
Abstract
Background
Lonicera japonica Thunb. is widely used in traditional Chinese medicine. Medicinal L. japonica mainly consists of dried flower buds and partially opened flowers, thus flowers are an important quality indicator. MADS-box genes encode transcription factors that regulate flower development. However, little is known about these genes in L. japonica.
Results
In this study, 48 MADS-box genes were identified in L. japonica, including 20 Type-I genes (8 Mα, 2 Mβ, and 10 Mγ) and 28 Type-II genes (26 MIKCc and 2 MIKC*). The Type-I and Type-II genes differed significantly in gene structure, conserved domains, protein structure, chromosomal distribution, phylogenesis, and expression pattern. Type-I genes had a simpler gene structure, lacked the K domain, had low protein structure conservation, were tandemly distributed on the chromosomes, had more frequent lineage-specific duplications, and were expressed at low levels. In contrast, Type-II genes had a more complex gene structure; contained conserved M, I, K, and C domains; had highly conserved protein structure; and were expressed at high levels throughout the flowering period. Eleven floral homeotic MADS-box genes that are orthologous to the proposed Arabidopsis ABCDE model of floral organ identity determination, were identified in L. japonica. By integrating expression pattern and protein interaction data for these genes, we developed a possible model for floral organ identity determination.
Conclusion
This study genome-widely identified and characterized the MADS-box gene family in L. japonica. Eleven floral homeotic MADS-box genes were identified and a possible model for floral organ identity determination was also developed. This study contributes to our understanding of the MADS-box gene family and its possible involvement in floral organ development in L. japonica.
Similar content being viewed by others


Background
Lonicera japonica Thunb., which belongs to the Caprifoliaceae family, is a perennial and evergreen vine that is widely used in traditional Chinese medicine [1]. It is rich in various active ingredients, including chlorogenic acid, luteolin, triterpenoid saponins, iridoids, and essential oils [2, 3]. Pharmacological studies have shown that L. japonica extract has multiple biological activities, including antioxidant, antiviral, anti-inflammatory, antibacterial, and hepatoprotective activities [1, 4, 5]. Medicinal L. japonica mainly consists of dried flower buds and partially opened flowers. The flowering processes of L. japonica could be divided into seven developmental stages: the young bud, three-green, two-white, great-white, silver, golden, and fade stages; the first four stages belong to the floral bud stage, and the last three stages belong to flower stage. It has been reported that specific floral developmental stages affect the concentrations of active components and volatile compounds. For example, chlorogenic acid (CGA) and luteolin are main compounds to evaluate the quality of medicinal L. japonica; the content of CGA is higher during three-green stage to two-white stage, while the content of luteolin is higher during two-white stage to great-white stage [6]. The relationship between these compounds and floral stages suggests that flowers are an important quality indicator [7]. During development, the flower buds of L. japonica gradually increase in size, reaching a maximum length of ~ 5 cm, and change from green to white. The flower buds then open and turn yellow approximately 2 days later, the budding period is both strictly fixed and not synchronized among individual plants, making it problematic for mass harvest. Most research on L. japonica has focused on the isolation, identification and analysis of its pharmacologically active components, as well as their biosynthetic pathways [8,9,10]. However, the genetic mechanism of flower development in L. japonica remains elusive.
During the plant cycle, flowering is a crucial transition from vegetative to reproductive growth. In the widely used ABCDE model, floral organ development is controlled by five classes of floral homeotic genes, called A, B, C, D, and E, which have distinct expression patterns in floral organs [11,12,13]. The encoded proteins form floral organ-specific tetramers that specify floral organ identity. In Arabidopsis, classes A and E determine first whorl sepal identity; classes A, B, and E determine second whorl petal identity; classes B, C, and E determine third whorl stamen identity; classes C and E determine fourth whorl carpel identity; and classes C, D, and E determine ovule identity within the fourth whorl [14, 15]. A lot of ABCDE model genes have been characterized in Arabidopsis, including AP1 and AP2 (class A); AP3 and PI (class B); AG (class C); STK, SHP1, and 2 (class D); SEP1, 2, 3, and 4, (class E) [16]. Notably, most floral homeotic genes (except for AP2) belong to the MADS-box family, which encodes a class of transcription factors that form tetramers and bind to two adjacent cis-regulatory DNA binding sites called CArG-boxes, thereby regulating floral organ formation [16].
The MADS-box gene family encodes transcription factors that are characterized by the presence of a conserved MADS-box (M) domain in the N-terminal region [17,18,19]. Based on their phylogenetic relationships, plant MADS-box genes can be classified into two type lineages known as Type-I and Type-II. Type-I genes can be further classified into the Mα, Mβ, and Mγ subgroups, and Type-II genes can be further classified into the MIKCc and MIKC* subgroups [20, 21]. The two types of genes encode proteins with distinct conserved domains; each Type-I gene encodes a conserved M domain and a variable C-terminal (C) domain, while each Type-II gene encodes the M and C domains as well as an intervening (I) domain and a keratin-like (K) domain [22]. The functional roles of the two types of MADS-box genes are extremely distinct. Type-I genes are rarely studied, while extensive studies have demonstrated that Type-II genes play essential roles in numerous physiological processes, including flower development [23,24,25]. In addition to the floral homeotic genes in the ABCDE model, several other Type-II MADS-box genes are also involved in the regulation of flower development. In Arabidopsis, several MADS-box genes, such as SOC1 [26], SVP [27], FLC [28], FLM [29], AGL15 [30], AGL18 [30], and AGL24 [31], participate in flower development. In wheat, two MADS-box transcription factor, TaVrt2 and TaVrn1, interact and promote flowering via the vernalization pathway [32]. In soybean, overexpression of a MADS-box gene GmAGL1 can regulate the expression of photoperiodic pathway related genes and promote flowering [33]. In recent years, MADS-box family genes have been identified and characterized in many plants, including Arabidopsis [21], rice [34], grapevine [35, 36], poplar [37], Brassica rapa [38], tomato [39], alfalfa [40], and soybean [41]. These studies provide useful information for understanding the function of MADS-box genes during plant growth and development. However, the role of L. japonica MADS-box genes remains elusive.
To clarify the regulatory mechanism of L. japonica flower development, in the present study, we sought to identify MADS-box family genes. The identified genes were characterized through analyses of phylogenesis, gene structure, conserved domains, protein structure, chromosomal location, and expression. Orthologs of Arabidopsis floral homeotic genes were identified, and their expression profiles in floral organs and interactions were analyzed to propose the mechanism of L. japonica floral organ specification. This study contributes to our understanding of the MADS-box gene family and its possible involvement in floral organ development in L. japonica.
Results
Identification of MADS-box genes in L. japonica
To identify MADS-box genes in L. japonica, HMM and BLAST searches were performed. A comparison of the results obtained using the two methods showed that some candidate genes identified using BLAST were not identified using the HMM method because of partial domain deletions. Therefore, gene-specific primers were designed to amplify these genes for sequence confirmation, and the sequences of 15 genes were revised (GenBank accession numbers: OP903000–OP903014) (File S1). The searches identified 36 MADS-box genes in L. japonica (LjMADS01–LjMADS36 in Table 1). Further, we conducted a HMMER search of the whole genome to identify MADS domain to mitigate the influence of genome annotation on MADS-box gene identification, and identified 12 new MADS-box genes (LjMADS37n–LjMADS48n in Table 1). Finally, 48 MADS-box genes were identified in L. japonica. Sequence analysis indicated that the lengths of the 48 encoded MADS-box proteins varied from 110 to 452 amino acids, and most of them (44/48) were 156–296 amino acids. The theoretical Mw and pI of L. japonica MADS-box proteins were in the range of 12140.29 to 47569.16 Da and 5.11 to 10.24, respectively (Table 1).
