63.54% were T deletions. On this chromosome arm, we also found 68 additions, were G additions accounted for 75% of total additions. We found substitutions too, but this type of mutation is not as common as deletions and additions on 2H-L, 3H-L and 5H-L barley chromosome arms.
G-Quadruplexes (G4), structures rich in guanine, were also studied in the telomere. Very different densities of G4s were found. 2H-L was the chromosome arm that presented a higher frequency of G4s (6.9/1000 bp) in comparison with 3H-L and 5H-L, which presented 1,7/1000 bp and 4.2/1000 bp, respectively (Table 3). A higher number of G4s were detected within the most distal and the most proximal parts of telomeres in all chromosome arms 2H-L, 3H-L and 5H-L (Fig. 3).

Localization of quadruplexes (G4s) within the terminal sequence of 2H-L, 3H-L and 5H-L barley chromosome arms using a threshold of 1.8 in (a) telomeric sequence, (b) subtelomeric sequence, and (c) three different chromosome regions from the telomeric sequence
A characterization of the distal subtelomeric region (500 Kb) has been carried out in barley chromosome arms 2H-L, 3H-L and 5H-L, focusing on different features that could be related to chromosome recognition and pairing within the subtelomeric region: genes, transposable elements, repeat sequences, CG content and CpG island, distribution of binding sites of proteins putatively involved in chromosome pairing and recombination, as well as predicted hot/cold recombination spots. For some of the most relevant features, the analysis was extended to a larger 5 Mb sequence.
We also analyzed the abundance and distribution of G-Quadruplexes (G4) within the 500-kb distal subtelomere region of chromosome arm 2H-L, 3H-L and 5H-L. A higher concentration of G4s was found near the start of the telomere, and different abundances and distributions were found among the three different chromosome arms examined (Fig. 3). Chromosome arm 3H-L was the one that presented less frequency of G4s in its subtelomeric sequence, 0.27/1000 bp. Chromosome arm 5H-L showed the higher frequency, 0.30/1000 bp, and chromosome arm 2H-L presented a frequency of 0.28/1000 bp (Table 4). The analysis was also done on 500-kb stretches located 5 Mb and 50 Mb from the telomere, and this analysis revealed a significantly lower abundance of G4s within the 500-kb distal subtelomere in comparison with the regions closer to the centromere (Table 4, Fig. 3).
The location of gene sequences was also studied in barley subtelomeres (Fig. 4a: protein coding genes; Fig. 4b: RNA genes; direct and reverse complementary sequences were considered in both cases) within the distal subtelomeres of chromosomes 2, 3 and 5 long arms. A total of 43 genes were predicted in these three regions altogether within the 500 kb distal subtelomere, 30 were coding genes and 13 non-coding genes. A total of 16 genes were predicted in 2H-L barley chromosome arm, 12 of these genes were coding genes and 4 were non-coding genes. On 3H-L chromosome arm we found 18 genes, 11 were coding genes and 7 non-coding genes. Further, 9 genes were located on 5H barley chromosome arm, 7 coding genes and 2 non-coding genes. Gene density varies among the three barley chromosome ends studied. The analysis was extended to a larger region, and we identified genes within the 5 Mb distal subtelomeric sequence. In this larger region we predicted 946 genes, 255 genes in 2H-L chromosome arm, 333 in 3H-L chromosome arm and 358 in 5H-L chromosome arm (Table 5). A distinct and unique gene distribution pattern was observed across the three analyzed chromosome arms. A greater density of genes was identified in the most proximal region of the subtelomeric sequence in all three barley chromosome arms examined in this study (see Fig. 1). Conversely, the farthest portion of the subtelomere (adjacent to the telomeric sequence) exhibited a reduced gene count.

Localization of the genes included in distal subtelomeric region (500 Kb) of barley chromosome arms 2H-L, 3H-L and 5H-L. Direct and reverse complementary sequences were considered. a Distribution of coding genes on the three barley chromosomes, detected by IPK database (Leibniz Institute of Plant Genetics and Crop Plant Research) using Ensembl Plants (https://plants.ensembl.org/index.html). b Distribution of non-coding genes detected by EoRNA database (Barley Expression Database that displays gene and transcript abundance using Barley Reference Transcript (BaRTv1.0) from The James Hutton Institute) using Ensembl plants (https://plants.ensembl.org/index.html)
We also analyzed the distal subtelomeric region of 2H-L, 3H-L and 5H-L barley chromosome arms, searching for the presence and distribution of TEs (transposable elements), including both retrotransposons (SINE, LINE and LTR elements) and DNA transposable elements (Fig. 5). 2H-L chromosome arm contained a high percentage of transposable elements in the 500 Kb distal subtelomere region (56.74%) compared to 3H-L chromosome arm, which only presented a 18.29% fraction of transposable elements. 5H-L chromosome arm has an intermediate percentage (36.1%). As a relevant differential attribute, transposable elements distribution pattern is chromosome specific (Fig. 5). LTR elements were analyzed. 2H-L chromosome arm presented a higher percent of LTR (52.22%), in contrast with 3H-L (17.41%) and 5H-L (31.69%) chromosome arms (Table 6). It is important to remark that only 2H-L and 5H-L chromosome arms present LINEs in a small proportion (1.73 and 0.0232% respectively), while 3H-L chromosome arm doesn’t present this type of transposable element. SINEs have not been detected on this study in any of these chromosomes (Table 6). We also studied DNA transposons. These transposable elements have been detected in greater number on 5H-L chromosome arm, taking a 4.39% of the 500 Kb sequence (Table 6).

Distribution of transposable elements in barley chromosomes included within the 500 Kb distal subtelomeric region adjacent to telomeric repeats of chromosome arms 2H-L, 3H-L and 5H-L. No SINEs retrotransposons were detected in these barley subtelomeric sequences
The study investigated the presence and distribution of DNA repeats within the distal subtelomeric region spanning 500 Kb on barley chromosome arms 2H-L, 3H-L, and 5H-L. A quantitative analysis of these DNA repeats is shown in Table 7. Satellite repeats, consisting of multiple copies of the same DNA sequence with varying lengths, ranging from a single base to several thousand bases, were examined. Our findings indicate that these satellite repeats are primarily clustered near the telomere, especially on chromosome arms 2H-L and 5H-L. In contrast, chromosome arm 3H-L exhibited dense blocks of satellite repeats, distinguishing it from chromosome arms 2H-L and 5H-L (Fig. 6). In contrast, simple repeats, characterized by duplications of short DNA nucleotide sequences (2–5 bp), such as A, CA, CGG, and so on, display a distinct distribution pattern. Within chromosome arms 2H-L, 3H-L, and 5H-L, these simple repeats are predominantly found in the proximal region of the subtelomeric sequence (Fig. 6). Poly-purine or poly-pyrimidine stretches and regions of extremely high AT or CG content, also known as low complexity regions, were detected in all three barley chromosome arms (Fig. 6). Indeed, we noted a substantial prevalence of these repeats within the distal region of the subtelomeric sequence, in close proximity to the telomere. These low complexity regions encompass areas enriched with adenine, adenine-guanine, and guanine sequences. The occurrence of these bases within these low complexity regions exceeds 85%. Distal subtelomere sequence (500 Kb) of barley chromosome arms 2H-L, 3H-L and 5H-L long arms were analyzed for the GC content and the identification of predicted CpG islands (Fig. 7). The CG content and predicted CpG islands showed uniformity across all chromosomes. Notably, a dense concentration of CG content was observed throughout the entire set of chromosomes (see Fig. 7a, b). It's noteworthy that specific regions, located in the most distal portions of 2H-L and 5H-L chromosome arms, exhibited an approximate CG content of 50%. 3H-L chromosome arm presented also these 50% CG zones, but in this case, these zones were extended throughout the whole region analyzed. CpG islands have been observed within the whole sequence of 2H-L, 3H-L and 5H-L chromosome arm ends (Fig. 7a, b). Furthermore, we identified substantial, contiguous clusters of CpG islands in close proximity to the telomere in all three instances. These clusters coincide with satellite regions and encompass roughly 50% of the sequence (refer to Fig. 7a, b). It's worth noting that chromosome arm 3H-L contains an unsequenced stretch spanning 50 Kb, from positions 130 Kb to 180 Kb, rendering it ineligible for analysis. Moreover, none of the examined barley chromosome arms displayed noteworthy features like a high density of CG-rich DNA segments or CpG islands.

