
Abstract
Background
Drug resistant Mycobacterium tuberculosis prevention and care is a major challenge in Ethiopia. The World health organization has designated Ethiopia as one of the 30 high burden multi-drug resistant tuberculosis (MDR-TB) countries. There is limited information regarding genetic diversity and transmission dynamics of MDR-TB in Ethiopia.
Objective
To investigate the molecular epidemiology and transmission dynamics of MDR-TB strains using whole genome sequence (WGS) in the Amhara region.
Methods
Forty-five MDR-TB clinical isolates from Amhara region were collected between 2016 and 2018, and characterized using WGS and 24-loci Mycobacterium Interspersed Repetitive Units Variable Number of Tandem Repeats (MIRU-VNTR) typing. Clusters were defined based on the maximum distance of 12 single nucleotide polymorphisms (SNPs) or alleles as the upper threshold of genomic relatedness. Five or less SNPs or alleles distance or identical 24-loci VNTR typing is denoted as surrogate marker for recent transmission.
Results
Forty-one of the 45 isolates were analyzed by WGS and 44% (18/41) of the isolates were distributed into 4 clusters. Of the 41 MDR-TB isolates, 58.5% were classified as lineage 4, 36.5% lineage 3 and 5% lineage 1. Overall, TUR genotype (54%) was the predominant in MDR-TB strains. 41% (17/41) of the isolates were clustered into four WGS groups and the remaining isolates were unique strains. The predominant cluster (Cluster 1) was composed of nine isolates belonging to lineage 4 and of these, four isolates were in the recent transmission links.
Conclusions
Majority of MDR-TB strain cluster and predominance of TUR lineage in the Amhara region give rise to concerns for possible ongoing transmission. Efforts to strengthen TB laboratory to advance diagnosis, intensified active case finding, and expanded contact tracing activities are needed in order to improve rapid diagnosis and initiate early treatment. This would lead to the interruption of the transmission chain and stop the spread of MDR-TB in the Amhara region.
Similar content being viewed by others


Background
Emerging drug-resistant (DR) Mycobacterium tuberculosis threatens tuberculosis (TB) prevention and care efforts globally [1]. Multi-drug resistant (MDR)-TB is defined as resistance to both isoniazid and rifampicin, and rifampicin resistance (RR) requires similar management as MDR-TB [2]. The new World Health Organization (WHO) definition for pre-extensively drug resistance (pre-XDR)-TB is defined as MDR/RR-TB plus resistance to any fluoroquinolones and extensively drug-resistance (XDR)-TB is defined as MDR/RR-TB plus resistance to both fluoroquinolones and either bedaquiline or linezolid [3].
Patients with undiagnosed DR-TB have higher morbidity and mortality than patients with drug-susceptible (DS)-TB disease. Without proper diagnosis and treatment, similar to DS-TB, DR-TB may continue to spread in their communities [4]. Ethiopia is one of the 30 high burden MDR/RR-TB countries globally and ranks 5th among the African countries after South Africa, Nigeria, Mozambique and Democratic Republic of Congo [2]. Management of MDR/RR-TB in Ethiopia is complicated in many areas due to the lack of rapid diagnostic methods drug susceptibility testing (DST) and access to second line drugs (SLDs) for MDR/RR-TB.
It is known that the proportion of MDR-TB is higher among patients who received previous treatment for TB compared to those who are TB treatment-naïve. This disparity is assumed to be due to large proportions of MDR-TB arises from selection of drug resistant mutations during previous ineffective anti-TB treatment period, in contrast to new transmission of pre-existing MDR-TB strains [5,6,7]. Over time, however, a shift from acquisition of drug resistance during treatment to direct person-to-person transmission of MDR-TB strains occurs [8, 9]. A meta-analysis study on transmissibility of DR-TB compared to DS-TB found that DR-TB results in fewer infections or cases in contacts than DS-TB [10]. It is important to know the transmission dynamics in order to target public health and improve TB prevention and care for the region.