Classification and phylogenetic analysis of L. japonica MADS-box genes
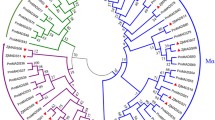
MADS-box genes can be grouped into two types according to their evolutionary relationships [20, 21]. The identified L. japonica MADS-box genes included 20 Type-I genes and 28 Type-II genes (Fig. 1). Based on a phylogenetic analysis of A. thaliana and L. japonica, the Type-I genes could be further classified into the Mα, Mβ, and Mγ subgroups, and most Type-I genes were present in species-specific monophyletic lineages. The numbers of L. japonica MADS-box genes in the Mα, Mβ, and Mγ subgroups were eight, two, and 10, respectively (Fig. 1A). Type-II MADS-box genes can also be classified into the MIKCc and MIKC* subgroups. There were 26 MIKCc and two MIKC* subgroup genes in L. japonica. MIKCc genes can be clustered into 12 evolutionary clades based on the known groups of A. thaliana: Bsister, AP3/PI, ANR1, AGL15/18, SVP, SOC1, AG, AGL12, FLC, AP1/FUL, SEP, and AGL6 [20]. Although L. japonica has fewer MIKCc genes (28) than Arabidopsis (39), it still contained genes in 12 clades (Fig. 1B and Fig. S1).

Phylogenetic analysis of Type I (A) and Type II (B) MADS-box genes in A. thaliana and L. japonica. MADS-box genes in A. thaliana and L. japonica are highlighted with blue and red squares, respectively
For the comparative genomic analysis of MADS-box genes, 22 plant species from the major evolutionary groups were selected, and the numbers of MADS-box genes were compared. As shown in Fig. 2, the number of MADS-box genes (48 genes) in L. japonica is quite small when compared to other plants, which merely more than that of E. breviscapus among the 13 dicotyledonous plants analyzed. Similar patterns were observed for the Type-I and -II genes; the number of Type-I genes was the lowest among the 13 dicotyledonous plants, and the number of Type-II genes was only greater than that of E. breviscapus and I. batatas (Fig. 2).

The evolutionary relationships of 22 plant species and the number detail of the MADS-box gene family of each species. The green and orange circles represent whole genome duplication and triplication during the evolution, respectively. The pink circle represents the species studied in this work
The structure and conserved domains of L. japonica MADS-box genes
The gene structure of the L. japonica MADS-box genes was analyzed by comparing the coding and genomic sequences. The results indicated that the exon-intron structures of the Type-I and Type-II MADS-box genes were extremely distinct (Table 1; Fig. 3). Most Type-I genes (18/20) had no introns, and the remaining two genes (LjMADS01 and LjMADS02) had only one intron (Fig. 3). All Type-II genes contained between six and ten introns (Fig. 2). The length of the first exon of the Type-II genes, which encodes the DNA-binding M domain, was well conserved (182–188 bp, except for LjMADS21). The conserved domains of L. japonica MADS-box proteins were also predicted, and the results revealed that the conserved domains of Type-I and Type-II MADS-box proteins were very different. Type-I proteins contained the M and C domains, while Type-II proteins contained the M, I, K, and C domains (Fig. 3). However, recent studies have indicated that Type-I proteins contain an I-like domain [42] and we also identified an I-like domain in the Type-I MADS-box proteins of L. japonica (Fig. 3 and Fig. S2).

Phylogenetic analysis, subfamily classification, conserved domains and gene structure of MADS-box genes in L. japonica. For conserved domains, the M domain, I domain, I-like domain, K domain, and C domain are represented in red, yellow, orange, light green, and green, respectively. For gene structure, the exons and introns are represented by black rectangles and black lines, respectively
Structural prediction of L. japonica MADS-box proteins
To study the structure of the proteins encoded by the L. japonica MADS-box genes, the secondary and three-dimensional structures were predicted using the NetSurfP-3.0 tool and AlphaFold2, respectively. The results showed that the secondary structure of the M domain in all MADS-box proteins was similar, and each included one α-helix and two β-strands. In addition, the secondary structures of the Type-I proteins (LjMADS01–LjMADS19 & LjMADS37n) were varied, whereas the Type-II proteins (LjMADS20–LjMADS36 & LjMADS38n–LjMADS48n) were more conserved, especially the MIKCc group proteins. Using LjMADS28 as example, the protein contained the conserved M domain, the I domain of this MIKCc group protein contained one helix, the K domain contained two helices, and the C domain consisted of random coils (Fig. 4A and Fig. S3). The three-dimensional structural predictions were similar to those for the secondary structure; the structural conservation of the Type-I proteins was low, whereas the Type-II proteins, particularly the MIKCc group proteins, had high structural conservation (Fig. 4B and Fig. S4). In addition, an alignment of the three-dimensional structure of LjMADS28 and the DNA-binding domain or keratin-like domain of Arabidopsis SEP3 showed high similarity (Fig. 4C and D).

Protein structure prediction of LjMADS28. A Secondary structure and conserved domains prediction of LjMADS28. From top to bottom are the amino acid sequence, relative surface accessibility, secondary structure, disorder, scale bar, and conserved domains. For secondary structure, the orange helical lines, purple arrows and purple lines indicate helices, strands and coils, respectively. B Three-dimensional structure prediction of LjMADS28 and its correspondence with conserved domains. C Alignment of three-dimensional structure of LjMADS28 and DNA-binding domain of Arabidopsis SEPALLATA 3 (PDB accession number: 7NB0). D Alignment of three-dimensional structure of LjMADS28 and keratin-like domain of Arabidopsis SEPALLATA 3 (PDB accession number: 4OX0)
Chromosomal localization and gene duplications of L. japonica MADS-box genes
The chromosomal distribution of the MADS-box genes in L. japonica was obtained from the genome annotation. Most of the genes (47/48) were mapped to the nine chromosomes of L. japonica except for LjMADS32 (Fig. 5). The distribution of MADS-box genes on the chromosomes was uneven; chromosomes 1 and 9 carried the largest number of genes, with ten genes each, whereas chromosomes 3 and 8 carried only two genes each. Gene duplication analysis detected two groups of tandemly duplicated MADS-box genes in L. japonica. The first group contained LjMADS08 and LjMADS09, and the second group contained LjMADS11, LjMADS12, and LjMADS13, and both groups were on chromosome 1 (Fig. 5). No segmental duplication events were found.

Chromosomal localization of L. japonica MADS-box genes. Genes of the Mα, Mβ and Mγ, MIKCc, and MIKC* groups are shown on blue, purple, red, orange, and green backgrounds, respectively. The vertical coordinate represents the length of the chromosomes. Tandem duplicated genes are indicated by red lines
Expression profiles of L. japonica MADS-box genes in different tissues and flowers at different developmental stages
To clarify the tissue expression profiles of MADS-box genes in L. japonica, RNA-seq data from nine tissues, including youngest leaves, second leaves, mature leaves, shoot apices, stems, green floral buds, white floral buds, white flowers, and yellow flowers, were downloaded from NCBI, and TPM values were calculated to evaluate the expression levels. Hierarchical clustering results showed that the expression profiles of the Type-I and Type-II MADS-box genes in the nine tissues differed (Fig. 6A). Of the 20 Type-I genes, only LjMADS02, LjMADS05, LjMADS15, LjMADS19 and LjMADS37n were expressed in some tissues; the other 15 genes were either not expressed or expressed at very low levels (Fig. 6A). In contrast to the Type-I MADS-box genes, Type-II genes had relatively higher expressions. Of the 28 Type-II genes, only one, LjMADS33, was not expressed; the other 27 genes were expressed at high levels in multiple tissues (Fig. 6A). Interestingly, the clustering results of the nine tissues showed that the genes expressed in the four flower-related tissues were clustered into one clade, and most Type-II genes had relatively higher expressions in flower buds and flowers than in other tissues (Fig. 6A).