Distribution of repeat elements in barley chromosomes included within the distal (500 Kb) subtelomeric region adjacent to the telomeric repeats of chromosome arms 2H-L, 3H-L and 5H-L. (a) satellites, (b) simple repeats, (c) GA, A, G rich domains

CG content and predicted CpG islands included identified in the distal subtelomeric sequence (500 Kb) of chromosome arms 2H-L, 3H-L and 5H-L. Emboss CpG plot was used for CG content calculation (a) and CpG island prediction (b). Predicted CpG islands are represented in black
Distal subtelomere sequence (500 Kb) of long arm chromosomes 2H-L, 3H-L and 5H-L were analyzed for the distribution of predicted hot and cold recombination regions (Fig. 8).

Distribution of predicted hot and cold recombination spots and associated sequences within the distal region of 2H-L, 3H-L and 5H-L barley chromosome arms. a Distal subtelomere sequence (500 Kb) of chromosomes were analyzed for the distribution of predicted hot (red) and cold (blue) recombination regions and sequences associated to them. iRSpot-EL was used for prediction of hot and cold recombination spots. Sequences associated with hot recombination spots were identified by MAST (MEME Suite 5.0.5). b An extension of the study over the 5 Mb distal subtelomeric region
We also examined the distribution of three short sequence motifs (CCGCCGCCG; CTCCCTCC; TTAGTCCCGGTT) that are potentially linked to regions of intense recombination. This analysis covered both the 500 Kb distal subtelomeric region and a broader 5 Mb region (see Fig. 8a, b). Within the 500 Kb distal subtelomere, our analysis revealed a decrease in the occurrence of these motifs in specific regions of the chromosome sequences. Notably, these particular regions align with areas abundant in CG and CpG content, as well as regions enriched in satellite repeats. It is important to mention that a different feature was detected on 2H-L chromosome arm; a high number of TTAGTCCCGGTT motif was found on these zones. We did not see a correlation between the location of these motifs and recombination hot spots. 3H-L chromosome arm presented the lowest number of these motifs, in comparison with the other chromosomes. We also extended our analysis up to 5 Mb of the distal subtelomeric region. A reduction of the number of CCGCCGCCG, CTCCCTCC, TTAGTCCCGGTT motifs was found toward the most distal part of the subtelomere of chromosome arms 3H-L and 5H-L, but we did not see the same reduction of the frequency of CCGCCGCCG motif on 2H-L chromosome arm (Fig. 8 b).
In this study, we also investigated the distribution of predicted binding sites for pertinent DNA-binding, regulatory, or structural proteins. This analysis was initially conducted within the 500 Kb distal subtelomeric region and subsequently expanded to encompass a 5 Mb region, which included a more proximal subtelomeric section.These proteins were chosen because of their potential implication in chromosome architecture and their putative function in chromosome dynamics such as chromosome approaching and homologous chromosome interactions occurring during early meiosis. DNA binding sites for SMC1β meiosis-specific cohesion protein [81], Ying Yang 1 protein (YY1) [9] and high mobility group proteins HMG [94] were analyzed in the distal subtelomere sequences (500 kb) (Fig. 9). The distribution of potential binding sites for the investigated proteins exhibited specificity unique to each barley chromosome end under examination. This observation lends support to their likely significance as critical factors governing the specificity of chromosome recognition and pairing. We observed that SMC1β, YY1 and HMG proteins biding sites were distributed in the same regions in the chromosome, finding differences in the distribution pattern among 2H-l, 3H-L and 5H-L chromosome arms.

Distribution of predicted binding sites of relevant DNA-binding proteins. a Distal subtelomere sequence (500 Kb) of chromosome arms 2H-L, 3H-L and 5H-L were analyzed for the distribution of predicted DNA-binding sites of putative wheat proteins homologous to SMC1β, YY1 and HMG proteins. Sequences were identified and plotted using MAST (MEME Suite 5.0.5). b A similar study on the 5 Mb distal subtelomeric region
In all the barley chromosome ends we examined, it was evident that regions characterized by a high density of transposable elements and coding genes also exhibited a heightened concentration of binding sites for SMC1β, YY1, and HMG proteins. Conversely, regions with a scarcity of genes and transposable elements displayed a lower density of these binding sites. To further explore this phenomenon, we extended our analysis to encompass a 5 Mb subtelomeric region. Our objective was to investigate whether any variations in the distribution pattern of these binding site proteins could be discerned. Notably, we observed a notable reduction in the density of binding sites in the most distal portion of the subtelomere, near the telomere boundary (Fig. 9b).
Discussion
The analysis of available barley sequences (NCBI, RefSeq: MorexV3) has shown that most chromosome ends are not sequenced completely or accurately assembled yet, considering that the recent assemblies of most chromosomes do not have the plant terminal telomeric repeat (5´-TTTAGGG-3´, or 3´-AAATCCC-5´ on the complementary strand) [70], as it was observed in bread wheat (polyploid) [1]. Rather than related to ploidy level, miss-assembly must be attributable to the complexity of all chromosome ends, where there are numerous repeats that have not been correctly assembled yet. It is remarkable that telomeric sequences were much longer in barley than in wheat [1]. In plants, telomere length differs depending on the chromosome and the species. These variations imply that telomere length in plants is genetically regulated [24]. Wide variations in telomere length were also seen in differentiating or aging cells in barley (H. vulgare) and rice [37, 63].
