
Abstract
Background
Elucidating genome-wide structural variants including copy number variations (CNVs) have gained increased significance in recent times owing to their contribution to genetic diversity and association with important pathophysiological states. The present study aimed to elucidate the high-resolution CNV map of six different global buffalo breeds using whole genome resequencing data at two coverages (10X and 30X). Post-quality control, the sequence reads were aligned to the latest draft release of the Bubaline genome. The genome-wide CNVs were elucidated using a read-depth approach in CNVnator with different bin sizes. Adjacent CNVs were concatenated into copy number variation regions (CNVRs) in different breeds and their genomic coverage was elucidated.
Results
Overall, the average size of CNVR was lower at 30X coverage, providing finer details. Most of the CNVRs were either deletion or duplication type while the occurrence of mixed events was lesser in number on a comparative basis in all breeds. The average CNVR size was lower at 30X coverage (0.201 Mb) as compared to 10X (0.013 Mb) with the finest variants in Banni buffaloes. The maximum number of CNVs was observed in Murrah (2627) and Pandharpuri (25,688) at 10X and 30X coverages, respectively. Whereas the minimum number of CNVs were scored in Surti at both coverages (2092 and 17,373). On the other hand, the highest and lowest number of CNVRs were scored in Jaffarabadi (833 and 10,179 events) and Surti (783 and 7553 events) at both coverages. Deletion events overnumbered duplications in all breeds at both coverages. Gene profiling of common overlapped genes and longest CNVRs provided important insights into the evolutionary history of these breeds and indicate the genomic regions under selection in respective breeds.
Conclusion
The present study is the first of its kind to elucidate the high-resolution CNV map in major buffalo populations using a read-depth approach on whole genome resequencing data. The results revealed important insights into the divergence of major global buffalo breeds along the evolutionary timescale.
Similar content being viewed by others


Background
Livestock contributes significantly to the national gross domestic product (GDP) of developing countries, including India. India is home to a large inventory of multiple farm animal species including more than 300 million bovines i.e., cattle and buffaloes. Buffaloes contribute significantly to the national GDP of India and its milk production and farmers’ profitability. It ensures the nutritional security of the masses in developing countries. More than half of the global bubaline population is reared in India with two main subspecies, riverine (Bubalus bubalis bubalis) and the swamp (Bubalus bubalis carabanensis). The two sub-species of buffaloes are characterized by distinct morphology and karyotype characteristics [1, 2]. Globally, the buffalo population represents 208 million heads [3]. India is the hotspot for buffalo biodiversity, mirrored by 20 recognized buffalo breeds [4]. Various Indian buffalo breeds, with improved genetic merit and performance vis-à-vis economic traits, have reached transboundary distribution across various global nations. Murrah, widely known as ‘Black gold’, is a transboundary milch breed of buffaloes accounting for 42.8% of the total Indian buffalo population. It has high milk production potential and distribution across multiple nations. Bhadawari buffaloes produce milk with high-fat content, which can range up to 13%. Jaffarabadi is one of the best milking riverine breeds, well known for its higher milk fat content and heavy body suitable for draught purposes. Banni buffaloes, believed to have evolved by the efforts of the local ‘Maldhari’ community of Gujarat, possess a unique gene pool that allows it to thrive in harsh climatic conditions. On the other hand, Surti and Pandharpuri are medium-sized breeds that can be distinguished by their sickle and sword-shaped horns, respectively. Multiple populations of these buffalo breeds have been imported by different global nations for improvement of their animal genetic resource (AnGR) base [5].
Buffaloes have evolved through more than 5000 years of domestication, leading to the adoption of morpho-physiological, and adaptive features useful in harsh tropical and humid environments. Buffaloes are ideally considered the future animals of choice to meet the ever-growing animal protein demand [6]. Buffaloes contribute around 45% to India’s total milk production. Buffalo milk is nutritionally rich with high-fat content and essential minerals and is thus recommended to produce cheese, yogurt, and cream. Besides the meat (carabeef), they also provide horns and hides, though their maximum export potential is still untapped.
Globally, numerous studies have focused on the genetic analyses of economic traits and genetic diversity in buffaloes [7, 8]. Various molecular markers, especially microsatellites and single nucleotide polymorphism (SNP) have gained increased penetrance into modern animal breeding programs wherein different structural and functional genetic variants are used to select animals for improved performance in future generations [9]. Previously, researchers utilized microsatellite markers to analyze the divergence time between swamp and river buffalo and succeeded in confirming their distinct genetic origins [10]. However, the introduction of genome assemblies and SNP chips has facilitated studies at the genome level, enabling the detection of QTLs associated with performance traits and variant detection [11, 12]. The successful release of the first haplotype-phased reference genome assembly, NDDB_SH_1, for the riverine buffalo has been a significant milestone in buffalo genomics. Along with the emergence of second and third-generation sequencing platforms, whole genome resequencing (WGS) has become more accessible, allowing for the discovery of genetic variants (structural and functional) and molecular markers with higher accuracy. It is coupled with increased and easy accessibility to bioinformatics and statistical programs that are useful to analyze the WGS data in livestock species.
Copy number variations, as structural variants, have gained significance with respect to their inheritance and association with multiple traits of economic interest and pathophysiological states. Copy number variants are unbalanced structural variants, conventionally defined as the fragments of the genome with sizes ranging from a few kilobases to 5 megabases [13]. CNVs are distinct from SNPs and Indels and occupy larger genomic spaces than other variants. In humans, 4.8–9.5% of the genome consists of CNVs, unlike the contribution from SNPs, which comes to only around 0.1% [14]. Consequently, they alter the gene dosage and genomic regulation, or cause position effects [15], resulting in drastic changes in gene expression. Different approaches have been used to elucidate the copy number variations at the genome-wide level in livestock. Initially, lower-resolution comparative genomic hybridization arrays were used, but PCR-based methods improved the resolution during subsequent times. Currently, array genotyping data and whole genome/exome/amplicon sequencing data are routinely studied while using various algorithms including PennCNV [16], CNVnator [17], CNVcaller [18], cn.MOPS [19] and others.
Various studies have reported the elucidation of copy number variations at a genome-wide scale in farm animal species including cattle [20], buffaloes [21], equine [22], pigs [23], yak [24], and chicken [25]. Furthermore, the association of CNVs with traits of economic interest has also been reported in livestock populations [9, 26, 27]. Keeping in view the aforementioned points, the present was undertaken to elucidate the CNV maps in six breeds of Indian riverine buffaloes using WGS data at two coverages. Breed-differentiated CNVs were detected, and genes overlapping the CNVs were also identified.