Considering the important role of the MADS-box gene family in flower development, the expression profiles of the L. japonica MADS-box genes were also analyzed in flowers at seven developmental stages. TPM values were calculated using RNA-seq data from flowers at seven developmental stages: the young bud (S1), three-green (S2), two-white (S3), great-white (S4), silver (S5), golden (S6), and fade (S7) stages. The results indicated that most Type-I genes were not expressed throughout the flowering period, except for LjMADS05, LjMADS15 and LjMADS37n (Fig. 6B). In contrast, most Type-II genes were expressed at high levels throughout the flowering period, except for LjMADS33 and LjMADS38n (Fig. 6B). The expression profiles of MADS-box genes suggested that the Type-II genes might be involved in flower development in L. japonica.

Expression profiling of L. japonica MADS-box genes based on RNA-seq data. A Expression profiling of MADS-box genes in nine different tissues. YL-Youngest leaf, SL-Second leaf, ML-Mature leaf, SA-Shoot apex, ST-Stem, GB-Green floral bud, WB-White floral bud, WF-White flower, YF-Yellow flower. B Expression profiling of MADS-box genes in flowers at seven different developmental stages. S1-Young bud stage, S2-Three-green stage, S3-Two-white stage, S4-Great-white stage, S5-Silver stage, S6-Golden stage, S7-Fade stage. MADS-box genes of Type I and Type II are shown on purple and blue background, respectively
Expression pattern analysis of the floral homeotic MADS-box genes in different floral organs
To further clarify the role of MADS-box genes in L. japonica flower development, the Arabidopsis ABCDE model gene orthologs were identified, and their expression profiles in different floral organs at different developmental stages were analyzed using qRT-PCR. Two reference genes were employed to conduct the qRT-PCR, respectively; both of the results show the similar tendendy so the results of qPCR (using LjGAPDH as an internal control) were put in Additional file (Fig. S5). Eleven orthologous floral homeotic genes were identified in L. japonica, including two class A genes (LjMADS26 and LjMADS27, AP1 orthologs), three class B genes (LjMADS24 and LjMADS25, AP3 orthologs; LjMADS40n, PI ortholog), two class C genes (LjMADS21 and LjMADS22, AG orthologs), one class D gene (LjMADS46n, STK ortholog), and two class E genes (LjMADS28, LjMADS30, and LjMADS42n, SEP1, SEP2, and SEP3 orthologs, respectively). The qRT-PCR results showed two interesting features: genes of different classes had different expression profiles in different flower organs, and genes of the same class had similar expression patterns. As shown in Fig. 7 and Fig. S5, the class A genes LjMADS26 and LjMADS27 were highly expressed in calyxes, and LjMADS27 was also expressed at lower levels in petals; however, neither gene was expressed in stamens and pistils. The three class B genes, LjMADS24, LjMADS25 and LjMADS40n, were expressed mainly in petals and stamens but were almost undetectable in calyxes and pistils. The two class C genes, LjMADS21 and LjMADS22, were mainly expressed in calyxes, stamens, and pistils, but not in petals, and both genes were downregulated in stamens during flower development. The class D gene LjMADS46n was expressed in calyxes at higher level and barely expressed in other organs. The two genes of class E, LjMADS28 and LjMADS30, were consistently expressed throughout development in calyxes, but only at certain stages in petals and stamens. For example, LjMADS28 was expressed in petals at the great-white stage (S4) and in stamens at late flowering stages (S4-S6), whereas LjMADS30 was expressed in petals at the three-green stage (S2). Differs from LjMADS28 and LjMADS30, LjMADS42n was expressed in all four organs and its expression level remained relatively stable from the bud stage to the flowering stage.

Expression pattern analysis of the floral homeotic MADS-box genes in different floral organs using qRT-PCR. A-D calyxes (A), petals (B), stamens (C) and pistils (D) of L. japonica at six different developmental stages, respectively. E qRT-PCR results of the floral homeotic MADS-box genes in different floral organs at six different developmental stages. The vertical coordinates represent the relative expression levels of MADS-box genes. The length of scale represents 1 cm
Analysis of the interactions between the floral homeotic MADS-box proteins using Y2H
Previous studies have demonstrated that different tetramers of MIKC-type MADS-box transcription factors play crucial roles in regulating floral organ identity [16]. Therefore, analysis of protein interactions could provide important information for elucidating the molecular mechanism of floral organ identity. In this study, the interactions between pairs of floral homeotic MADS-box proteins in L. japonica were analyzed using Y2H. As shown in Fig. 8 and Fig. S6, homo- and heterodimers were formed between several floral homeotic MADS-box proteins of L. japonica. Five of the eleven MADS-box proteins, LjMADS26, LjMADS27, LjMADS30, LjMADS40n and LjMADS42n, formed homodimers through reciprocal interactions in yeast. Complex heterodimeric interactions were also observed between several pairs of MADS-box proteins. For example, LjMADS26 could interact with LjMADS27, LjMADS40n, LjMADS21, LjMADS46n, and LjMADS42n respectively to form heterodimers and the interactions were confirmed with reciprocal transformations. Meanwhile, LjMADS26 interacted only with LjMADS28 and LjMADS30 as BD vectors and not as AD vectors; but it interacted only with LjMADS22 as AD vectors and not as BD vectors. Based on the interaction results of proteins from the different classes, several members of the same class showed similar interaction patterns. For example, LjMADS26 and LjMADS27 (class A) formed homo- or heterodimers with multiple MADS-box proteins and exhibited similar interaction patterns, although there were some differences. LjMADS24 and LjMADS25 (class B) did not interact with other proteins when expressed from BD vectors. LjMADS21 and LjMADS22 (class C) formed heterodimers with MADS-box proteins of classes A and E, and similar results were also observed for LjMADS46n (class D). These results suggested that the floral homeotic MADS-box proteins in L. japonica formed homo- and heterodimers, and different dimers were formed by the different classes. However, the interaction pattern of LjMADS40n (class B) is inconsistent with that of the other two class B proteins, possibly because they belong to two different orthologs in class B (LjMADS24 and LjMADS25 are AP3 orthologs and LjMADS40n is PI orthologs).

Analysis of interaction between the floral homeotic MADS-box proteins using Y2H. The co-transformed yeast cells grown on SD/-Trp/-Leu (A) or SD/-Ade/-His/-Leu/-Trp (B) medium. Proteins of class A, B, C, D and E are represented in light steel blue, light blue, light green, light yellow and light orange, respectively
Characterization of the floral organ identity model in L. japonica
Integrating the expression pattern and protein interaction data for the floral homeotic MADS-box genes in L. japonica, a possible model for floral organ identity determination was developed (Fig. 9). In this model, classes A (LjMADS26 and LjMADS27), C (LjMADS21 and LjMADS22), D (LjMADS46n), and E (LjMADS28, LjMADS30 and LjMADS42n) proteins determined first whorl calyx identity; classes A (LjMADS27), B (LjMADS24, LjMADS25 and LjMADS40n), and E (LjMADS28, LjMADS30 and LjMADS42n) proteins determined second whorl petal identity; classes B (LjMADS24, LjMADS25 and LjMADS40n), C (LjMADS21 and LjMADS22), and E (LjMADS28 and LjMADS42n) proteins determined third whorl stamen identity; and classes C (LjMADS21 and LjMADS22) and E (LjMADS28, LjMADS30 and LjMADS42n) proteins determined fourth whorl pistil identity.