Our examination of proximal telomere sequences revealed a phenomenon reminiscent of what has been previously documented in rice by Mizuno et al. [64]. This phenomenon involves the addition, deletion, or chromosome-specific substitution of individual nucleotides within the repetitive telomere sequences. Consequently, we chose to commence our analysis by scrutinizing the alterations in the nucleotides constituting these repeating units. While the seven-nucleotide unit typically exhibited conservation, we observed single nucleotide variations throughout the entire telomere sequence. Notably, these variations were more densely concentrated near the junction of the telomere and the chromosome-specific region. All types of mutations, including deletions, additions and substitutions, were found in the three chromosome arms examined.
Across all of these chromosomes, deletions and additions were the predominant types of mutations, while substitutions were relatively rare. Each chromosome exhibited a distinct mutation density and a specific distribution pattern of mutations. In particular, we detected numerous instances of deletions involving one of the "T"s in "TTTAGGG," resulting in a sequence change from "TTTAGGG" to "TTAGGG." Additionally, a lesser number of deletions occurred within "GGG." Conversely, numerous additions of "G" were observed in "GGG," leading to a sequence alteration to "TTTAGGGG." While substitutions were also identified, they were less prevalent compared to deletions and additions in barley chromosome arms 2H-L, 3H-L, and 5H-L. In contrast, in rice, substitutions were discovered to be as prevalent as deletions and additions, as documented by Mizuno et al. [64]. Research on rice further revealed that deletions did not occur randomly,rather, this type of mutation exhibited a bias towards specific bases. Specifically, it was observed that the "T" in "TTT" experienced more deletions than the "G" in "GGG," indicating a preference for thymine mutations [64]. The low-fidelity synthesis of telomere arrays by the telomerase catalytic subunit, which produces 6 bp, or the variety of RNA templates inside telomerases are thought to be responsible for this alteration [88]. Our findings in barley are similar to what was found in rice.
The telomeres of both rice and barley exhibit nucleotide deletions or insertions within the "T" of the canonical "TTTAGGG" repeat. These telomere repeat variations in rice and barley may be attributed to alterations in the genomic sequence responsible for encoding telomerase RNA or changes in its catalytic component, as proposed by Sýkorová et al. [96]. The distribution of these mutations across the entire telomere sequence suggests that a portion of the genomic sequence encoding the RNA template may possess nucleotide deletions, accounting for the deletions observed in barley telomeres, as previously suggested for rice by Mizuno et al. [64]. Having described these frequent variations in telomere repeat sequences, it becomes intriguing to explore their impact on the specificity of interactions with telomeric proteins and their functional consequences. This inquiry holds particular relevance in the context of chromosome recognition and homologous pairing, considering the possibility that homologous chromosomes may initiate their pairing through their telomeres during the bouquet stage of meiosis.Tandem repeats of guanine sequences, denominated G-quadruplexes (G4s), have been analyzed too. G4s, which include four short runs of guanines, are non-canonical structures that can be formed in DNA and RNA sequences when monovalent cations are present. DNA sequences that link the four G-tracts form loops [27]. Technically, a G-quadruplex can be formed by four G-tract DNA sequences that are separated by small runs of non-G bases and each one includes at least three Gs. One consensus sequence of this type of G4s is G3–5N1–7G3–5N1–7G3–5N1–7G3–5 (stricter or more flexible models have also been suggested). Replication of DNA, telomere maintenance (they function as capping structures on this regions), and gene transcription are the three most critical processes carried out by G4s [93]. G4s can also bind specific proteins, such as Rif1, POT1-TPP1 complex, HMGB1 protein, TRF2, among others [4, 31, 58, 66, 80]. It is important to remember that proteins have an impact on the composition and operation of G4s [110].
It is worth noting that the genomes of all organisms contain sequences with the potential to form G-quadruplexes, as indicated by Brázda et al. [15]. This widespread presence suggests the potential significance of G-quadruplexes in various biological contexts. In barley, there is a notably higher prevalence of G4 quadruplexes in the telomeric region, in contrast to their frequency in the subtelomeric region and the rest of the chromosome, as detailed in Tables 3 and 4. This disparity may be attributed to the greater abundance of guanine residues comprising the telomeric sequence, as highlighted by Gu et al. [29]. Interestingly, G4s quadruplexes can form superstructures by intramolecular interactions, and more interestingly by intermolecular interactions [75], what could facilitate the direct interaction between chromosomes.
As previously discussed regarding telomere repeat variations, understanding the distribution of G4-quadruplexes along the telomeres, especially in light of variations around the canonical sequence motif, gains significance. Additionally, considering the numerous proteins that bind to telomeres via these G4-quadruplexes, and the potential for direct interactions between DNA molecules from different chromosomes, it becomes intriguing to investigate how these variations might influence the specificity of such interactions and their ensuing functional repercussions. This could be relevant in the context of chromosome recognition and homologue pairing, considering the possibility that homologues could initiate their pairing through their telomeres and distal subtelomeres during the bouquet stage of meiosis.
The subtelomeric region was defined as the stretch between the telomere and the farthest chromosome-specific sequence in previously studied organisms [40, 51, 57]. Before the first active gene at the distal region of subtelomeres, tens of kilobases of highly rearranged and repetitive DNA are a common characteristic thought to be shared by all plants [3, 71, 73, 87, 103, 109]. Nonetheless, with its smaller genome, the model plant A. thaliana, paints a different image. Arabidopsis subtelomeric regions are short (less than 5 Kb) and relatively simple, but these regions contain short stretches of BAAAA (where B = C, T, or G) and a 32-bp tract composed almost entirely of G [40]. In barley chromosomes, we have not found a sequence pattern that could function as a molecular marker of the limit between telomere and subtelomere or between the distal subtelomere region and the rest of the chromosome. Telomeric sequence and the subtelomere, as well as the distal subtelomere area and the rest of the chromosome, are not clearly delineated in barley by any consistent sequence pattern shared by all chromosomes. Within the 3-Kb ends, we also failed to identify a characteristic that would be common to all three examined chromosomal arms. Aguilar & Prieto [1] could not identify any relevant common sequence motif within these regions in bread wheat chromosomes either. However, we found the short stretches of DNA described in Arabidopsis in all three arms analyzed. BAAAA (where B = C, T, or G) was detected in all chromosome arms analyzed in this study and the tract compose of G was found as well in all chromosome arms, but with a smaller number of Gs. This could be related for the fact that barley is a diploid, the same as Arabidopsis. In a polyploid organism like bread wheat, these Arabidopsis DNA stretches could not be found [1].
For our analysis of the subtelomeric region, we proceeded as it was done in model cereal species like rice and wheat, and in other organisms like humans and Arabidopsis [1, 38, 53, 63]. We decided to start our study focusing on the 500 Kb distal region of subtelomeres, but we later expanded the analysis to a larger chromosome region (5 Mb). Subtelomeres are thought to be important for homologue-specific chromosomal pairing and recognition, according to previous studies. For instance, introgressed barley homologous chromosomes failed to detect and associate in pairs when subtelomeres were absent, indicating a crucial role for subtelomeres in these processes [16]. In rye, clustering heterochromatin blocks at the subtelomeres have also been observed, pointing to a potential role of these areas in chromosome connections [61]. In barley, we have focused our analysis on different features of subtelomeric regions that could contribute to pairing specificity. The results of this analysis have been compared with the results obtained in wheat [1].