The genome size of M. tuberculosis is 4.4 Mbp with low mutation rate and no evidence of horizontal gene transfer [11]. The M. tuberculosis complex comprises of seven lineages that vary in geographical distribution around the globe; the strain-types may be specifically adapted to people of different genetic backgrounds [11, 12]. Four lineages are predominant in humans: lineage 1, Indo-Oceanic; lineage 2, East Asian (Beijing spoligotype families); lineage 3, Central Asian Strain (CAS/Delhi spoligotype families); and lineage 4, Euro-American (Latin American–Mediterranean (LAM), Haarlem and the ‘ill-defined’ T spoligotype families) [11]. In addition to lineage 5 and 6, a new M. tuberculosis complex lineage, lineage 7 (Aethiops vetus), has identified among strains originated from Ethiopia [13]. Knowing the dominant lineages for particular region can be important to TB prevention and care since the strain type may play a role in disease outcome, variation in vaccine efficacy [14], development of drug resistance [15] and disease epidemiology as it is associated with the presence or absence of clustering due to recent transmission [16].
Classical genotyping tools such as spoligotyping and mycobacterial interspersed repetitive units variable number of tandem repeats (MIRU-VNTR), used alone, can lack sufficient discriminatory power due to high similarity of strains in particular lineages [17, 18]. Spoligotyping method alone is unable to analyze transmission cluster [19, 20]. MIRU-VNTR typing can be a powerful tool to discriminate M. tuberculosis lineages and estimation of transmission dynamics [21].
Whole genome sequencing (WGS) has the highest resolution in investigating the transmission chain, with detailed view of genetic diversity as well as determining drug resistant-conferring gene mutations. However, the implementation of WGS is limited especially in high burden countries, due to lack of international standard, shared approaches in universal software for analysis and storage database, as well as financial constraints [22].
The use of WGS gene-by-gene MLST (multi-locus sequence typing) methods and Ridom SeqSphere software has resulted in a more standardized and user-friendly approach than traditional WGS single nucleotide polymorphism (SNP) mapping for resolving and understanding outbreaks [23, 24].
The genotype cluster rate of M. tuberculosis strains has been used to assess the efficacy of TB prevention and care programs [25], since clustering is generally a marker for ongoing or recent transmission while unique patterns indicate reactivation of TB disease [25, 26]. In Ethiopia, classical genotyping methods performed on DS-TB have shown complex genetic diversity of M. tuberculosis strains [27]. Only a few studies on MDR-TB strain genotypes have been reported with investigations limited by sample size [28] or use of only spoligotyping method [19, 20]. Our finding will help to strengthen TB patients care and management, and revealed the need for improving contact tracing and TB prevention and control strategies in the country. To the best of our knowledge, this is the first in-depth study investigating the molecular epidemiology and transmission dynamics of MDR-TB strains using WGS and MIRU-VNTR in the Amhara region, Ethiopia.
Materials and methods
Study setting, design and period
A cohort study was conducted from January 2016 to September 2018 at nine MDR/RR-TB treatment centers in the Amhara region, Ethiopia. A total of 200 patients diagnosed with MDR/RR-TB and treatment failed pulmonary TB patients [29] were enrolled. All the study participants were ≥ 15 years of age. A single sputum sample was collected prior to commencing second-line drug TB treatment. A structured questionnaire was used to collect the following information; MDR/RR-TB contact history, previous history of anti-TB treatment, geographical location (region, zone, district and kebele) and other demographic characteristics of patients. A samples of Lowenstein-Jensen (LJ) culture positive isolates from 45/200 MDR/RR-TB (n = 38) and treatment failed (n = 3) patients were selected for MIRU-VNTR and WGS analyses due to limited funding. The selection criteria were based on the patient’s admission/ data collection year, molecular drug resistance patterns, MDR/RR-TB treatment center hospitals, and geographical location of patients.
HIV testing
HIV test was done for each study participants using rapid HIV test algorithm of the Federal Ministry of Health of Ethiopia (KHB/STAT-PAK®/Unigold™ or Wantai/Uni-Gold/Vikia) based on the manufacturer instructions. The test was done using finger stick blood sample at the Provider Initiative Counseling and Testing (PICT) services at the respective health institutions in the Amhara region.
Specimen processing, culture and drug susceptibility test
Sputum collection, transportation, M. tuberculosis isolation, identification and DST were performed as previously described [29]. The mycobacteria bacilli DNA was extracted from culture colonies using GenoLyse® DNA extraction kit (Hain Lifescience GmbH, Nehren, Germany). Molecular DST was performed using GenoType® MTBDRplus version 2 kit strips (to detect rifampicin and isoniazid resistance) and GenoType® MTBDRsl version 2 kit strips (to detect fluoroquinolones and aminoglycosides resistance) according to the manufacturer’s instructions (Hain Lifescience GmbH, Nehren, Germany).