The predicted floral quartet model and the underlying ABCDE model of organ identity determination in L. japonica. Genes of class A, B, C, D and E are represented in light steel blue, dark blue, light green, orange and purple, respectively
Discussion
Prediction of coding genes is an important part of genome annotation. Although the accuracy of gene prediction has gradually improved as prediction methods have continuously advanced, some errors still occur, especially for multi-exon genes [43, 44]. In the present study, we identified 48 MADS-box genes in the L. japonica genome, including 20 Type-I and 28 Type-II genes. Interestingly, the gene structures of the two types were extremely distinct; Type-I genes have no intron or only one intron, and Type-II genes contain multiple introns. In addition, some exons of Type-II genes are short, which also increased the difficulty of gene prediction. During MADS-box gene identification in L. japonica, we found that some Type-II genes had sequence deletions, so we cloned these candidate genes and found that 15 of the 28 Type-II genes had errors in their predicted sequences. These errors were mainly due to exon deletions. Therefore, we speculated that the complex multi-exon structure was the main reason for the low prediction accuracy of Type-II genes in L. japonica. After sequencing correction, accurate MADS-box gene sequences were obtained.
The structure of a protein is closely related to its function; therefore, determining the structure of a protein is useful for studying its function and mechanism of action. Traditional protein structure elucidation relies mainly on experiments, such as X-ray crystallography and nuclear magnetic resonance [45, 46]. AlphaFold is a novel protein structure prediction method based on machine learning that is known for its high prediction accuracy [47, 48]. In this study, we used AlphaFold to predict the structure of L. japonica MADS-box proteins, and the results indicated that the structures of the Type-I proteins were quite varied, while the Type-II proteins were more conserved, especially the MIKCc group proteins. To date, no complete MADS-box protein structure has been reported for plants. Only partial structures of the Arabidopsis MADS-box protein SEP3 have been solved using X-ray crystallography, which includes the DNA-binding domain (M and I domains) [42] and the K domain [49]. The alignment showed that the partial structure of SEP3 obtained by using X-ray crystallography was very similar to the structure of LjMADS28 (ortholog to Arabidopsis SEP2) predicted by AlphaFold in the present study. This result indicates that AlphaFold has high accuracy for predicting protein structure and provides new ideas for studying protein structure and function.
Although the Type-I and -II MADS-box genes arose from a single gene duplication before the divergence of plants and animals, they different in their phylogenesis, gene structure, conserved domains, protein structure, chromosomal distribution, and expression profiles [17]. In contrast to the intensively studied Type-II genes, the functions of many Type-I MADS-box genes remain uncharacterized. In this study, most Type-I genes were clustered into species-specific clades, suggesting that they arose through tandem duplication after the divergence of the Arabidopsis and L. japonica lineages. In contrast, Type-II genes appear to have the most recent common ancestors of Arabidopsis and L. japonica. Similar results were found in other plants, such as Arabidopsis [21], peach [50], and physic nut [51], suggesting that these two types of genes have significantly different evolutionary patterns. Previous studies showed that Type-I MADS-box transcription factors lack the I and K domains found in Type-II proteins [52, 53]. However, recent studies have indicated that Type-I proteins contain an I-like domain that is involved in DNA binding [42]. Here, we also discovered an I-like domain in the Type-I MADS-box proteins of L. japonica, and structure prediction indicated that the I-like sequences formed a short α-helix. Although few functional researches on Type-I genes have been reported, studies have shown that they are mainly expressed at extremely low levels in a tissue-specific pattern and may play important roles in female gametophyte, embryo, and endosperm development [54,55,56]. In this study, expression analysis based on RNA-seq data showed that most of the Type-I genes were expressed at undetectable levels. Some genes showed tissue-specific expression, such as LjMADS19, which was only expressed in flower buds, and LjMADS02 and LjMADS05, which were expressed in flower buds and flowers but not in other tissues. This flower bud- and flower-specific expression also suggests that some Type-I MADS-box genes are involved in the flower-related development in L. japonica.
MADS-box gene family is widely distributed throughout the eukaryotic section of the tree of life. The development of whole genome sequencing has promoted in-depth studies of this family. The number of MADS-box genes is varied in different plants, for example, A. thaliana, Gossypium hirsutum and blueberry (Vaccinium spp.) contain 105, 207 and 249 MADS-box genes, respectively [57, 58]. In this study, 48 MADS-box genes were identified in L. japonica. The gene number in L. japonica is relatively small compared with other plants, which may be due to the absence of recent genome duplication events during evolution. In addition, the type composition of MADS-box genes is also varied in different plants. For example, E. breviscapus also has a relatively small gene number of 44, but most of which belong to Type-I (38/44), and the expression of Type-I genes are relatively active than that of L. japonica [59]. In Erycina pusilla, there is only one Type-I gene [60]. Besides, no Type-I genes have been reported in gymnosperm Gnetum genmon [61]. The large differences in the number of these genes suggest that the MADS-box gene family has diverged in function during evolution.
Phylogenetic tree analysis showed that 25 of the 26 MIKCc genes of L. japonica could be subgrouped into 12 clades based on the known groups of A. thaliana, and similar results have been reported in many other plants. However, there is one MIKCc gene, LjMADS35, that cannot be classified into one of the clades. Similar genes have been reported in other plants, such as TM8, a MADS-box gene that seems be related to the correct differentiation of the tomato reproductive structures, cannot be classified into one of the 12 clades of A. thaliana [62]. These results suggest that the 12 evolutionary clades of Arabidopsis are not sufficient to contain all MIKCc genes, and that new clades will be identified as more MADS-box genes are identified.
In plants, the ABCDE model has been proposed to explain, to some extent, the molecular determination of floral organ identity. According to this model, a hierarchical combination of the five classes of floral homeotic genes regulates the identity of different floral organs [11,12,13]. However, the genes contained in the ABCDE model vary in different plants. In Arabidopsis, class D include STK, SHP1, and SHP2 [21]. However, no orthologs of SHP1 and SHP2 were identified in L. japonica. In rice [34] and Phyllostachys edulis [63], no SHP orthologs were identified, but in apple [64], peach [65] and tomato [39], at least one SHP gene were identified. Although the ABCDE model has a certain degree of conservation, it varies among plants. For example, in Arabidopsis, first whorl sepal identity is determined by classes A + E, second whorl petal identity is determined by classes A + B + E, third whorl stamen identity is determined by classes B + C + E, and fourth whorl carpel identity is determined by classes C + E [16]. However, in barley, first whorl paleae identity is determined by classes A + E, second whorl lodicule identity is determined by classes A + B + C + E, third whorl stamen identity is determined by classes A + B + C + E, and fourth whorl carpel identity is determined by classes A + C + D + E [66]. Here, the identity of the first whorl calyx identity was determined by classes A + C + D + E, second whorl petal identity was determined by classes A + B + D + E, third whorl stamen identity was determined by classes D + E, and fourth whorl pistil identity was determined by classes C + D + E. This variation may be due to differences in floral organs that developed during evolution [16, 67]. Possible regulatory models have been proposed based on the tissue-specific expression patterns of floral homeotic genes in different floral organs in some plants, such as grapevine [68], barley [66], and Phalaenopsis Aphrodite [69]. Because floral homeotic proteins function as tetramers, we considered both their expression patterns and protein interactions, so that the inferred model would be more reliable. However, due to huge differences between different plants and the complexity of the regulatory model, the ABCDE model of floral organ identity requires further refinement. In conclusion, this study contributes to our understanding of the MADS-box gene family in L. japonica, and the prediction of ABCDE model in L. japonica is helpful to explore the molecular mechanisms of flowering regulation. Also, our studies offering an opening for study on MADS-box gene family in Caprifoliaceae, which might be the new basis for further research in this family.