Concerning the identification of gene sequences within the 500 Kb distal subtelomeric regions of chromosome arms 2H-L-L, 3H and 5H-L, we predicted a total of 43 genes in these three regions (Ensembl plants). Unexpectedly, gene density was higher in all chromosome arms when a larger region (5 Mb) was analyzed, unlike what was found in wheat [1, 2]. A relevant feature is the fact that all chromosome arms present a specific pattern of gene distribution. We can see on the 500 Kb analysis that the only common feature is the fact that all three chromosome arms present a higher density of genes at the most proximal part of the subtelomere.
An important feature of TEs was discovered through this analysis: TEs have a chromosome-specific distribution pattern. From a minimum 17% in the 3H-L distal subtelomere and a maximum 54% in the 2H-L, the relative abundance of TEs varies among the chromosomal arms. A similar feature was found in maize and wheat [105, 1, 42, 90]. LTR-type retroelements (Ty1/Copia, Gypsy/DIRS1) are the most prevalent TEs within the distal subtelomeric regions. In general, the most prevalent TEs in plants are LTR-type retroelements [104]. In our study, we found that barley subtelomeres contain an average of approximately 37% TEs, including 33% retroelements and 3% DNA transposons. In a polyploid species like bread wheat, at a genomic scale, TEs represent more than 80% of the whole genome, including 70% retroelements and 13% DNA transposons [18, 46]. Among the retroelements, Gypsy and Copia LTR retroelements are predominant in wheat, while CACTA DNA elements are the most abundant DNA transposons [18]. In barley, we found a higher abundance of Gypsy and Copia LTR retroelements too. Similarities can be observed between these two species, being LTRs the most abundant TEs in both [111]. As suggested by Wicker et al. [106] in barley, TEs are major determinants of overall chromosome structure. The distribution of these TEs within the subtelomeric region could also influence the specificity of the first chromosomal contacts between homologous chromosomes at the start of meiosis.
Repetitive sequences vary in size and complexity among species, and they are more prevalent in species with larger genomes [36]. The subtelomeres of plants like Arabidopsis, tobacco, barley, wheat, and potato have a variety of repeat families [10, 32, 39, 54, 100]. In barley, our analysis includes all repeat sequences from satellites to simple repeats (1–5 bp long) and low complexity repeats (poly-purine or poly-pyrimidine stretches, or regions of extremely high AT or GC content). We analyzed these repeats along the 500-Kb distal subtelomeric region of chromosome arms 2H-L, 3H-L and 5H-L. Again, we found that repeat sequence distribution patterns are chromosome-specific, so that chromosomes may be distinguished by this feature in the subtelomeric region. In cereal chromosomes, repeat sequences can be seen as heterochromatic areas. Tandem repeats are relatively prevalent in maize, but they are predominantly found in knob areas and are less prevalent in subtelomeric regions [5, 42]. Rye, barley, and wheat, which are closely related species, exhibit remarkably distinct patterns. A specific characteristic of rye chromosome ends is that they have large heterochromatin blocks [102]. Although the distribution of heterochromatin in wheat and barley chromosomes is complex, their subtelomeres are devoid of significant amounts of heterochromatin [28, 48]. Besides, in wheat, 4AS and 7DS chromosome arms were the only ones that showed a satellite distribution that is similar to the distribution found in barley chromosomes. In all these cases, satellite regions were close to telomeres. It is important to mention that on barley chromosomes we found a higher density of simple repeats in contrast with wheat chromosomes, except for 4AS and 7DS wheat chromosome arms, where the repeats were close to the telomere as well [1].
Genes, transposable elements, and various forms of tandem repeat sequences work together to create a complex and dynamic structure of distal subtelomeres in barley, which appears to be chromosome specific and may contribute to the specificity of chromosome interactions at the start of meiosis. This intricate and dynamic structure of subtelomeres is shared by other plants, including rice with its small genome [102]. Subtelomeres can play a role during the first contacts and pairing of chromosomes, as was previously suggested, and also stabilize the chromosomal ends in the absence of canonical telomeric repeats or protect distal genes from active loss/gain processes within the terminal regions [25, 35, 51].
The functionality of subtelomeres may be determined by simple characteristics like the relative abundance of GC and AT. The whole distal subtelomere sequence (500 Kb) of the long arms of 2H, 3H, and 5H barley chromosome arms differed in GC content and predicted CpG islands. This fact supports the high polymorphism of these subtelomeric regions. It is important to highlight that we found a high density of GC-rich DNA stretches and CpG islands in the most distal part of the subtelomere, near the telomere, in the three chromosome arm ends examined. These solid blocks of GC-rich DNA and CpG islands correspond to areas where we found TREP37 and TREP38 barley satellites. Subtelomeric satellites, near the telomeric region of barley chromosomes, have been shown in previous studies [87]. The presence of genes in animals and plants was shown to be highly associated with GC content and CpG islands [6, 44, 67]. This association is obvious in our work. Most critical, however, is the correlation between GC content and the key processes of recombination and crossover, which mostly occur at the subtelomeric areas and are strongly influenced by the right homologous chromosome interaction earlier in meiosis. Recombination and GC concentration were found to be clearly correlated in maize [95], as in Triticeae [22, 30, 68]. Research on Brachypodium, maize, and rice found a strong relationship between high GC content, local recombination, and crossover rate [92]. Interestingly, in barley we could also find a good correlation between predicted G4 quadruplexes, GC content and predicted CpG islands, as previously described elsewhere. Apparently, with the species evolution, G4 motifs become more abundant within the promoter regions of genes, especially in genes coding for transcription factors, and there seems to be a negative correlation between methylation state and G4 density [108].
We also analyzed the distribution of short sequence motifs associated with hot recombination regions, previously studied in wheat by Aguilar and Prieto [1]. Hot recombination-related sequences were detected, but we could not find the same correlation between these motifs and hot recombination spots as the correlation found in wheat [1]. In our study and in wheat studies [1] there were obvious disparities among chromosomes in terms of location and size of these areas. Interestingly, an apparent positive correlation was found in barley between frequency of hot recombination spots, transposable elements and satellites, but not with genes.