DNA extraction
The genomic DNA of M. tuberculosis complex was extracted for MIRU-VNTR and WGS using Cetyl-trimethyl-ammonium Bromide (CTAB) method as described previously [30]. Briefly, sputum samples were inoculated on LJ medium and incubated at 37oC until growth becomes clearly visible. One loopful cultured bacterial cells was transferred into the 1.5 ml micro-centrifuge tube containing 400 𝜇L TE (Tris-EDTA) buffer and subjected for heating at 95oC water bath for 60 min to kill the cells and then cooled at room temperature. 50 𝜇L lysozyme (10 mg/ml) was then added and incubated at 37oC overnight. 70 𝜇L of 10% SDS (Sigma, USA) and 5 𝜇L proteinase K (10 mg/ml; Sigma, USA) were added to the thawed sample and incubated for 10 min at 65oC with intermittent mixing. This is followed by the addition of 100 𝜇L of 5 M NaCl and mixed, then 100 𝜇L cetyl-trimethyl-ammonium bromide (CTAB/NaCL) (pre-warmed to 65oC in water bath) was added and vortex mixed until the solution becomes milky and then incubated at 65oC in water bath for 10 min. Then, 700 𝜇L of chloroform/isoamyl alcohol was added to each tube and the mixture was centrifuged at 13,000 x g for 5 min. The upper aqueous phase of the preparation was transferred into a clean 1.5 ml tube containing 700 𝜇L of cold (-20oC) isopropanol and incubated at -20oC for 1 h. The solution was centrifuged at 14,000 xg for 20 min and the supernatant was discarded. The tube was washed with 1 ml ice cold (-20oC) 70% ethanol by centrifuging at 14,000 xg for 10 min. The supernatant was discarded and the DNA pellet was air dried before being re-suspended in 50 𝜇L TE buffer and kept at -20oC until use. The DNA extraction was done at University of Gondar TB culture laboratory and the DNA purity and concentration was checked using NanoDrop™ spectrophotometer machine. The extracted DNA pellet was shipped to The Ohio State University (Ohio, USA) and Michigan Department of Health and Human Services (MDHHS, Michigan, USA) for MIRU-VNTR and WGS typing.
Genotyping and molecular analysis
MIRU-VNTR analysis
MIRU-VNTR 24-loci was performed as described previously [31]. PCR amplification was performed in a total volume of 20 µl containing 2 µl DNA sample and 18 µl reaction mixtures. All reactions were subjected to GeneAmp 2700 thermo-cycler: 95oC for 5 min followed by 35 cycles of 1 min at 94oC, 1 min at 59oC, 1.5 min at 72oC and terminated by 10 min at 72oC. Genotyping was performed using multiplex PCR with a Rox-labeled MapMarker 1000 size standard (PE Applied BIosystem) for sizing of the PCR products. The PCR fragments were analyzed using ABI 3500xl capillary electrophoresis analyzer (Applied Biosystems, Foster City, California, USA), and the number of copies of each locus was determined by automated assignment using GeneMaper 4.0 software. The number of repeats present in each locus was determined and the results were presented as a set of digits, MIRU code, which reflects the number of MIRU sequence repeats/ alleles. Locus that did not amplify was repeated using singleplex PCR.
Lineage identification of M. tuberculosis complex strains was carried out first by best-match analysis with the reference strain with 0.17 maximum cut of distance, and followed by tree based identification on the MIRU-VNTRplus database (https://www.miru-vntrplus.org) [32].A dendrogram was generated using the unweighted pair group method with arithmetic average (UPGMA) and phylogenetic tree analysis was done using MIRU-VNTR typing database. A cluster was defined as two or more M. tuberculosis complex isolates sharing identical 24-loci MIRU-VNTR patterns. The recent transmission index was calculated as the number of clustered patients minus number of clusters divided by total number of patients [33]. The discriminatory power was determined by the Hunter-Gaston discriminatory index (HGDI) [34] calculated online using the discriminatory power calculator available at http://insilico.ehu.es. The allelic diversity of each 24-loci MIRU-VNTR was determined in the MIRU-VNTRplus database and grouped into highly (HGDI > 0.6), moderately (0.6 < HGDI > 0.3) and poorly (HGDI < 0.3) discriminatory.