Results and discussion
Descriptive statistics
The latest advancements in whole genome resequencing enable the accurate detection of both common and rare CNVs. It enables the identification of smaller and naive (previously unknown ones) genetic variants down to the level of individual base pairs. The whole genome resequencing approach presents many benefits for the elucidation of structural variants including CNVs. The read depth-based methods are recommended for Illumina NGS data [15] as they do not require the reference sample and express the exact CNV counts rather than their positions. This method is adopted in CNVnator, which uses a mean shift-based (MSB) approach for accurate variant calling [28], better sensitivity and a low false-discovery rate [17]. In this study, WGS data on multiple buffalo breeds were used to elucidate the CNV and CNVR maps using a read-depth-based approach in CNVnator. Bin sizes of 1000 and 100 were found to be optimal to assess the CNVs in different buffalo breeds at 10X and 30X coverages, respectively. The read-depth approach in CNVnator is based on binning read-depth signals which are dependent on sequencing coverage and read length [29]. Therefore, an integrated run of all the samples could not be attempted in this study. However, instead of merging the twin coverage datasets, an opportunity was explored to present the CNV maps at both these coverages and report CNV/ CNVR maps in these global buffalo breeds with separate downstream processing. Post-filtering, 14,368 CNVs (8924 deletions and 5444 duplications) were scored at 10X coverage covering six global buffalo breeds. The maximum number of CNVs was observed in Murrah (2627) while Surti scored the minimum number (2092). Yang et al. [30] found 21,152 CNV regions in a whole genome dataset of 20 buffalo breeds comprising 141 buffaloes using LUMPY and CNVnator. Strillacci et al. [31] elucidated the CNV profile of Iranian river buffaloes using 90K genotyping array data and reported lesser number of structural variants (9550 CNVs, representing 1.97% of the buffalo genome) with a loss-gain ratio of more than one. The inconsistency in CNV counts within the same species is attributable to differences in CNV detection algorithms; sample size; the evolutionary history of the population/ breed; and data used for calling the structural variants. Additionally, the elucidation of structural variants including CNVs from NGS data is greatly influenced by other factors such as read length, sequence coverage, GC bias and mappability of next-generation sequencing platforms [32]. The number and distribution of CNVs were elucidated at two coverages at whole genome level i.e., 10X and 30X. The breed-wise descriptive statistics of CNVs have been illustrated in Fig. 1(a) and Fig. 1(b).

(a): CNV distribution of six buffalo breeds using a read-depth based approach on whole genome resequencing data at 10X coverage. (b). CNV distribution of six buffalo breeds using a read-depth based approach on whole genome resequencing data at 30X coverage
On the other hand, 1,27,222 CNVs were obtained across all the breeds at 30X coverage. Similar estimates have been reported in previous studies using WGS data in other species including 1,82,823 CNVs in cattle [33], 2,08,649 CNVs in goats [34] and 1,64,733 CNVs in mink [35]. Interestingly, the maximum number of total CNVs was found in Pandharpuri (25,688); however, a minimum number of CNVs were scored in Surti (17,373), consistent with the results at lower (10X) coverage. The differences in the number of CNVs explain the genetic variability between the species or breeds, especially in terms of their evolutionary history, effective population size and other similar attributes [36]. In terms of overlapping (at least 1 bp overlap) and unique (no overlap) occurrences of CNVs within and across breeds (Table 1), Pandharpuri had the maximum number with 43,788 hits (10X versus 30X comparison), while Murrah had the highest count for unique with 335 hits (10X versus 30X comparison).
The proportion of CNV types was also estimated besides the assessment of the CNV counts. In each of the breeds assessed at both the coverages, deletion events overnumbered the duplications, as depicted in Fig. 1(a) and Fig. 1(b). The finding was concurrent with previous NGS studies reported in cattle [33] and buffaloes [30]. Moreover, the deletion events were abundantly reported from aCGH arrays due to ascertainment bias, pinpointing that deletions are effectively captured by them as compared to the other analytical methods [37]. In addition, Turner et al. [38] have reported that non-allelic homologous recombination (NAHR), which is one of the potential mechanisms of CNV generation, is more likely to result in deletions as compared to duplications. However, an opposite trend has also been reported in horse populations with duplications exceeding the deletion events [22, 39].
In this study, the percentage of loss and gain events were estimated separately for all six breeds, among which Pandharpuri scored the maximum percentage of deletions at both coverages (65.25% and 69.46% at 10X and 30X, respectively). On the other hand, Surti showed the highest percentage of duplications (41.87%) at 10X while Banni scored the highest proportion estimate (33.82%) at 30X coverage. The breeds under study showed a loss/gain ratio ranging from 1.39 to 1.88 at lower sequencing coverage. When the coverage was increased to 30X, the ratio varied from 1.96 to 2.27, with Pandharpuri having the highest ratio at both the coverages. Similar estimates have been reported by Strillacci et al. [31] in Iranian buffaloes, where the Mazandarani breed had the highest ratio of 1.32. However, in the polled yak, Jia et al. [24] reported a very high ratio of loss to gain events (15.56:1) using Bovine HD bead chip genotyping data for CNV detection. The differences in loss and gain events inferred that there was a net loss of genetic material in all the breeds being studied.
The disparity in CNV length is also evident from different studies. Considering the 10X coverage, the size varied from 5 kb to a maximum of 4.9 Mb. Whereas the minimum length of CNVs in the present study was 1.1 kb for 30X, which was close to the cut-off set for filtering. This also indicated that at higher coverage, the breakpoint resolution is more, yielding comparatively smaller segments with higher accuracy [40, 41]. The largest CNV length that was documented exceeded those found in cattle: 28 kb [42] and 129.9 kb [43]. Nonetheless, other researchers have reported maximum sizes in the Mb range in chicken, horses, and buffaloes that were in line with the above findings. For instance, in chicken, the maximum size was 4.3 Mb [44], it was 4.55 Mb in horses [22], and the longest CNV of 3.48 Mb size has been reported in buffaloes [31].
Detection of CNVRs
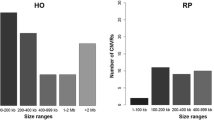
The CNVR diversity among buffalo breeds, as elucidated in the present study, has been presented in Fig. 2(a) and Fig. 2(b). At 10X coverage, the total number of CNVRs across all the breeds was 4878 with estimates ranging from 783 (Surti) to 833 (Jaffarabadi). A systematic investigation of CNVs by Liu et al. [21] identified only 1344 CNVRs in 14 water buffaloes using the read depth-based approach.