Methods
Plant materials
L. japonica used for plant material collection was planted in the Germplasm Nursery at Nanjing Botanical Garden Mem. Sun Yat-Sen, Nanjing, China. Flower buds and flowers collected at six different stages as described previously [70] were dissected and divided into four floral organs: calyxes, petals, stamens, and pistils. Plant materials collection and dissection were completed as quickly as possible, and the tissues were frozen in liquid nitrogen and stored at − 80 °C until use.
Data collection
L. japonica genome and protein sequences were obtained from the National Genomics Data Center (http://bigd.big.ac.cn/gwh/) with a BioProject ID of PRJCA001719 [71]. A. thaliana MADS-box protein sequences were retrieved from TAIR (https://www.arabidopsis.org/). MADS-box gene information of Selaginella moellendorffii [72], Gnetum gnemon [61], Pinus tabulaeformis [73], Cycas panzhihuaensis [74], Oryza sativa [34], Phyllostachys edulis [63], Erycina pusilla [60], Bletilla striata [75], Ananas comosus [76], Ipomoea batatas [77], Cucumis sativus [78], Solanum lycopersicum [39], Solanum tuberosum [79], Lactuca sativa [80], Erigeron breviscapus [59], Gossypium hirsutum [57], Malus pumila [64], Pyrus bretschneideri [65], Medicago sativa [40], Glycine max [41], and Humulus lupulus [81] were retrieved from previous studies.
Identification and cloning of MADS-box genes inL. japonica.
Hidden Markov model (HMM) and BLAST methods were carried out to identify MADS-box genes in L. japonica. For the HMM method, the MADS-box SFR family domain (PF00319) was downloaded from Pfam (http://www.pfam.org/) and then searched against the L. japonica protein database using HMMER3 (v3.3.2) [82]. For the BLAST method, A. thaliana MADS-box sequences were used as BLASTP queries against the L. japonica protein database. In order to mitigate the influence of genome annotation on MADS gene identification, we further conducted a HMMER search of the whole genome to identify MADS domain. Then, the identified regions were extracted and then imported to Genscan for gene prediction [83]. After prediction, previous transcriptome data was used to confirm the accuracy of results.
After comparing the results of the two methods, we found that several candidate sequences were missing domains. Therefore, we designed gene-specific primers to amplify these genes. The sequences of the primers used to amplify the candidate genes are showed in Supplementary Table 1. The amplified PCR products were extracted and cloned into the pCE2 TA/Blunt-Zero Vector (Vazyme, Nanjing, China). Positive clones were screened and sequenced (Sangon Biotech, Shanghai, China).
Phylogenetic analysis of MADS-box sequences
The full-length MADS-box protein sequences of the A. thaliana and L. japonica were aligned using the G-INS-I method in MAFFT (v7.505) [84]. The aligned sequence file was then imported to IQ-TREE (v1.6.12) [85] to construct the maximum likelihood tree. Furthermore, the newick file was uploaded to iTOL (https://itol.embl.de/) [86] to modify the phylogenetic tree.
Characterization of L. japonica MADS-box genes
The theoretical molecular weight (Mw) and isoelectric point (pI) of the L. japonica MADS-box proteins were calculated using the ProtParam tool (https://web.expasy.org/protparam/). Conserved domains were predicted using SMART (http://smart.embl-heidelberg.de/) [87]. The gene structure was identified by comparing the coding sequence to the genomic sequence, and then illustrated using Exon-Intron Graphic Maker (http://wormweb.org/exonintron). Secondary structure was predicted using the NetSurfP-3.0 tool (https://services.healthtech.dtu.dk/service.php?NetSurfP-3.0) [88] and modified using Adobe Illustrator 2020. Three-dimensional structures were predicted using the AlphaFold2 (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb) [89]. PyMOL 2.5 software (https://pymol.org/2/) was used to view the PDB file and align the three-dimensional structures. The three-dimensional structures of the DNA-binding domain and keratin-like domain of Arabidopsis SEP3 were retrieved from the RCSB PDB database (https://www.rcsb.org/, accession numbers 7NB0 and 4OX0, respectively).
Chromosomal distribution and gene duplication
The chromosomal locations of the MADS-box genes in L. japonica were obtained from the genome annotation and were illustrated using TBtools (v1.098661) [90]. To identify gene duplication events in L. japonica MADS-box genes, the sequence similarity matrix was analyzed using BioEdit (v7.0.9.0), and tandemly and segmentally duplicated genes were identified based on sequence similarity and chromosomal distribution.
Expression analysis based on RNA-seq data
To obtain gene expression data for L. japonica, RNA-seq data from nine tissues [9] as well as flowers at seven different developmental stages [91] were obtained from the Sequence Read Archive (SRA) of NCBI (https://www.ncbi.nlm.nih.gov/). The SRA accession numbers are listed in Table S2. SRA data were converted to fastq format using the SRA Toolkit (v2.11.0). FastQC (v0.11.9) was used to assess the quality of the sequencing data, and Trimmomatic (v0.39) [92] was used to filter the raw data. After filtering low-quality data, clean reads were aligned to L. japonica reference genes using Kallisto (v0.46.1) [93] to calculate transcripts per million (TPM). To obtain more accurate expression levels for MADS-box genes, the corresponding sequences in the original reference data were replaced with the corrected sequences. A heatmap was then generated with TBtools (v1.098661) [90] using the relative expression values. The expression levels of seven developmental stages were the average of three biological replications.
RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA from different floral organs of L. japonica was extracted using the FlaPure Plant RNA Extraction Kit (Genesand, Beijing, China). The quality and concentration of the total RNA were examined using a NanoDrop 2000 spectrophotometer (Thermo Scientific, MA, USA). Reverse transcription was conducted using 1 µg of total RNA and the UnionScript First-strand cDNA Synthesis Kit (with dsDNase) (Genesand). qRT-PCR was conducted using GS AntiQ qPCR SYBR Green Master Mix (Genesand) according to the manufacturer’s instructions and the ABI QuantStudio™ 6 Flex System (Applied Biosystems, CA, USA). The PCR cycling were carried out with 95 °C for 1 min, followed by 40 cycles of 95 °C for 20 s and 60 °C for 30 s. LjActin and LjGAPDH were employed as internal controls to calculate relative transcriptional levels using the 2−ΔCt method, respectively [94]. The sequences of the primers used for qRT-PCR are listed in Table S3. All qRT-PCRs were performed with three biological replications. IBM SPSS Statistics 26 was used for statistical analyses. GraphPad Prism (v9.1.0.221) and Adode Illustrator 2020 were used to illustrate the figures.