For all the DNA features analyzed, the subtelomeric areas of the barley chromosome arms investigated here showed a significant degree of polymorphism, which could account for the specificity of the initial chromosome associations in a diploid organism like barley. However, DNA-binding proteins or protein complexes may also be required for the earliest physical interactions of chromosomes to ensure appropriate pairing between homologues [20]. For this reason, we also examined the distribution of DNA-binding proteins that may be involved in chromosome architecture. Candidate proteins were chosen based on their potential involvement in the early meiotic events previously covered in the literature, as well as the presence of known DNA-binding sites. We used putative wheat proteins homologous to human SMC1β meiosis-specific cohesin [81], Ying Yang 1 protein [9] and HMG proteins [94] and looked for their putative DNA-binding sites at the distal subtelomeric sequences on barley chromosomes. Cohesins play a crucial part in sister chromatid cohesion in addition to other meiosis-specific processes such the creation of chromosomal axes, synaptonemal complexes, and reciprocal recombination [20, 33]. Ying Yang 1, an architectural protein, is essential for connecting higher-order chromatin folding in both mammals and Arabidopsis [9, 107]. According to previous studies, HMG proteins may interact with AT-rich regions to play a role during the first interactions between homologues before proper pairing [94]. Our observations showed that the distribution of potential binding sites for the proteins under investigation was chromosome-specific, with clear variations in density and distribution among chromosomes. We found an unusual differential distribution of putative binding sites for cohesins, with a very low concentration of these putative sites in comparison with YY1 and HMG binding sites. We found a lower quantity of cohesins, YY1 and HMG binding sites on satellite-rich zones and a higher quantity on gene-rich areas. When comparing the 500 Kb distal region, we could see that binding-site densities varied among chromosomes, but when a larger region of 5 Mb was taken into consideration, these densities were more similar. In barley, a reduction of the number of binding-sites on the most distal part of the subtelomere, near the limit with the telomeric region, can be observed. This reduction is not detectable in wheat studies. In bread wheat, near the telomere region, a higher density of binding-sites was found [1].
The distribution pattern of genes, transposable elements, repeats, GC content, predicted CpG islands, recombination hotspots, G4 quadruplexes, and targeted sequence motifs for key DNA-binding proteins, described in this study, show a high variability both in telomeres and subtelomeres. The molecular basis for the specificity of homologous recognition and pairing in the early chromosomal interactions at the start of meiosis in barley may be provided by these polymorphisms. Our finding of a higher polymorphism in most distal chromosome ends suggest that the most distal part of subtelomeres and telomeres themselves might be particularly relevant for homologue pairing.
Availability of data and materials
All raw data underlying this article are available in the article.
Change history
20 November 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12864-023-09797-1
References
Aguilar M, Prieto P. Sequence analysis of wheat subtelomeres reveals a high polymorphism among homoeologous chromosomes. The Plant Genome [Internet]. 2020;13(3). Available from: https://acsess.onlinelibrary.wiley.com/doi/10.1002/tpg2.20065.
Alaux M, Rogers J, Letellier T, Flores R, Alfama F, Pommier C, et al. Linking the International Wheat Genome Sequencing Consortium bread wheat reference genome sequence to wheat genetic and phenomic data. Genome Biology [Internet]. 2018;19(1). Available from: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1491-4#citeas.
Alkhimova OG, Mazurok NA, Potapova TA, Zakian SM, Heslop-Harrison JS, Vershinin AV. Diverse patterns of the tandem repeats organization in rye chromosomes. Chromosoma. 2004;113(1):42–52. https://doi.org/10.1007/s00412-004-0294-4.
Amato J, Cerofolini L, Brancaccio D, Giuntini S, Iaccarino N, Zizza P, et al. Insights into telomeric G-quadruplex DNA recognition by HMGB1 protein. Nucleic Acids Res. 2019;47(18):9950–66. https://doi.org/10.1093/nar/gkz727.
Ananiev E V, Phillips RL, Rines HW. A knob-associated tandem repeat in maize capable of forming fold-back DNA segments: Are chromosome knobs megatransposons? Genetics. 1998: 95. Available from: www.pnas.org.
Ashikawa I. Gene-associated CpG islands in plants as revealed by analyses of genomic sequences. Plant J. 2001;26(6):617–25. https://doi.org/10.1046/j.1365-313x.2001.01062.x.
Bailey T, Gribskov M. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics. 1998;1(14):48–54.
Bass HW, Lizarazu OR, Ananiev EV, Bordoli SJ, Rines HW, Phillips RL, et al. Evidence for the coincident initiation of homolog pairing and synapsis during the telomere-clustering (bouquet) stage of meiotic prophase. J Cell Sci. 2000;113:1033–42.
Beagan JA, Duong MT, Titus KR, Zhou L, Cao Z, Ma J, et al. YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res. 2017;27(7):1139–52 Available from: http://genome.cshlp.org/content/27/7/1139.abstract.
Belostotsky DA, Ananiev EV. Characterization of relic DNA from barley genome. Theor Appl Genet. 1990;80(3):380–74.
Blackburn EH. Telomere states and cell fates. Nature. 2000;408:53–6 Available from: www.nature.com.
Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106:661–73.
Blasio F, Prieto P, Pradillo M, Naranjo T. Genomic and Meiotic Changes Accompanying Polyploidization. Plants. 2022;11(1):1–32.
Blokhina YP, Nguyen AD, Draper BW, Burgess SM. The telomere bouquet is a hub where meiotic double-strand breaks, synapsis, and stable homolog juxtaposition are coordinated in the zebrafish, Danio rerio. PLOS Genetics [Internet]. 2019;15(1). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6336226/.
Brázda V, Hároníková L, Liao JCC, Fojta M. DNA and RNA Quadruplex-Binding Proteins. Int J Mol Sci. 2014;15(10):17493–517. Available from: https://www.mdpi.com/1422-0067/15/10/17493.
Calderón MDC, Rey MD, Cabrera A, Prieto P. The subtelomeric region is important for chromosome recognition and pairing during meiosis. Sci Rep. 2014;1:4.
Calderón MC, Rey MD, Martín A, Prieto P. Homoeologous chromosomes from two Hordeum species can recognize and associate during meiosis in wheat in the presence of the Ph1 locus. Front Plant Sci. 2018;9:585. https://doi.org/10.3389/fpls.2018.00585.
Clavijo BJ, Venturini L, Schudoma C, Accinelli GG, Kaithakottil G, Wright J, et al. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017;27(5):885–96.
Darrier B, Rimbert H, Balfourier F, Pingault L, Josselin AA, Servin B, et al. High-resolution mapping of crossover events in the hexaploid wheat genome suggests a universal recombination mechanism. Genetics. 2017;206(3):1373–88. https://doi.org/10.1534/genetics.116.196014.
Ding DQ, Matsuda A, Okamasa K, Nagahama Y, Haraguchi T, Hiraoka Y. Meiotic cohesin-based chromosome structure is essential for homologous chromosome pairing in Schizosaccharomyces pombe. Chromosoma. 2016;125(2):205–14.
van Emden TS, Forn M, Forné I, Sarkadi Z, Capella M, Martín Caballero L, et al. Shelterin and subtelomeric DNA sequences control nucleosome maintenance and genome stability. EMBO Reports [Internet]. 2018;20(1). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6322387/.
Escobar JS, Cenci A, Bolognini J, Haudry A, Laurent S, David J, et al. An integrative test of the dead-end hypothesis of selfing evolution in triticeae (POACEAE). Evolution (N Y). 2010;64(10):2855–72.
Fan C, Zhang Y, Yu Y, Rounsley S, Long M, Wing RA. The subtelomere of Oryza sativa chromosome 3 short arm as a hot bed of new gene origination in rice. Mol Plant. 2008;1(5):839–50.