(a): The distribution pattern of CNVRs across six buffalo breeds on the concatenation of overlapping CNVs at 10X coverage. (b): The distribution pattern of CNVRs across six buffalo breeds on the concatenation of overlapping CNVs at 30X coverage
In the present study, a total of 55,002 CNVRs were recorded across all breeds at 30X coverage. The highest number of CNVRs was scored in Jaffarabadi (10,179) while the estimate was lowest in Surti (7553). Kumar et al. [45] found that despite the proximity of their breeding tracts, Jaffarabadi and Surti exhibit genetic distinctness, which may be indicated by their different CNVR count. Prior studies have also suggested that variation in CNVRs might have facilitated rapid adaptation during the domestication process and expansion of the population [46].
The CNVRs, elucidated in the present study, were classified into three categories; deletion, duplication and mixed (containing both deletions and duplications). A total of 3182, 1551, and 145 CNVRs with deletion, duplication and mixed events were scored at 10X coverage. On breed-wise examination, Pandharpuri (554) and Jaffarabadi (552) showed to have the most deletion-based CNVRs at 10X coverage. On the other hand, Bhadawari showed the highest number of duplications (270), while Banni obtained the highest count in mixed events (31). In another study, Zhang et al. [47] studied the nature of CNVRs in water buffaloes (n = 106), and recorded similar counts, with 2245 loss, 1289 gain, and 200 mixed events out of total 3734 CNV regions. It equated to only 0.88% of the reference genome assembly of Mediterranean riverine buffalo (UOA_WB_1).
The number of CNVRs within each type increased almost 10 times when the coverage was changed from 10X to 30X (3182 versus 39,144 deletion types, 1551 versus 14,558 duplication types, and 145 versus 1300 mixed types at 10X and 30X, respectively). Among different breeds, the maximum number of deletions, and duplications were observed in Jaffarabadi (7281) and Pandharpuri (3076), respectively. The latter also had the highest score for mixed-type CNVRs. Intriguingly, Surti exhibited the least counts for all three CNVR types. The average loss-gain ratio observed across all the breeds was 2.06 at 10X (varying from 1.86 to 2.41). On the other hand, the ratio was found to be higher at 30X (2.74), ranging between 2.15 and 3.25.
The unique and overlapping CNVRs were analyzed for all the breeds, and the data have been summarised in Table 2. Jaffarabadi had the highest number of overlapping hits on within-breed 10X versus 30X comparison, while Surti showed the lowest estimate. Importantly, the highest number of overlapping CNVRs were shared between Jaffarabadi and Pandharpuri, which might be indicative of possible genetic relatedness. In concurrence with the above findings, Kumar et al. [45] reported that Jaffarabadi and Pandharpuri belonged to the same lineage of buffalo breeds. Besides, the greatest fraction of overlapping CNVRs were documented for Bhadawari with 31.6% similarity between the coverages. In terms of unique hits, Murrah scored the highest (n = 114), while Pandharpuri (n = 31) showed the lowest hits.
Table 3 presents additional information on the average size of CNVRs, genomic coverage and the perfect overlap in CNVRs (%) at two levels of sequencing coverage. The average length of CNVRs indicated that longer segments (~ 0.2 Mb) were detected at 10X while a wide variation in size (6 kb − 24.4 kb) was noted at 30X. Furthermore, the proportion of CNVR size with respect to the reference genome (2.77Gb) suggested that Murrah (6.00% at 10X) and Pandharpuri (8.79% at 30X) made the most significant contributions in the genomic overlap of structural variants among the assessed breeds, reflecting their differential representation in the reference assembly. It is noteworthy that the genome coverage from other CNV studies in cattle (6%) and yak (5.7–6%) also falls within the same range [48,49,50]. Recently, Yang et al. [30] identified 21,152 CNVRs, representing 1.99% of the bubaline autosome length using multiple algorithms. Additionally, a couple of studies have discussed the chromosomal distribution of CNVRs including sex chromosomes, X and Y [51].
Remarkably, a higher level of sequence coverage or deeper sequencing was found to be beneficial for the precise detection of CNVs in buffaloes. This is in line with the previous research showing that greater sequencing coverage being linked to better sensitivity in CNV detection [52]. Prior studies have also suggested that coverage of 4X may be sufficient for detecting CNVs using the read depth method [53]. The average size of CNVRs scored in different breeds at 10X and 30X coverages was 0.201 Mb and 0.013 Mb, respectively. Table 4 depicts the distribution and relations of CNVRs in relation to gene density in different buffalo breeds across autosomes at twin coverages. Chr_13 was found to be gene-poor (in terms of density) but with the highest number of CNVRs (63–85 CNVRs at 10X coverage and 717–812 CNVRs at 30X coverage) in different breeds. Most of the clustering of CNVRs was evident in telomeric and sub-telomeric regions across different autosomes in most breeds. The least number of CNVRs were present on the smallest autosome (Chr_23) at both sequencing coverages, except Banni (10X) and Pandharpuri (30X).
Overall, the distribution of CNVRs at both the coverages was uneven, which was expected given the functionality of corresponding genes or regulatory genetic elements. Figure 3 represents the distribution of CNVRs on different chromosomes of the bubaline genome in relation to the gene density, using ideograms, across different breeds at 10X coverage.