Yeast two-hybrid assay
A yeast two-hybrid (Y2H) assay was conducted to analyze the interactions between the L. japonica MADS-box proteins. The coding sequences were cloned into the AD and BD fusion vectors pGADT7 and pGBKT7, respectively. The primer sequences used to construct the recombinant vectors are listed in Table S4. Self-activation verification assay was conducted before the Y2H assay. The coding sequences were cloned into the BD vectors and the recombinant vectors were transformed into Saccharomyces cerevisiae AH109 competent cells (Weidi, Shanghai, China), and the transformed cells were cultured on SD/-Trp media for 24 h at 28 ℃ and then transferred to SD/-Trp/-Ade/-His media for 3–4 days at 28 ℃ to detect self-activation. Then, the AD and BD vectors were co-transformed into the yeast competent cells and cultured on SD/-Trp/-Leu medium. Subsequently, the yeast cells were screened on SD/-Trp/-Leu or SD/-Ade/-His/-Leu/-Trp medium to identify interactions between MADS-box proteins.
Data availability
The datasets generated in this study have been deposited in the NCBI GenBank with accession numbers of OP903000 - OP903014 (File S2). L. japonica genome and protein sequences were obtained from the National Genomics Data Center with a BioProject ID of PRJCA001719 (https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA001719). A. thaliana MADS-box protein sequences were retrieved from TAIR (https://www.arabidopsis.org/browse/genefamily/mads_tffamily.jsp). RNA-seq data from nine tissues and flowers at seven different developmental stages were obtained from the Sequence Read Archive (SRA) of NCBI. The SRA accession numbers are listed in Table S2. The three-dimensional structures of the DNA-binding domain and keratin-like domain of Arabidopsis SEP3 were retrieved from the RCSB PDB database (https://www.rcsb.org/, accession numbers 7NB0 and 4OX0, respectively).
Abbreviations
- AP1:
-
APETALA1
- AP2:
-
APETALA2
- AP3:
-
APETALA3
- PI:
-
PISTILLATA
- AG:
-
AGAMOUS
- STK:
-
SEEDSTICK
- SHP1:
-
SHATTERPROOF 1
- SHP2:
-
SHATTERPROOF 2
- SEP2:
-
SEPALLATA2
- SEP3:
-
SEPALLATA3
- SEP4:
-
SEPALLATA4
- SOC1:
-
SUPPRESSOR OF OVEREXPRESSION OF CO 1
- SVP:
-
SHORT VEGETATIVE PHASE
- FLC:
-
FLOWERING LOCUS C
- FLM:
-
FLOWERING LOCUS M
- AGL6:
-
AGAMOUS-LIKE 6
- AGL12:
-
AGAMOUS-LIKE 12
- AGL15:
-
AGAMOUS-LIKE 15
- AGL18:
-
AGAMOUS-LIKE 18
- AGL24:
-
AGAMOUS-LIKE 24
- TT16:
-
TRANSPARENT TESTA16
- ANR1:
-
ARABIDOPSIS NITRATE REGULATED 1
- FUL:
-
FRUITFULL
- M domain/M:
-
MADS-box domain
- I domain/I:
-
Intervening domain
- I-like domain:
-
Intervening-like domain
- K domain/K:
-
Keratin-like domain
- C domain/C:
-
C-terminal domain
- HMM:
-
Hidden Markov model
- Mw:
-
Molecular weight
- pI:
-
Isoelectric point
- SMART:
-
Simple Modular Architecture Research Tool
- PDB:
-
Protein Data Bank
- NCBI:
-
National Genomics Data Center
- SRA:
-
Sequence Read Archive
- qRT-PCR:
-
Quantitative real-time PCR
- Y2H:
-
Yeast two-hybrid
- TPM:
-
transcripts per million
- bp:
-
Base pair
- Da:
-
Dalton
- Chr:
-
Chromosome
References
Shang X, Pan H, Li M, Miao X, Ding H. Lonicera japonica Thunb.: ethnopharmacology, phytochemistry and pharmacology of an important traditional chinese medicine. J Ethnopharmacol. 2011;138(1):1–21.
Li RJ, Kuang XP, Wang WJ, Wan CP, Li WX. Comparison of chemical constitution and bioactivity among different parts of Lonicera japonica Thunb. J Sci Food Agric. 2020;100(2):614–22.
Yang R, Min K, Wang Y, Chen S, Ma M, Li M, et al. Rapid semi-quantitative analysis of hemolytic triterpenoid saponins in Lonicerae Flos crude drugs and preparations by paper spray mass spectrometry. Talanta. 2022;239:123148.
Yoo HJ, Kang HJ, Song YS, Park EH, Lim CJ. Anti-angiogenic, antinociceptive and anti-inflammatory activities of Lonicera japonica extract. J Pharm Pharmacol. 2008;60(6):779–86.
Tang X, Liu X, Zhong J, Fang R. Potential application of Lonicera japonica extracts in animal production: from the perspective of intestinal health. Front Microbiol. 2021;12:719877.
Li X, Li Q, Zhang F, Zhang Y, Lin Y, Zhang D. L jiang. Study on harvest time and origin processing method of Lonicerae Japonicae Flos. Lishizhen Medicine and Materia Medica. 2020;31(01):97–99. (in Chinese).
Wang X, Wang F, Wang Z, Wang X, Shi S, Shen M, et al. The Pharmacopoeia of the people’s Republic of China. Part. 2020;1:230.
Li Y, Kong D, Bai M, He H, Wang H, Wu H. Correlation of the temporal and spatial expression patterns of HQT with the biosynthesis and accumulation of chlorogenic acid in Lonicera japonica flowers. Hortic Res. 2019;6:73.
Rai A, Kamochi H, Suzuki H, Nakamura M, Takahashi H, Hatada T, et al. De novo transcriptome assembly and characterization of nine tissues of Lonicera japonica to identify potential candidate genes involved in chlorogenic acid, luteolosides, and secoiridoid biosynthesis pathways. J Nat Med. 2017;71(1):1–15.
Yuan Y, Song L, Li M, Liu G, Chu Y, Ma L, et al. Genetic variation and metabolic pathway intricacy govern the active compound content and quality of the chinese medicinal plant Lonicera japonica thunb. BMC Genomics. 2012;13:195.
Ma H, dePamphilis C. The ABCs of floral evolution. Cell. 2000;101(1):5–8.
Theissen G, Saedler H. Plant biology. Floral quartets. Nature. 2001;409(6819):469–71.
Coen ES, Meyerowitz EM. The war of the whorls: genetic interactions controlling flower development. Nature. 1991;353(6339):31–7.
Honma T, Goto K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature. 2001;409(6819):525–29.
Theissen G. Development of floral organ identity: stories from the MADS house. Curr Opin Plant Biol. 2001;4(1):75–85.
Theißen G, Melzer R, Rümpler F. MADS-domain transcription factors and the floral quartet model of flower development: linking plant development and evolution. Development. 2016;143(18):3259–71.
Alvarez-Buylla ER, Pelaz S, Liljegren SJ, Gold SE, Burgeff C, Ditta GS, et al. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc Natl Acad Sci U S A. 2000;97(10):5328–33.
De Bodt S, Raes J, Van de Peer Y, Theissen G. And then there were many: MADS goes genomic. Trends Plant Sci. 2003;8(10):475–83.
Gramzow L, Theissen G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010;11(6):214.
Henschel K, Kofuji R, Hasebe M, Saedler H, Münster T, Theissen G. Two ancient classes of MIKC-type MADS-box genes are present in the moss Physcomitrella patens. Mol Biol Evol. 2002;19(6):801–14.