Fulcher N, Teubenbacher A, Kerdaffrec E, Farlow A, Nordborg M, Riha K. Genetic architecture of natural variation of telomere length in Arabidopsis thaliana. Genetics. 2015;199(2):625–35.
Garrido-Ramos MA. Satellite DNA in plants: More than just rubbish. Cytogenet Genome Res. 2015;146:153–70 S. Karger AG.
Gaut BS, Wright SI, Rizzon C, Dvorak J, Anderson LK. Recombination: an underappreciated factor in the evolution of plant genomes. Nat Rev Genet. 2007;8(1):77–84. https://doi.org/10.1038/nrg1970.
Gellert M, Lipsett MN, Davies DR. Helix Formation by Guanylic Acid. Chem Gellerd al. 1962;48(12):2018–3.
Gill BS, Kimber G. Giemsa C-Banding and the Evolution of Wheat (polyploid/chromosomes/heterochromatin/genomes). 1974; 71. Available from: https://www.pnas.org.
Gu P, Lu Y, Li S, Ma C. A Label-Free Fluorescence Aptasensor Based on G-Quadruplex/Thioflavin T Complex for the Detection of Trypsin. Molecules. 2022;27(18). Available from: https://www.mdpi.com/1420-3049/27/18/6093.
Haudry A, Cenci A, Guilhaumon C, Paux E, Poirier S, Santoni S, et al. Mating system and recombination affect molecular evolution in four Triticeae species. Genet Res (Camb). 2008;90(1):97–109.
He Q, Zeng P, Tan JH, Ou TM, Gu LQ, Huang ZS, et al. G-quadruplex-mediated regulation of telomere binding protein POT1 gene expression. Biochim Biophys Acta - Gen Subj. 2014;1840(7):2222–33 Available from: https://www.sciencedirect.com/science/article/pii/S0304416514001056.
Heacock M, Spangler E, Riha K, Puizina J, Shippen DE. Molecular analysis of telomere fusions in Arabidopsis: multiple pathways for chromosome end-joining. EMBO J. 2004;23(11):2304–13. https://doi.org/10.1038/sj.emboj.7600236.
Ishiguro KI, Kim J, Fujiyama-Nakamura S, Kato S, Watanabe Y. A new meiosis-specific cohesin complex implicated in the cohesin code for homologous pairing. EMBO Rep. 2011;12(3):267–75.
Islam AKMR, Shepherd KW, Sparrow DHB. Isolation and characterization of euplasmic wheat-barley chromosome addition lines. Heredity (Edinb). 1981;46(2):161–74.
Jain D, Hebden AK, Nakamura TM, Miller KM, Cooper JP. HAATI survivors replace canonical telomeres with blocks of generic heterochromatin. Nature. 2010;467(7312):223–7.
Kidwell MG. Transposable elements and the evolution of genome size in eukaryotes. Genetica. 2002;115(1):63–49.
Kilian A, Stiff C, Kleinhofs A. Barley telomeres shorten during differentiation but grow in callus culture. Genetics. 1995; 92. Available from: https://www.pnas.org.
Kotani H, Hosouchi T, Tsuruoka H. Structural Analysis and Complete Physical Map of Arabidopsis thaliana Chromosome 5 Including Centromeric and Telomeric Regions. DNA RESEARCH. 1999; 6. Available from: www.stanford.edu/Arabidopsis/agi.html.
Koukalowi B, Reich J, Matyfiek R, Kuhrovh V, Bezd~k M. A BamHI family of highly repeated DNA sequences of Nicotiana tabacum. Theor Appl Genet. 1989;78(1):80–77.
Kuo HF, Olsen KM, Richards EJ. Natural variation in a subtelomeric region of arabidopsis: Implications for the genomic dynamics of a chromosome end. Genetics. 2006;173(1):401–17.
Kuo P, Da Ines O, Lambing C. Rewiring Meiosis for Crop Improvement. Front Plant Sci [Internet]. 2021;12. DE ESTA NO ESTOY SEGURA.
Lamb JC, Meyer JM, Corcoran B, Kato A, Han F, Birchler JA. Distinct chromosomal distributions of highly repetitive sequences in maize. Chromosom Res. 2007;15(1):33–49.
Lambing C, Franklin FCH, Wang CJR. Understanding and manipulating meiotic recombination in plants. Plant Physiol. 2017;173(3):1530–42.
Larsen F, Gundersen G, Lopez R, Prydz’ H. CpG Islands as Gene Markers in the Human Genome. 1991.
Lese CM, Fantes JA, Riethman HC, Ledbetter DH. Characterization of Physical Gap Sizes at Human Telomeres. Genome Res. 1999;9:888–94 Available from: www.genome.org.
Li W, Zhang P, Fellers JP, Friebe B, Gill BS. Sequence composition, organization, and evolution of the core Triticeae genome. Plant J. 2004;40(4):500–11.
Linardopoulou EV, Williams EM, Fan Y, Friedman C, Young JM, Trask BJ. Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature. 2005;437(7055):94–100.
Linde-Laursen I. Giemsa C-banding of barley chromosomes: I Banding pattern polymorphism. Hereditas. 1978;88(1):55–64.
Lingner J, Cecht TR. Telomerase and chromosome end maintenance. Curr Opin GenetDev. 1998; 8. Available from: http://biomednet.com/elecref/O959437XO0800226.
Liu B, Wang S, Long R, Chou KC. iRSpot-EL: identify recombination spots with an ensemble learning approach. Bioinformatics. 2017;33(1):35–41. https://doi.org/10.1093/bioinformatics/btw539.
Louis EJ, Vershinin AV. Chromosome ends: Different sequences may provide conserved functions. BioEssays. 2005;27:685–97.
Macina RA, Morii K, Hu XL, Negorev DG, Spais C, Ruthig LA, et al. Molecular cloning and RARE cleavage mapping of human 2p, 6q, 8q, 12q, and 18q telomeres. Genome Res. 1995;5(3):225–32 Available from: http://genome.cshlp.org/content/5/3/225.abstract .
Macina RA, Negorev DG, Spais C, Ruthig LA, Hu XL, Riethman HC. Sequence organization of the human chromosome 2q telomere. Hum Mol Genet. 1994;3(10):1847–53. https://doi.org/10.1093/hmg/3.10.1847.
Mao L, Devos KM, Zhu L, Gale MD. Cloning and genetic mapping of wheat telomere-associated sequences. Mol Gen Genet. 1997;254:584–91.
McEachern MJ, Krauskopf A, Blackburn EH. Telomeres and their control. Annu Rev Genet. 2000;34(1):331–58. https://doi.org/10.1146/annurev.genet.34.1.331.
McKnight TD, Shippen DE. Plant telomere biology. Plant Cell. 2004;16(4):794–803. https://doi.org/10.1105/tpc.160470.
Mefford HC, Trask BJ. The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet. 2002;3:91–102.
Mei Y, Deng Z, Vladimirova O, Gulve N, Johnson FB, Drosopoulos WC, et al. TERRA G-quadruplex RNA interaction with TRF2 GAR domain is required for telomere integrity. Sci Rep. 2021;11(1):3509. https://doi.org/10.1038/s41598-021-82406-x.