The ideograms depicting the distribution of CNVRs in different bubaline breeds at 10X WGS coverage
Gene profiling
The longest CNVRs of each breed at both the coverages were evaluated, and the genes harbored on the top five CNVRs were shortlisted. The longest CNVR in Banni was located on autosome 16 at 10X, spanning 0.573 Mb. It harboured several genes such as SRGAP2, IKBKE, RASSF5, ELF2D, DYRK3, MAPKAPK, IL-10, IL-19, IL-20 and IL-24. Interestingly, some of the genes overlapped with those observed in other breeds as well. For example, the ZEB2 was shared among all the breeds, which is known to play a crucial role in the transforming growth factors β-signaling pathway, and its effects on growth, weight traits, and horn ontogenesis in cattle [54, 55]. IL-10 gene is responsible for maintaining body homeostasis by resolving acute inflammation [56]. IKBKE, on the other hand, has been reported to regulate the maternal immune response during conceptus implantation in cattle [57]. Similarly, Oliveira et al. [58] reported the regulatory role of SDC1 gene in controlling the milk yield in Ayrshire cattle. In addition, the largest CNVRs of Bhadawari encompassed different genes such as KHDRBS2, DYNC2I1, and VIPR2. Among these genes, KHDRBS2 has been associated with reproductive traits in goats and Brahman cows, as well as adaptability in Colombian cattle [59]. In Jaffarabadi, the longest CNVR (0.573 Mb) was present on chromosome 16. The unique genes that showed overlap with the largest (top five) CNV regions were LDAH, GDF7, HS1BP3, U4, OSR1, and 7SK. Studies have shown that LDAH promotes triglyceride production [60] while GDF7 plays a role in seminal growth and neuronal development [61]. In Surti, the longest CNVRs harboured genes like DYNC2I2, VIPR3, COA1, STK17A, and HECW1, with COA1 being implicated in mitochondrial translation thereby contributing to fitness and longevity. In mammals, the genomic region of COA1 is recognized as a prominent evolutionary breakpoint area in which a combined deletion of STK17A and COA1 genes was studied in rodents [62]. The largest CNVR in this breed was found on chromosome 2, which spanned a size of 0.668 Mb. Furthermore, SFMBT2 gene was observed in Murrah, with the longest CNVR on chromosome 2 similar to Surti, covering a size of 1.17 Mb. This gene is important for trophoblast maintenance, placental development, and regulation of chondrocyte proliferation [63]. Finally, in Pandharpuri, the greatest CNVR spanned 0.729 Mb of buffalo genome with genomic coordinates spreading across chromosome 2. PARD3 gene was identified in one of the top CNVRs, which is involved in cell growth and division as well as the formation of tight junctions in epithelial cells [64].
On the other hand, at 30X coverage, Banni showed the largest CNVR on chromosome 3, with a length of 0.337 Mb. The region contained several genes, including TMEM45A, ZBTB12, EHMT2, SLC44A4, NEU1, SNORD52, SNORD48, and two heat shock proteins namely HSPA1A and HSPA1L, which are involved in normal cell growth and survival, as well as protecting cytotoxic conditions [65]. HSPA1A and HSPA1L genes are reported to have sequence similarity arising from the duplication of genes offering thermotolerance at the cellular level [66]. Similarly, Bhadawari and Surti also showed large CNV regions on the same chromosome, with comparatively smaller sizes of around 0.285 Mb for both breeds. In Bhadawari, the genes such as TRAPPC3, COL8A2, ADPRS, TEKT2, and genes of the Argonaute family (AGO1, 3 and 4) showed overlap with CNVRs. The eukaryotic AGO proteins are active mediators in RNA silencing and other cellular processes [67]. For Surti, the genes exactly overlapped with that of the Bhadawari breed. Among the reported genes, Collagen VIII (COL8) is reported to play a major role in vascular integrity [68]. Jaffarabadi carried the longest CNVR on chromosome 14, which was the smallest one among all the breeds (0.246 Mb) contributing only 0.0089% of the genomic size. RGS7 gene was observed in Jaffarabadi, belonging to the G-protein signaling family, that are significant in regulating a wide range of neuronal processes, such as vision, and nociception in mammalian species [69]. This gene was also identified as a candidate for milk production in Holstein cattle [70]. Considering the top five CNVRs of Murrah, the following genes were identified: COA1, STK17A, RGS7, DYNC2I1, VIPR2, and HECW1. Among these, STK17A gene is significantly involved in apoptosis, which has functions in immune response and disease resistance [71]. In contrast, VIPR2 gene encodes a receptor that responds to vasoactive intestinal peptide (VIP), which helps with smooth muscle relaxation and the secretion of exocrine and endocrine glands. Mahoney et al. [72] also found that interfering with VIP production leads to a delay and reduction in the luteinizing hormone (LH) surge. Another gene, HECW1, is highly active in nerve cells and participates in the regulation of protein homeostasis, which has implications for both longevity and conditions related to ageing [73]. In sheep, studies have also reported this gene to be involved in variation with regard to the number of lambs born and have identified it as a target of selection in the Luzhong mutton sheep breed [74, 75]. Strikingly, the longest CNVR among all the breeds was present on chromosome 5 in Pandharpuri (0.475 Mb), covering 0.0171% of the genome. Consequently, this region showed overlap with multiple genes, including olfactory receptor genes (OR10J5, OR10J1, OR10J4, and OR10J3), KANSL2, SNORA2C, LALBA, CRP, APCS, TUBA1B, LMBR1L, DHH, RHEBL1, and KMT2D. Zhou et al. [76] described the potential role of olfactory genes in yak for the perception of chemical stimuli, which is very crucial for reproduction, acquiring basic needs such as food and mate, high altitude adaptation, and ultimately the survival of the animal. Another gene, Tubulin alpha 1b (TUBA1B) is an important component in the formation of the cytoskeleton, which is involved in immune cell infiltration, cell movement and within-cell transport [77]. Additionally, LALBA gene polymorphisms have been shown to influence milk production traits and somatic cell count in Polish Holstein-Friesian cows [78]. Interestingly, many of the genes identified at 30X in different breeds were similar to those scored at 10X coverage.
The genes overlapping the CNVs provided useful insights into the evolutionary history of these breeds. The genes present in all six populations were elucidated in DAVID. These genes were involved in the significant enrichment of pathways including oxidative phosphorylation, thermogenesis and pentose phosphate pathway. Significant enrichment was also noticed for biological processes like mitochondrial electron transport, regulation of presynapse assembly and actin filament organization. Similarly, cellular components including the respiratory chain, mitochondrial respiratory chain complex I and mitochondrial inner membrane were mainly involved while molecular functions like NADH dehydrogenase activity and metal ion binding were significantly enriched. The network of hub genes as identified using STRING-DB has been presented in supplementary Fig. 1.
Conclusions
The present study provides new insights into the genetic variations among six important buffalo breeds. The findings from the present study elaborate on the evolutionary differences of six global buffalo breeds in terms of structural variants i.e., CNVs and CNVRs. Interestingly, a higher level of sequence coverage or deeper sequencing was found to be beneficial for the precise detection of CNVs in buffaloes with finer details. The genomic coverage of CNVs and CNVRs in these buffalo breeds varied amongst themselves. The highest genomic coverage of CNVRs was found for Murrah (6.00%) and Pandharpuri (8.79%) breeds. The results offered potential candidate genes linked to performance differences that could be used for selective breeding in the future. The CNV and CNVR maps for different buffalo breeds may be useful for the association of these structural variants with important (re)production and adaptability traits.