Parenicová L, de Folter S, Kieffer M, Horner DS, Favalli C, Busscher J, et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell. 2003;15(7):1538–51.
Lai X, Daher H, Galien A, Hugouvieux V, Zubieta C. Structural basis for plant MADS transcription factor oligomerization. Comput Struct Biotechnol J. 2019;17:946–53.
Heijmans K, Morel P, Vandenbussche M. MADS-box genes and floral development: the dark side. J Exp Bot. 2012;63(15):5397–404.
Ng M, Yanofsky MF. Function and evolution of the plant MADS-box gene family. Nat Rev Genet. 2001;2(3):186–95.
Callens C, Tucker MR, Zhang D, Wilson ZA. Dissecting the role of MADS-box genes in monocot floral development and diversity. J Exp Bot. 2018;69(10):2435–59.
Lee J, Lee I. Regulation and function of SOC1, a flowering pathway integrator. J Exp Bot. 2010;61(9):2247–54.
Lee JH, Yoo SJ, Park SH, Hwang I, Lee JS, Ahn JH. Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes Dev. 2007;21(4):397–402.
Sharma N, Geuten K, Giri BS, Varma A. The molecular mechanism of vernalization in Arabidopsis and cereals: role of flowering locus C and its homologs. Physiol Plant. 2020;170(3):373–83.
Jin S, Kim SY, Susila H, Nasim Z, Youn G, Ahn JH. FLOWERING LOCUS M isoforms differentially affect the subcellular localization and stability of SHORT VEGETATIVE PHASE to regulate temperature-responsive flowering in Arabidopsis. Mol Plant. 2022;15(11):1696–709.
Adamczyk BJ, Lehti-Shiu MD, Fernandez DE. The MADS domain factors AGL15 and AGL18 act redundantly as repressors of the floral transition in Arabidopsis. Plant J. 2007;50(6):1007–19.
Michaels SD, Ditta G, Gustafson-Brown C, Pelaz S, Yanofsky M, Amasino RM. AGL24 acts as a promoter of flowering in Arabidopsis and is positively regulated by vernalization. Plant J. 2003;33(5):867–74.
Xie L, Zhang Y, Wang K, Luo X, Xu D, Tian X, et al. TaVrt2, an SVP-like gene, cooperates with TaVrn1 to regulate vernalization-induced flowering in wheat. New Phytol. 2021;231(2):834–48.
Zeng X, Liu H, Du H, Wang S, Yang W, Chi Y, et al. Soybean MADS-box gene GmAGL1 promotes flowering via the photoperiod pathway. BMC Genomics. 2018;19(1):51.
Arora R, Agarwal P, Ray S, Singh AK, Singh VP, Tyagi AK, et al. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics. 2007;8:242.
Grimplet J, Martínez-Zapater JM, Carmona MJ. Structural and functional annotation of the MADS-box transcription factor family in grapevine. BMC Genomics. 2016;17:80.
Díaz-Riquelme J, Lijavetzky D, Martínez-Zapater JM, Carmona MJ. Genome-wide analysis of MIKCC-type MADS box genes in grapevine. Plant Physiol. 2009;149(1):354–69.
Leseberg CH, Li A, Kang H, Duvall M, Mao L. Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene. 2006;378:84–94.
Saha G, Park JI, Jung HJ, Ahmed NU, Kayum MA, Chung MY, et al. Genome-wide identification and characterization of MADS-box family genes related to organ development and stress resistance in Brassica rapa. BMC Genomics. 2015;16(1):178.
Wang Y, Zhang J, Hu Z, Guo X, Tian S, Chen G. Genome-wide analysis of the MADS-Box transcription factor family in Solanum lycopersicum. Int J Mol Sci. 2019;20(12):2961.
Dong X, Deng H, Ma W, Zhou Q, Liu Z. Genome-wide identification of the MADS-box transcription factor family in autotetraploid cultivated alfalfa (Medicago sativa L.) and expression analysis under abiotic stress. BMC Genomics. 2021;22(1):603.
Fan CM, Wang X, Wang YW, Hu RB, Zhang XM, Chen JX, et al. Genome-wide expression analysis of soybean MADS genes showing potential function in the seed development. PLoS ONE. 2013;8(4):e62288.
Lai X, Vega-Léon R, Hugouvieux V, Blanc-Mathieu R, van der Wal F, Lucas J, et al. The intervening domain is required for DNA-binding and functional identity of plant MADS transcription factors. Nat Commun. 2021;12(1):4760.
Brent MR, Guigó R. Recent advances in gene structure prediction. Curr Opin Struct Biol. 2004;14(3):264–72.
Aromolaran O, Aromolaran D, Isewon I, Oyelade J. Machine learning approach to gene essentiality prediction: a review. Brief Bioinform. 2021;22(5):bbab128.
Huang C, Kalodimos CG. Structures of large protein complexes determined by nuclear magnetic resonance spectroscopy. Annu Rev Biophys. 2017;46:317–36.
Carugo O. Protein hydration: investigation of globular protein crystal structures. Int J Biol Macromol. 2017;99:160–65.
Jones DT, Thornton JM. The impact of AlphaFold2 one year on. Nat Methods. 2022;19(1):15–20.
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–89.
Puranik S, Acajjaoui S, Conn S, Costa L, Conn V, Vial A, et al. Structural basis for the oligomerization of the MADS domain transcription factor SEPALLATA3 in Arabidopsis. Plant Cell. 2014;26(9):3603–15.
Wells CE, Vendramin E, Jimenez Tarodo S, Verde I, Bielenberg DG. A genome-wide analysis of MADS-box genes in peach [Prunus persica (L.) Batsch]. BMC Plant Biol. 2015;15:41.
Tang Y, Wang J, Bao X, Wu Q, Yang T, Li H, et al. Genome-wide analysis of Jatropha curcas MADS-box gene family and functional characterization of the JcMADS40 gene in transgenic rice. BMC Genomics. 2020;21(1):325.
Nam J, Kim J, Lee S, An G, Ma H, Nei M. Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc Natl Acad Sci U S A. 2004;101(7):1910–15.
De Bodt S, Raes J, Florquin K, Rombauts S, Rouzé P, Theissen G, et al. Genomewide structural annotation and evolutionary analysis of the type I MADS-box genes in plants. J Mol Evol. 2003;56(5):573–86.
Qiu Y, Köhler C. Endosperm evolution by duplicated and neofunctionalized type I MADS-Box transcription factors. Mol Biol Evol. 2022;39(1):msab355.
Masiero S, Colombo L, Grini PE, Schnittger A, Kater MM. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell. 2011;23(3):865–72.
Bemer M, Heijmans K, Airoldi C, Davies B, Angenent GC. An atlas of type I MADS box gene expression during female gametophyte and seed development in Arabidopsis. Plant Physiol. 2010;154(1):287–300.
Nardeli SM, Artico S, Aoyagi GM, de Moura SM, da Franca Silva T, Grossi-de-Sa MF, et al. Genome-wide analysis of the MADS-box gene family in polyploid cotton (Gossypium hirsutum) and in its diploid parental species (Gossypium arboreum and Gossypium raimondii). Plant Physiol Biochem. 2018;127:169–84.
Wang X, Huang Q, Shen Z, Baron GC, Li X, Lu X et al. Genome-wide identification and analysis of the mads-box transcription factor genes in blueberry (Vaccinium spp) and their expression pattern during fruit ripening. Plants (Basel). 2023;12(7).
Tang W, Tu Y, Cheng X, Zhang L, Meng H, Zhao X, et al. Genome-wide identification and expression profile of the MADS-box gene family in Erigeron breviscapus. PLoS ONE. 2019;14(12):e0226599.