Meints B, Vallejos C, Hayes P. Multi-use naked barley: a new frontier. J Cereal Sci. 2021;1:102.
Mewborn SK, Lese Martin C, Ledbetter DH. The dynamic nature and evolutionary history of subtelomeric and pericentromeric regions. Cytogenetic Genome Res. 2005;108:22–5.
Mikhailova El, Sosnikhina SP, Kirillova GA, Tikholiz OA, Smirnov VG, Jones RN, et al. Nuclear dispositions of subtelomeric andpericentromeric chromosomal domains duringmeiosis in asynaptic mutants of rye (Secale cereale L.). J Cell Sci. 2001;114:1875–82.
Miller TE, Reader SM, Chapman V. The addition of Hordeum chilense chromosomes to wheat. 1982.
Mizuno H, Wu J, Kanamori H, Fujisawa M, Namiki N, Saji S, et al. Sequencing and characterization of telomere and subtelomere regions on rice chromosomes 1S, 2S, 2L, 6L, 7S, 7L and 8S. Plant J. 2006;46(2):206–17.
Mizuno H, Wu J, Katayose Y, Kanamori H, Sasaki T, Matsumoto T. Characterization of chromosome ends on the basis of the structure of TrsA subtelomeric repeats in rice (Oryza sativa L.). Mol Genet Genomics. 2008;280(1):19–24.
Molnár-Láng M, Linc G, Szakács É. Wheat-barley hybridization: The last 40 years. Euphytica. 2014;195:315–29 Kluwer Academic Publishers.
Moriyama K, Lai MS, Masai H. Interaction of Rif1 Protein with G-Quadruplex in Control of Chromosome Transactions. In: Masai H, Foiani M, editors. DNA Replication: From Old Principles to New Discoveries. Singapore: Springer Singapore; 2017. 287–310. https://doi.org/10.1007/978-981-10-6955-0_14.
Mouchiroud D, D’onofrio G, Aissani B, Macaya G, Gautier C, Bernardi G. The distribution of genes in the human genome. Gene. 1991;100:187–1.
Muyle A, Serres-Giardi L, Ressayre A, Escobar J, Glémin S. GC-biased gene conversion and selection affect GC content in the oryza genus (rice). Mol Biol Evol. 2011;28(9):2695–706.
Naranjo T. Dynamics of rye telomeres in a wheat background during early meiosis. Cytogenet Genome Res. 2014;143(1–3):60–8.
Navrátilová P, Toegelová H, Tulpová Z, Kuo YT, Stein N, Doležel J, et al. Prospects of telomere-to-telomere assembly in barley: Analysis of sequence gaps in the MorexV3 reference genome. Plant Biotechnol J. 2022;20(7):1373–86. https://doi.org/10.1111/pbi.13816.
Ohmido N, Kijima K, Ashikawa I, de Jong JH, Fukui K. Visualization of the terminal structure of rice chromosomes 6 and 12 with multicolor FISH to chromosomes and extended DNA fibers. Plant Mol Biol. 2001;47(3):413–21. https://doi.org/10.1023/A:1011632111845.
Page SL, Hawley RS. Chromosome choreography: The meiotic ballet. Science. 2003;301:785–9.
Pearce SR, Pich U, Harrison G, Flavell AJ, Heslop-Harrison JS(, Schubert I, et al. TheTy1-copia group retrotransposons ofAllium cepa are distributed throughout the chromosomes but are enriched in the terminal heterochromatin. Chromosom Res. 1996;4(5):357–64. https://doi.org/10.1007/BF02257271.
Pedersen C, Rasmussen SK, Linde-Laursen I. Genome and chromosome identification in cultivated barley and related species of the Triticeae (Poaceae) by in situ hybridization with the GAA-satellite sequence. Genome. 1996;39(1):93–104. https://doi.org/10.1139/g96-013.
Pedroso IM, Duarte LF, Yanez G, Burkewitz K, Fletcher TM. Sequence specificity of inter- and intramolecular G-quadruplex formation by human telomeric DNA. Biopolymers. 2007;87(1):74–84. https://doi.org/10.1002/bip.20790.
Perrod S, Gasser SM. Long-range silencing and position effects at telomeres and centromeres: Parallels and differences. Cell Mol Life Sci. 2003;60:2303–18.
Prieto P, Ramíarez MC, Ballesteros J, Cabrera A. Identification of intergenomic translocations involving wheat, hordeum vulgare and hordeum chilense chromosomes by FISH. Hereditas. 2001;135(2–3):171–4. https://doi.org/10.1111/j.1601-5223.2001.t01-1-00171.x.
Prieto P, Martín A, Cabrera A. Chromosomal distribution of telomeric and telomeric-associated sequences in Hordeum chilense by in situ hybridization. Hereditas. 2004;141(2):122–7. https://doi.org/10.1111/j.1601-5223.2004.01825.x.
Prieto P, Shaw P, Moore G. Homologue recognition during meiosis is associated with a change in chromatin conformation. Nat Cell Biol. 2004;6(9):906–8.
Ray S, Bandaria JN, Qureshi MH, Yildiz A, Balci H. G-quadruplex formation in telomeres enhances POT1/TPP1 protection against RPA binding. Proc Natl Acad Sci. 2014;111(8):2990–5. https://doi.org/10.1073/pnas.1321436111.
Revenkova E, Eijpe M, Heyting C, Gross B, Jessberger R. Novel Meiosis-Specific Isoform of Mammalian SMC1. Mol Cell Biol. 2001;21(20):6984–98.
Rey MD, Calderón MC, Rodrigo MJ, Zacarías L, Alós E, Prieto P. Novel bread wheat lines enriched in carotenoids carrying Hordeum chilense chromosome arms in the ph1b background. PLoS ONE. 2015;10(8):e0134598. https://doi.org/10.1371/journal.pone.0134598.
Riaz A, Kanwal F, Börner A, Pillen K, Dai F, Alqudah AM. Advances in Genomics-Based Breeding of Barley: Molecular Tools and Genomic Databases. Agronomy. 2021;11(5):894–4. Available from: https://www.mdpi.com/2073-4395/11/5/894.
Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–7 Available from: https://www.sciencedirect.com/science/article/pii/S0168952500020242.
Richards EJ, Ausubel FM. Isolation of a higher eukaryotic telomere from Arabidopsis thaliana. Cell. 1988;53(1):127–36 Available from: https://www.sciencedirect.com/science/article/pii/0092867488904941 .
Riethman H, Ambrosini A, Paul S. Human subtelomere structure and variation. Chromosome Res. 2005;13:505–15.
Röder MS, Lapitan NLV, Sorrells ME, Tanksley SD. Genetic and physical mapping of barley telomeres. Mol Gen Genet MGG. 1993;238(1):294–303. https://doi.org/10.1007/BF00279558.