Methods
Sampling and whole genome resequencing data
The present study was undertaken on whole genome resequencing data on 75 buffaloes (Bubalus bubalis) representing six distinct breeds, with 12 animals each from Murrah, Surti, and Banni and 13 animals each from Bhadawari, Jaffarabadi, and Pandharpuri populations (Table 5). These breeds have been imported by different nations courtesy their high genetic merit and proven performance with respect to economic traits and have been integrated into the breeding policy for improvement of buffaloes in these countries [5]. The sequencing data was retrieved from an online database and was based on an earlier publication report [79]. The samples were obtained from breeding tracts of respective buffalo breeds (as given in Table 5) and sequenced using two different platforms as described by Dutta et al. [79]. Briefly, paired-end sequencing data were generated at two sequencing centres using different coverages and sequence read lengths. One set of samples was sequenced using NEBNext Ultra DNA Library Prep Kit (library preparation) and Illumina HiSeq 2500 sequencing platform at SciGenom Labs (India) at 10X coverage with a read length of 250 bp. Whereas, the rest of the samples were sequenced using Illumina TruSeq Nano DNA Library Prep Kit (library preparation) and Illumina HiSeq X sequencing platform at Edinburgh Genomics (United Kingdom) at 30X coverage with a read length of 150 bp. The average sequencing coverage for samples at two centres (SciGenom Labs, India, and Edinburgh Genomics, United Kingdom) was 8X and 37X, respectively. In nutshell, whole genome resequencing was retrieved on these animals across two different coverages: 10X for six animals from each breed and 30X for the rest, as indicated in Table 5.
The quality of the sequence reads was assessed with FastQC v0.12.1 [80] and poor-quality reads and adapters were removed via TrimGalore v0.6.5 [81] using default parameters. After trimming and reassessing the read quality, the Burrows-Wheeler aligner v0.7.12 [82] was used to index the genome NDDB_SH_1 of water buffaloes (release date: September 2021), which has a total sequence length of 2,622,460,639 bp [83]. The sequence reads, after quality control were mapped against the genome index using BWA-MEM algorithm with default settings [82].
Post-alignment processing
Following alignment to the genome assembly, the individual sequence alignment map (SAM) files were converted into a more efficient binary alignment map format (BAM) using SAMtools v1.17 [84]. The output files were validated by Picard tools v2.25.1 [85] build under GATK v4.0.2.0 and sorted in concordance with genomic coordinates. Subsequently, the SM read tags were added to the mapped reads. Furthermore, the PCR duplicates in BAM files were marked for removal using the ‘MarkDuplicates’ function of Picard tools. This effectively minimizes the PCR amplification bias by clipping on 5′ read ends.
CNV detection, filtering and concatenation
CNV detection and analysis were undertaken using read-depth-based software CNVnator v.0.4.1 [17]. The core principle behind CNVnator involves dividing the genome into non-overlapping bins set by the user and calculating the RD signal by counting the mapped reads within each bin. Following this, CNVnator performs statistical significance tests for CNV predictions [17]. In the present study, copy number histograms were generated from PCR duplicate-free Picard-BAM files for partitioning the CNV calls and for downstream statistical analysis. Subsequently, the optimal bin size for read depth analysis was chosen as the multiple of 100s in all the samples by considering the read depth, read length, distribution, and quality. The optimal bin size was selected based on the ratio of the read depth and its variance; fitting the recommended ratio between 4 and 5. Correction for GC waves was done within CNVnator, which is necessary to eliminate the GC bias resulting from reduced depth coverage at GC-rich regions [86, 87]. Subsequently, the CNV variants were individually called using the ‘call’ command in CNVnator.
After CNV detection, the quality control and post-pruning were done based on p-value, zero mapping quality (q0), and CNV size. The study considered the following parameters for filtering of CNVs: p-value calculated by t-test statistics < 0.01, variants with mapping quality < 0.5, and the size of CNVs < 1 kb and > 5 Mb, as suggested by previous studies [88]. The q0 filter (fraction of mapped reads with zero quality) of 0.5 was used in the present study that was indicative of the degree of certainty that a read comes from the location to which it is aligned.
A customized Python script was used to concatenate the overlapping CNVs (with minimum of one bp overlap) into copy number variation regions (CNVRs). The merged CNVRs were marked as deletion, duplication, or mixed depending on whether the events in proximal CNVs were all deletion, all duplication, or a mix of deletion and duplication. The whole analysis was done using the high-performance computing facility of Indiana University and Purdue University, Indianapolis (IUPUI), USA (now Indiana University Indianapolis).
The impact of sequence coverage on CNV detection was also investigated in study animals with two levels of sequence coverage (10X and 30X). Changes in the CNV count were attributable to the changes in sequencing coverage and read length. RIdeogram package [89] was used to plot the distribution of CNVRs in relation to the gene density. Briefly, the gene density parameters were elucidated from gff file corresponding to the genome assembly used in the study using a window of 1 Mb. The chromosomal coordinates were retrieved from assembly metadata. RIdeogram package was used to plot the CNVR data overlaid by the gene density parameters.
Unique and overlapping CNVs and CNVRs
The chromosomal coordinates of structural variants from different populations were used in BEDTools v2.26.0 [90] to elucidate the common (with the same chromosomal coordinates), overlapping (with at least one bp overlap) and unique CNVs and CNVRs. Furthermore, the genomic coverage of CNVRs against the reference genome assembly was elucidated in each population.
Gene profiling
The information on genes and genetic variants overlapping CNVRs in different breeds was extracted by mining data corresponding to their genomic coordinates from the genome annotation file (gtf file, corresponding to the NDDB_SH_1 genome assembly) using BEDTools program. The common genes found to overlap CNVRs in all six populations were used for profiling and functional annotation in Database for Annotation, Visualization and Integrated Discovery (DAVID). Furthermore, the longest CNVRs in terms of chromosomal coordinates in each breed were identified and overlapping genes were elucidated in Ensembl. The information on the functioning of these genes was retrieved by mining the information from PubMed and other relevant databases. Additionally, the common genes that showed overlap with CNVRs were processed in STRING-DB for identification of hub genes that are involved in various pathways in buffaloes.
Data Availability
The original datasets supporting the conclusions of this article are available at (https://www.ebi.ac.uk/ena/browser/view/PRJEB39591) with accession number PRJEB39591.