Lin CS, Hsu CT, Liao DC, Chang WJ, Chou ML, Huang YT, et al. Transcriptome-wide analysis of the MADS-box gene family in the orchid Erycina pusilla. Plant Biotechnol J. 2016;14(1):284–98.
Gramzow L, Weilandt L, Theißen G. MADS goes genomic in conifers: towards determining the ancestral set of MADS-box genes in seed plants. Ann Bot. 2014;114(7):1407–29.
Daminato M, Masiero S, Resentini F, Lovisetto A, Casadoro G. Characterization of TM8, a MADS-box gene expressed in tomato flowers. BMC Plant Biol. 2014;14:319.
Cheng Z, Ge W, Li L, Hou D, Ma Y, Liu J, et al. Analysis of MADS-Box gene family reveals conservation in floral organ abcde model of moso bamboo (Phyllostachys edulis). Front Plant Sci. 2017;8:656.
Tian Y, Dong Q, Ji Z, Chi F, Cong P, Zhou Z. Genome-wide identification and analysis of the MADS-box gene family in apple. Gene. 2015;555(2):277–90.
Meng D, Cao Y, Chen T, Abdullah M, Jin Q, Fan H, et al. Evolution and functional divergence of MADS-box genes in Pyrus. Sci Rep. 2019;9(1):1266.
Kuijer HNJ, Shirley NJ, Khor SF, Shi J, Schwerdt J, Zhang D, et al. Transcript profiling of MIKCc MADS-Box genes reveals conserved and novel roles in barley inflorescence development. Front Plant Sci. 2021;12:705286.
Ali Z, Raza Q, Atif RM, Aslam U, Ajmal M, Chung G. Genetic and molecular control of floral organ identity in cereals. Int J Mol Sci. 2019;20(11):2743.
Palumbo F, Vannozzi A, Magon G, Lucchin M, Barcaccia G. Genomics of Flower Identity in Grapevine (Vitis vinifera L). Front Plant Sci. 2019;10:316.
Su CL, Chen WC, Lee AY, Chen CY, Chang YC, Chao YT, et al. A modified ABCDE model of flowering in orchids based on gene expression profiling studies of the moth orchid Phalaenopsis aphrodite. PLoS ONE. 2013;8(11):e80462.
Qi X, Yu X, Xu D, Fang H, Dong K, Li W, et al. Identification and analysis of CYP450 genes from transcriptome of Lonicera japonica and expression analysis of chlorogenic acid biosynthesis related CYP450s. PeerJ. 2017;5:e3781.
Pu X, Li Z, Tian Y, Gao R, Hao L, Hu Y, et al. The honeysuckle genome provides insight into the molecular mechanism of carotenoid metabolism underlying dynamic flower coloration. New Phytol. 2020;227(3):930–43.
Gramzow L, Barker E, Schulz C, Ambrose B, Ashton N, Theißen G, et al. Selaginella genome analysis - entering the “Homoplasy Heaven” of the MADS world. Front Plant Sci. 2012;3:214.
Niu S, Li J, Bo W, Yang W, Zuccolo A, Giacomello S, et al. The chinese pine genome and methylome unveil key features of conifer evolution. Cell. 2022;185(1):204–17e214.
Liu Y, Wang S, Li L, Yang T, Dong S, Wei T, et al. The Cycas genome and the early evolution of seed plants. Nat Plants. 2022;8(4):389–401.
Mi ZY, Zhao Q, Lu C, Zhang Q, Li L, Liu S, et al. Genome-wide analysis and the expression pattern of the MADS-Box Gene Family in Bletilla striata. Plants (Basel). 2021;10(10):2184.
Hu J, Chang X, Zhang Y, Yu X, Qin Y, Sun Y, et al. The pineapple MADS-box gene family and the evolution of early monocot flower. Sci Rep. 2021;11(1):849.
Shao Z, He M, Zeng Z, Chen Y, Hanna AD, Zhu H. Genome-wide identification and expression analysis of the MADS-Box gene family in sweet potato [Ipomoea batatas (L.) Lam]. Front Genet. 2021;12:750137.
Hu L, Liu S. Genome-wide analysis of the MADS-box gene family in cucumber. Genome. 2012;55(3):245–56.
Gao H, Wang Z, Li S, Hou M, Zhou Y, Zhao Y, et al. Genome-wide survey of potato MADS-box genes reveals that StMADS1 and StMADS13 are putative downstream targets of tuberigen StSP6A. BMC Genomics. 2018;19(1):726.
Ning K, Han Y, Chen Z, Luo C, Wang S, Zhang W, et al. Genome-wide analysis of MADS-box family genes during flower development in lettuce. Plant Cell Environ. 2019;42(6):1868–81.
Márquez Gutiérrez R, Cherubino Ribeiro TH, de Oliveira RR, Benedito VA, Chalfun-Junior A. Genome-wide analyses of MADS-Box genes in Humulus lupulus L. reveal potential participation in plant development, Floral Architecture, and Lupulin Gland Metabolism. Plants (Basel). 2022;11(9):1237.
Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46(W1):W200–4.
Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 1997; 26878–94.
Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 2019;47(W1):W5–10.
Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016;44(W1):W232–5.
Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–6.
Letunic I, Khedkar S, Bork P. SMART: recent updates, new developments and status in 2020. Nucleic Acids Res. 2021;49(D1):D458–60.
Høie MH, Kiehl EN, Petersen B, Nielsen M, Winther O, Nielsen H, et al. NetSurfP-3.0: accurate and fast prediction of protein structural features by protein language models and deep learning. Nucleic Acids Res. 2022;50(W1):W510–5.
Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. 2022;19(6):679–82.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Li J, Ye C, Chang C. Comparative transcriptomics analysis revealing flower trichome development during flower development in two Lonicera japonica Thunb. Cultivars using RNA-seq. BMC Plant Biol. 2020;20(1):341.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34(5):525–7.
Qi X, Fang H, Chen Z, Liu Z, Yu X, Liang C. Ectopic expression of a R2R3-MYB transcription factor gene LjaMYB12 from Lonicera japonica increases flavonoid accumulation in Arabidopsis thaliana. Int J Mol Sci. 2019;20(18):4494.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (32000262), the Science and Technology Support Project Fund of Changzhou (CE20202006), and the Independent Research Fund of Public Research Institutes in Jiangsu Province (BM2018021-4).
Author information
Authors and Affiliations
Contributions
C. Liang and X. Qi conceived and designed the experiments. Y. Lin, Z. Chen, and Y. Wan performed the experiments. Y. Lin, X. Qi, and H. Fang analyzed the data. Y. Lin wrote the manuscript, and X. Qi and C. Liang revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Experimental research and field studies on plants including the collection of plant material are comply with relevant guidelines and regulation.
Consent for publication
Not applicable.
Statement
Plant materials of Lonicera japonica Thunb. were collected from Germplasm Nursery at Nanjing Botanical Garden Mem. Sun Yat-Sen, Nanjing, China. All the required permissions have been obtained from Institute of Botany, Jiangsu Province and Chinese Academy of Sciences. The plant materials don’t include any wild species at risk of extinction. We comply with relevant institutional, national, and international guidelines and legislation for plant study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, Y., Qi, X., Wan, Y. et al. Genome-wide analysis of the MADS-box gene family in Lonicera japonica and a proposed floral organ identity model. BMC Genomics 24, 447 (2023). https://doi.org/10.1186/s12864-023-09509-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09509-9