Rotková G, Skleničková M, Dvořáčková M, Sýkorová E, Leitch AR, Fajkus J. An evolutionary change in telomere sequence motif within the plant section Asparagales had significance for telomere nucleoprotein complexes. Cytogenet Genome Res. 2004;107(1–2):132–8 Available from: https://www.karger.com/DOI/10.1159/000079584 .
Rudd MK, Friedman C, Parghi SS, Linardopoulou EV, Hsu L, Trask BJ. Elevated rates of sister chromatid exchange at chromosome ends. PLoS Genet. 2007;3(2):0319–23.
SanMiguel P, Tikhonov A, Jin YK, Motchoulskaia N, Zakharov D, Melake-Berhan A, et al. Nested Retrotransposons in the Intergenic Regions of the Maize Genome. Science (80-). 1996;274:765–8.
Scherthan H. A bouquet makes ends meet. Mol Cell Biol. 2001;2:621–7.
Serres-Giardi L, Belkhir K, David J, Glémin S. Patterns and evolution of nucleotide landscapes in seed plants. Plant Cell. 2012;24(4):1379–97.
Stefos GC, Theodorou G, Politis I. DNA G-quadruplexes: functional significance in plant and farm animal science. Anim Biotechnol. 2019;32:262–71 Bellwether Publishing, Ltd.
Subirana JA, Messeguer X. The distribution of alternating AT sequences in eukaryotic genomes suggests a role in homologous chromosome recognition in meiosis. J Theor Biol. 2011;283(1):28–34.
Sundararajan A, Dukowic-Schulze S, Kwicklis M, Engstrom K, Garcia N, Oviedo OJ, et al. Gene Evolutionary Trajectories and GC Patterns Driven by Recombination in Zea mays. Front Plant Sci. 2016;7. Available from: https://www.frontiersin.org/articles/10.3389/fpls.2016.01433.
Sýkorová E, Lim KY, Kunická Z, Chase MW, Bennett MD, Fajkus J, et al. Telomere variability in the monocotyledonous plant order Asparagales. Proc R Soc B Biol Sci. 2003;270(1527):1893–904.
Taddei A, Gasser SM. Multiple pathways for telomere tethering: Functional implications of subnuclear position for heterochromatin formation. Biochimica et Biophysica Acta - Gene Struct Express. 2004;1677:120–8.
Tham WH, Zakian VA. Transcriptional silencing at Saccharomyces telomeres: Implications for other organisms. Oncogene. 2002;21:512–21.
Tomita K, Cooper JP. The meiotic chromosomal bouquet: SUN collects flowers. Cell. 2006;125:19–21 (Elsevier B.V).
Torres GA, Gong Z, Iovene M, Hirsch CD, Robin Buell C, Bryan GJ, et al. Organization and evolution of subtelomeric satellite repeats in the potato genome. G3. 2011;1(2):85–92.
Valenzuela NT, Perera E, Naranjo T. Identifying crossover-rich regions and their effect on meiotic homologous interactions by partitioning chromosome arms of wheat and rye. Chromosom Res. 2013;21(5):433–45.
Vershinin A V, Evtushenko E V. What is the specificity of plant subtelomeres? In: Subtelomeres. Berlin: Springer; 2014. p. 195–209.
Vershinin AV, Schwarzacher T, Heslop-Harrison JS. The large-scale genomic organization of repetitive DNA families at the telomeres of rye chromosomes. Plant Cell. 1995;7(11):1823–33. https://doi.org/10.1105/tpc.7.11.1823.
Vicient CM, Casacuberta JM. Impact of transposable elements on polyploid plant genomes. Ann Botany. 2017;120:195–207 (Oxford University Press).
Weil’ CF, Wessle SR. Molecular Evidence That Chromosome Breakage by Ds Elements 1s Caused by Aberrant Transposition. PlantCell. 1993;5(5):522–15.
Wicker T, Schulman AH, Tanskanen J, et al. The repetitive landscape of the 5100 Mbp barley genome. Mob DNA. 2017;8:22. https://doi.org/10.1186/s13100-017-0102-3.
Wu X, Cheng Y, Li T, Wang Z, Liu JY. In vitro identification of DNA-binding motif for the new zinc finger protein AtYY1. Acta Biochim Biophys Sin (Shanghai). 2012;44(6):483–9.
Wu F, Niu K, Cui Y, Li C, Lyu M, Ren Y, et al. Genome-wide analysis of DNA G-quadruplex motifs across 37 species provides insights into G4 evolution. Commun Biol. 2021;4(1):98. https://doi.org/10.1038/s42003-020-01643-4.
Wu KS, Tanksley SD. Genetic and physical mapping of telomeres and macrosatellites of rice. Plant Mol Biol. 1993;22(5):861–72. https://doi.org/10.1007/BF00027371.
Yuan WF, Wan LY, Peng H, Zhong YM, Cai WL, Zhang YQ, et al. The influencing factors and functions of DNA G-quadruplexes. Cell Biochem Funct. 2020;38:524–32 (John Wiley and Sons Ltd).
Zhao D, Ferguson AA, Jiang N. What makes up plant genomes: The vanishing line between transposable elements and genes. Biochimica et Biophysica Acta - Gene Regul Mech. 2016;1859:366–80 (Elsevier).
Zickler D. From early homologue recognition to synaptonemal complex formation. Chromosoma. 2006;115:158–74.
Zickler D, Kleckner N. Recombination, pairing, and synapsis of homologs during meiosis. Cold Spring Harb Perspect Biol. 2015;7(6):1–28.
Acknowledgments
I.M.S.L. is grateful for her contract (Nº expediente administrativo subvención: POEJ00028/ N.º expediente: AND21_IAS_M2_064) from Garantía Juvenil-Andalucía 2022, Consejería de Universidad, Investigación e Innovación, Secretaría General de Investigación e Innovación, Junta de Andalucía/Cofinanciación FEDER 91%. Authors deeply appreciate Borja Rojas Panadero his support and expertise with the bioinformatics analysis.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work has been supported by P20_00971 grant and the Qualifica Project QUAL21_023 IAS both from Consejería de Transformación Económica, Industria, Conocimiento y Universidades/Cofinanciación FEDER 80%—Programa Operativo FEDER de Andalucía 2014-2020. I.M.S.L. is also grateful for her contract (Nº expediente administrativo subvención: POEJ00028/ N.º expediente: AND21_IAS_M2_064) from Garantía Juvenil-Andalucía 2022, Consejería de Universidad, Investigación e Innovación, Secretaría General de Investigación e Innovación, Junta de Andalucía/Cofinanciación FEDER 91%.
Author information
Authors and Affiliations
Contributions
M.A. and P.P. conceived the work. I.M.S.L. performed the experiments. M.A., I.M.S.L. and P.P. wrote the manuscript. All authors read and approved the final version of the manuscript before submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent to publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: Table 1 was missing highlighting and underlined text.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Serrano-León, I.M., Prieto, P. & Aguilar, M. Telomere and subtelomere high polymorphism might contribute to the specificity of homologous recognition and pairing during meiosis in barley in the context of breeding. BMC Genomics 24, 642 (2023). https://doi.org/10.1186/s12864-023-09738-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09738-y