Abbreviations
- BAM:
-
Binary alignment map
- CNV:
-
Copy number variation
- CNVR:
-
Copy number variation region
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- GDP:
-
Gross domestic product
- LH:
-
Luteinizing hormone
- MSB:
-
Mean shift based
- SAM:
-
Sequence alignment map
- SNP:
-
Single nucleotide polymorphism
- WGS:
-
Whole genome (re)sequencing
References
Yindee M, Vlamings BH, Wajjwalku W, Techakumphu M, Lohachit C, Sirivaidyapong S, et al. Y-chromosomal variation confirms independent domestications of swamp and river buffalo. Anim Genet. 2010;41:433–5.
Degrandi TM, Pita S, Panzera Y, de Oliveira EHC, Marques JRF, Figueiró MR, et al. Karyotypic evolution of ribosomal sites in buffalo subspecies and their crossbreed. Genet Mol Biol. 2014;37:375.
Minervino AHH, Zava M, Vecchio D, Borghese A. Bubalus bubalis: a short story. Front Vet Sci. 2020;7:971.
New Breeds / Lines - ICAR- National Bureau of Animal Genetic Resources. https://nbagr.icar.gov.in/en/new-breeds-lines/. Accessed 6 May 2023.
Bastianetto E, de Oliveira DAA, McManus C, Bagolin D, de Leite J, Melo RC. CB de. Genetic material from buffalo and cattle: crucial importance in the formalization of bilateral trade between India and Brazil. Anim Reprod. 2020;17:e20200031.
Selokar NL, Singh MK, Kumar D, Sharma R, Chauhan MS, Yadav PS. Buffalo Cloning: Multiplying India’s Black Gold. Genomic, Proteomics, and Biotechnology. 2022;:239–48.
Rehman SU, Hassan FU, Luo X, Li Z, Liu Q. Whole-genome sequencing and characterization of Buffalo Genetic Resources: recent advances and Future Challenges. Anim (Basel). 2021;11:1–20.
Chen Z, Zhu M, Wu Q, Lu H, Lei C, Ahmed Z, et al. Analysis of genetic diversity and selection characteristics using the whole genome sequencing data of five buffaloes, including Xilin buffalo, in Guangxi, China. Front Genet. 2023;13:1084824.
Ahmad SF, Singh A, Panda S, Malla WA, Kumar A, Dutt T. Genome-wide elucidation of CNV regions and their association with production and reproduction traits in composite Vrindavani cattle. Gene. 2022;830:146510.
Zhang Y, Sun D, Yu Y, Zhang Y. Genetic variation and divergence among Swamp Buffalo, River Buffalo and cattle: a microsatellite survey on five populations in China. Asian-Australas J Anim Sci. 2008;21:1238–43.
Du C, Deng T, Zhou Y, Ye T, Zhou Z, Zhang S, et al. Systematic analyses for candidate genes of milk production traits in water buffalo (Bubalus Bubalis). Anim Genet. 2019;50:207–16.
Ravi Kumar D, Nandhini PB, Joel Devadasan M, Sivalingam J, Mengistu DW, Verma A et al. Genome-wide association study revealed suggestive QTLs for production and reproduction traits in indian Murrah buffalo. 3 Biotech. 2023;13.
Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, et al. Mapping copy number variation by population-scale genome sequencing. Nat 2011. 2011;470:7332.
Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16:172–83.
Pirooznia M, Goes F, Zandi PP. Whole-genome CNV analysis: Advances in computational approaches. Front Genet. 2015;6 MAR:138.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SFA, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–74.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–84.
Wang X, Zheng Z, Cai Y, Chen T, Li C, Fu W, et al. CNVcaller: highly efficient and widely applicable software for detecting copy number variations in large populations. Gigascience. 2017;6:1–12.
Klambauer G, Schwarzbauer K, Mayr A, Clevert DA, Mitterecker A, Bodenhofer U et al. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012;40.
Kommadath A, Grant JR, Krivushin K, Butty AM, Baes CF, Carthy TR, et al. A large interactive visual database of copy number variants discovered in taurine cattle. Gigascience. 2019;8:1–12.
Liu S, Kang X, Catacchio CR, Liu M, Fang L, Schroeder SG, et al. Computational detection and experimental validation of segmental duplications and associated copy number variations in water buffalo (Bubalus bubalis). Funct Integr Genomics. 2019;19:409–19.
Laseca N, Molina A, Valera M, Antonini A, Demyda-Peyrás S. Copy number variation (CNV): a new genomic insight in horses. Animals. 2022;12:1435.
Stafuzza NB, Silva RMDO, Fragomeni BDO, Masuda Y, Huang Y, Gray K, et al. A genome-wide single nucleotide polymorphism and copy number variation analysis for number of piglets born alive. BMC Genomics. 2019;20:1–11.
Jia C, Wang H, Li C, Wu X, Zan L, Ding X, et al. Genome-wide detection of copy number variations in polled yak using the Illumina BovineHD BeadChip. BMC Genomics. 2019;20:1–14.
Sohrabi SS, Mohammadabadi M, Wu DD, Esmailizadeh A. Detection of breed-specific copy number variations in domestic chicken genome. Genome. 2018;61:7–14.
Shi SY, Li LJ, Zhang ZJ, Wang EY, Wang J, Xu JW, et al. Copy number variation of MYLK4 gene and its growth traits of Capra hircus (goat). Anim Biotechnol. 2020;31:532–7.
Huang Y, Li Y, Wang X, Yu J, Cai Y, Zheng Z, et al. An atlas of CNV maps in cattle, goat and sheep. Sci China Life Sci. 2021;64:1747–64.
Wang LY, Abyzov A, Korbel JO, Snyder M, Gerstein M. MSB: a mean-shift-based approach for the analysis of structural variation in the genome. Genome Res. 2009;19:106–17.
Suvakov M, Panda A, Diesh C, Holmes I, Abyzov A. CNVpytor: a tool for copy number variation detection and analysis from read depth and allele imbalance in whole-genome sequencing. Gigascience. 2021. 10.
Yang L, Han J, Deng T, Li F, Han X, Xia H, et al. Comparative analyses of copy number variations between swamp buffaloes and river buffaloes. Anim Genet. 2023;54:199–206.
Strillacci MG, Moradi-Shahrbabak H, Davoudi P, Ghoreishifar SM, Mokhber M, Masroure AJ, et al. A genome-wide scan of copy number variants in three iranian indigenous river buffaloes. BMC Genomics. 2021;22:1–14.
Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Reviews Genet 2011. 2011;12:5.
Hu Y, Xia H, Li M, Xu C, Ye X, Su R, et al. Comparative analyses of copy number variations between Bos taurus and Bos indicus. BMC Genomics. 2020;21:1–11.
Guo J, Zhong J, Liu GE, Yang L, Li L, Chen G, et al. Identification and population genetic analyses of copy number variations in six domestic goat breeds and Bezoar ibexes using next-generation sequencing. BMC Genomics. 2020;21:1–13.
Davoudi P, Do DN, Rathgeber B, Colombo SM, Sargolzaei M, Plastow G et al. Genome-wide detection of copy number variation in american mink using whole-genome sequencing. BMC Genomics. 2022;23.
Kim YM, Ha SJ, Seong HS, Choi JY, Baek HJ, Yang BC, et al. Identification of Copy Number Variations in four horse breed populations in South Korea. Animals. 2022;12:3501.
Jenkins GM, Goddard ME, Black MA, Brauning R, Auvray B, Dodds KG, et al. Copy number variants in the sheep genome detected using multiple approaches. BMC Genomics. 2016;17:1–14.
Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Blayney ML, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–5.
Solé M, Ablondi M, Binzer-Panchal A, Velie BD, Hollfelder N, Buys N, et al. Inter- A nd intra-breed genome-wide copy number diversity in a large cohort of european equine breeds. BMC Genomics. 2019;20:1–12.
Duan J, Zhang JG, Deng HW, Wang YP. Comparative studies of Copy Number Variation detection methods for next-generation sequencing Technologies. PLoS ONE. 2013;8:e59128.
Gross AM, Ajay SS, Rajan V, Brown C, Bluske K, Burns NJ, et al. Copy-number variants in clinical genome sequencing: deployment and interpretation for rare and undiagnosed disease. Genet Med. 2019;21:1121–30.
Stothard P, Choi JW, Basu U, Sumner-Thomson JM, Meng Y, Liao X et al. Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genomics. 2011;12.
Zhan B, Fadista J, Thomsen B, Hedegaard J, Panitz F, Bendixen C. Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping. BMC Genomics. 2011;12:1–20.
Crooijmans RPMA, Fife MS, Fitzgerald TW, Strickland S, Cheng HH, Kaiser P, et al. Large scale variation in DNA copy number in chicken breeds. BMC Genomics. 2013;14:1–10.
S K, J G, N K, K D, N N, P J, et al. Genetic variation and relationships among eight indian riverine buffalo breeds. Mol Ecol. 2006;15:593–600.
Lye ZN, Purugganan MD. Copy Number Variation in Domestication. Trends Plant Sci. 2019;24:352–65.
Zhang X, Chen N, Chen H, Lei C, Sun T. Comparative analyses of copy number variations between swamp and river buffalo. Gene. 2022;830:146509.
Letaief R, Rebours E, Grohs C, Meersseman C, Fritz S, Trouilh L, et al. Identification of copy number variation in french dairy and beef breeds using next-generation sequencing. Genet Sel Evol. 2017;49:77.
Wang H, Chai Z, Hu D, Ji Q, Xin J, Zhang C, et al. A global analysis of CNVs in diverse yak populations using whole-genome resequencing. BMC Genomics. 2019;20:1–12.
Zhang X, Wang K, Wang L, Yang Y, Ni Z, Xie X, et al. Genome-wide patterns of copy number variation in the chinese yak genome. BMC Genomics. 2016;17:1–12.
Sun T, Hanif Q, Chen H, Lei C, Dang R. Copy number variations of four Y-Linked genes in Swamp Buffaloes. Anim (Basel). 2020;10.
Zhang L, Bai W, Yuan N, Du Z. Comprehensively benchmarking applications for detecting copy number variation. PLoS Comput Biol. 2019;15:e1007069.
Gao Y, Jiang J, Yang S, Hou Y, Liu GE, Zhang S, et al. CNV discovery for milk composition traits in dairy cattle using whole genome resequencing. BMC Genomics. 2017;18:1–12.
Gehrke LJ, Upadhyay M, Heidrich K, Kunz E, Klaus-Halla D, Weber F et al. A de novo frameshift mutation in ZEB2 causes polledness, abnormal skull shape, small body stature and subfertility in Fleckvieh cattle. Scientific Reports 2020 10:1. 2020;10:1–14.
Zhang D, Zhang X, Li F, La Y, Li G, Zhang Y, et al. The association of polymorphisms in the ovine PPARGC1B and ZEB2 genes with body weight in Hu sheep. Anim Biotechnol. 2022;33:90–7.
Hussain T, Shah SZA, Zhao D, Sreevatsan S, Zhou X. The role of IL-10 in Mycobacterium avium subsp. paratuberculosis infection. Cell Communication and Signaling. 2016;2016 14:1.
Nakamura K, Kusama K, Ideta A, Kimura K, Hori M, Imakawa K. Effects of miR-98 in intrauterine extracellular vesicles on maternal immune regulation during the peri-implantation period in cattle. Sci Rep. 2019;9.
Oliveira HR, Cant JP, Brito LF, Feitosa FLB, Chud TCS, Fonseca PAS, et al. Genome-wide association for milk production traits and somatic cell score in different lactation stages of Ayrshire, Holstein, and Jersey dairy cattle. J Dairy Sci. 2019;102:8159–74.
Strillacci MG, Vevey M, Blanchet V, Mantovani R, Sartori C, Bagnato A. The genomic variation in the Aosta cattle breeds raised in an extensive Alpine Farming System. Anim (Basel). 2020;10:1–18.
Goo YH, Son SH, Paul A. Lipid Droplet-Associated Hydrolase promotes lipid Droplet Fusion and enhances ATGL degradation and triglyceride Accumulation. Sci Rep. 2017;7.
Settle S, Marker P, Gurley K, Sinha A, Thacker A, Wang Y, et al. The BMP family Member Gdf7 is required for seminal vesicle growth, branching morphogenesis, and cytodifferentiation. Dev Biol. 2001;234:138–50.
Shinde SS, Sharma S, Teekas L, Sharma A, Vijay N. Recurrent erosion of COA1/MITRAC15 exemplifies conditional gene dispensability in oxidative phosphorylation. Scientific Reports 2021 11:1. 2021;11:1–16.
Hussain S, Sun M, Guo Y, Mushtaq N, Zhao Y, Yuan Y, et al. SFMBT2 positively regulates SOX9 and chondrocyte proliferation. Int J Mol Med. 2018;42:3503–12.
Cui R, Chen D, Li N, Cai M, Wan T, Zhang X, et al. PARD3 gene variation as candidate cause of nonsyndromic cleft palate only. J Cell Mol Med. 2022;26:4292–304.
Lanneau D, Wettstein G, Bonniaud P, Garrido C. Heat shock proteins: cell protection through protein triage. ScientificWorldJournal. 2010;10:1543–52.
Hansen PJ. Prospects for gene introgression or gene editing as a strategy for reduction of the impact of heat stress on production and reproduction in cattle. Theriogenology. 2020;154:190–202.
Müller M, Fazi F, Ciaudo C. Argonaute Proteins: from structure to function in Development and pathological cell fate determination. Front Cell Dev Biol. 2020;7:360.
Mohabeer AL, Kroetsch JT, McFadden M, Khosraviani N, Broekelmann TJ, Hou G, et al. Deletion of type VIII collagen reduces blood pressure, increases carotid artery functional distensibility and promotes elastin deposition. Matrix Biol Plus. 2021;12:100085.
Anderson GR, Posokhova E, Martemyanov KA. The R7 RGS protein family: multi-subunit regulators of neuronal G protein signaling. Cell Biochem Biophys. 2009;54:33–46.
Teng J, Wang D, Zhao C, Zhang X, Chen Z, Liu J et al. Longitudinal genome-wide association studies of milk production traits in Holstein cattle using whole-genome sequence data imputed from medium-density chip data. J Dairy Sci. 2023;106.
Xu Y, Feng Y, Li S, Sun J. Identification and characterization of apoptosis-related gene serine/threonine kinase 17A (STK17A) from japanese flounder Paralichthys olivaceus. Fish Shellfish Immunol. 2020;98:1008–16.
Mahoney MM, Ramanathan C, Hagenauer MH, Thompson RC, Smale L, Lee T. Daily rhythms and sex differences in vasoactive intestinal polypeptide, VIPR2 receptor and arginine vasopressin mRNA in the suprachiasmatic nucleus of a diurnal rodent, Arvicanthis niloticus. Eur J Neurosci. 2009;30:1537–43.
Erdman VV, Karimov DD, Tuktarova IA, Timasheva YR, Nasibullin TR, Korytina GF. Alu Deletions in LAMA2 and CDH4 genes are Key Components of Polygenic Predictors of Longevity. Int J Mol Sci. 2022;23.
Hernández-Montiel W, Martínez-Núñez MA, Ramón-Ugalde JP, Román-Ponce SI, Calderón-Chagoya R, Zamora-Bustillos R. Genome-wide Association Study reveals candidate genes for litter size traits in Pelibuey Sheep. Anim (Basel). 2020;10.
Tao L, He XY, Wang FY, Pan LX, Wang XY, Gan SQ, et al. Identification of genes associated with litter size combining genomic approaches in Luzhong mutton sheep. Anim Genet. 2021;52:545–9.
Zhou C, Liu Y, Zheng X, Shang K, Cheng M, Wang L, et al. Characterization of olfactory receptor repertoires provides insights into the high-altitude adaptation of the yak based on the chromosome-level genome. Int J Biol Macromol. 2022;209:220–30.
Hu X, Zhu H, Chen B, He X, Shen Y, Zhang X et al. Tubulin alpha 1b is Associated with the Immune Cell Infiltration and the response of HCC Patients to Immunotherapy. Diagnostics. 2022;12.
Ostrowska M, Zwierzchowski L, Brzozowska P, Kawecka-Grochocka E, Żelazowska B, Bagnicka E. The effect of single-nucleotide polymorphism in the promoter region of bovine alpha-lactalbumin (LALBA) gene on LALBA expression in milk cells and milk traits of cows. J Anim Sci. 2021;99:1–9.
Dutta P, Talenti A, Young R, Jayaraman S, Callaby R, Jadhav SK et al. Whole genome analysis of water buffalo and global cattle breeds highlights convergent signatures of domestication. Nature Communications 2020 11:1. 2020;11:1–13.
Andrews S, FastQC. A Quality Control tool for High Throughput Sequence Data. Soil. 2010;5. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 21 Dec 2021.
Krueger F. Babraham Bioinformatics - Trim Galore! Version 0.4.4. 2017. https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/. Accessed 24 Dec 2021.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013.
NDDB_SH_1 - Genome. - Assembly - NCBI. https://www.ncbi.nlm.nih.gov/assembly/GCF_019923935.1. Accessed 6 May 2023.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10:1–4.
Picard Tools - By Broad Institute. https://broadinstitute.github.io/picard/. Accessed 6 May 2023.
Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–92.
Mallory XF, Edrisi M, Navin N, Nakhleh L. Methods for copy number aberration detection from single-cell DNA-sequencing data. Genome Biol. 2020;21:1–22.
Jang J, Kim K, Lee YH, Kim H. Population differentiated copy number variation of Bos taurus, Bos indicus and their african hybrids. BMC Genomics. 2021;22:1–11.
Hao Z, Lv D, Ge Y, Shi J, Weijers D, Yu G, et al. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput Sci. 2020;6:1–11.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Acknowledgements
The authors acknowledge the support from James G.D. Prendergast, corresponding author of publication under which the data was originally generated, who was kind enough to provide timely clarifications whenever required. The first author gratefully acknowledges the support provided by Janga Lab, IUPUI, Indianapolis, USA (now Indiana University Indianapolis) throughout the study. We are grateful to Arnav Mehrotra, Animal Genomics, ETH Zurich for support during the analysis and helpful comments on the manuscript. The authors would like to express appreciation and sincere gratitude to the editors and reviewers for their thoughtful and constructive comments which helped to significantly improve various aspects of the manuscript.
Funding
The study was supported financially by the CAAST-ACLH project of NAHEP and CABin scheme.
Author information
Authors and Affiliations
Contributions
SFA, AK, GKG and SCJ: Design of the work; SFA, CCS, SV and WAM: Analysis; SCJ, AK, SMA: Interpretation of data; SFA, CCS, SV: Drafted the work or substantively revised it; TD: Resources and supervision. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
It is declared that the current study was conducted without any commercial or financial interest that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ahmad, S.F., Chandrababu Shailaja, C., Vaishnav, S. et al. Read-depth based approach on whole genome resequencing data reveals important insights into the copy number variation (CNV) map of major global buffalo breeds. BMC Genomics 24, 616 (2023). https://doi.org/10.1186/s12864-023-09720-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09720-8