WGS analysis
The WGS was performed using NextSeq 550 desktop sequencer (Illumina, San Diego, CA, USA) at MDHHS. Isolated genomic DNA of individual strains was prepared for sequencing using Illumina Nextera XT library preparation kits according to the manufacturer’s instruction (Illumina, San Diego, CA, USA).
SNP analysis
Each isolate was sequenced using paired-end sequencing with 4 replicates for each fastq read file, resulting in two sequencing read files (designated by R1 and R2), with 4 replicate each designated by L001, L002, L003 and L004. The final R1 and R2 fastq files of each isolate were created by concatenating their respective 4 sequencing replicates. Variant SNP (vSNP) calling of the M. tuberculosis samples was performed using the open source software tool vSNP (https://github.com/USDA-VS/vSNP). The variant SNP pipeline takes as input the two raw reads (R1 and R2) and aligns them to the M. tuberculosis H37Rv reference genome (GenBank accession number: NC_000962.3) using Burrows Wheeler Alignment (BWA) [35] and Genome Analysis Toolkit 2.5.2 (GATK) [36,37,38]. The tool Picard Mark Duplicates was used to remove mapped reads that are redundant and that could potentially lead to erroneous SNP calling (http://broadinstitute.github.io/picard/). The Variant Call Format (VCF) files with the detected SNPs for each isolate was created [39, 40], and VCF filter kept isolates with a Phred quality score above 20 (https://github.com/vcflib/vcflib#vcflib). Proline-glutamate (PE) and proline‐proline‐glutamate (PPE)‐ polymorphic GC‐repetitive sequences (PGRS) were filtered from the analysis [39], as well as SNP positions that had missing data ‘N’, gaps or ambiguous nucleotides. Integrated Genomics Viewer (IGV) [41] provided a way to visually validate the detected SNP sites, and samples that showed no variation in IGV were excluded from the analysis. Maximum likelihood phylogenetic trees were calculated after the curation of SNP sites using RAxML software [42].
Core genome MLST (cgMLST) analysis
Raw reads of each samples were aligned to M. tuberculosis reference genome H37Rv (RefSeq ID: NC_000962.3) using Burrows Wheeler Alignment. Assembled genomes were uploaded onto the Ridom SeqSphere software version 6.0.2 (Ridom; Münster, Germany). Each isolate sequence was aligned to the Ridom SeqSphere M. tuberculosis cgMLST scheme, previously defined consisting of 2891 core genes (GenBank accession number NC_000962.3), for alignment and subsequent genomic analysis [24, 43]. Successful alignments to the cgMLST were defined as good targets by the Ridom SeqSphere software, and full cgMLST analysis was carried out on isolate sequences that conferred > 98% good targets. The cgMLST scheme was also used to compare the 41 M. tuberculosis isolates. The resulting phylogeny comparison was made using a Neighbor Joining tree by the Ridom SeqSphere. Minimum spanning tree was created using the same software. The reference genome of M. tuberculosis H37Rv (NC_000962.3) was used to root the tree.
Genomic relatedness cluster analysis
Pairwise SNP distances were calculated between all sequences using the hamming distance metric, which calculates the number of elements that are dissimilar between two sequences [44]. Genotype clusters were inferred based on how genetically similar two isolates were from each other. The upper thresholds of genomic relatedness or cluster is defined as 12 SNPs or alleles cut off or less and a recent transmission event is defined as 5 or less SNPs or alleles [43, 45]. If two isolates exhibited a distance of more than 12 SNPs or alleles, they were called unique strains.
Drug resistance gene mutations analysis
Antimicrobial resistance prediction genetic markers are listed in the TB Profiler database (http://tbdr.lshtm.ac.uk) such as rpoB, rpoC, ahpC, embA, embB, embC, ethA, gidB, gyrA, gyrB, inhA, katG, pncA, rpsL, and rrs genes [46]. We used the phylo-resistance-search-engine (PhyReSE: http://phyresse.org) and TBprofiler (http://tbdr.lshtm.ac.uk) software to analyze the mutations involved in drug resistance and lineages determination [46, 47].
Statistical analysis
Data were entered using Epi-Data version 3.1 and exported to SPSS version 20 (SPSS Inc., Chicago, Illinois, USA) for analysis. Data completeness and consistency was checked by running frequencies of each variable. Descriptive statistics and cluster analysis were done.
Results
Demographic and clinical characteristics of patients
Among the 45 study participants, the median age was 29 years (IQR: 25–37) and 73% were between 15 and 34 years of age, 60% were male and 20% were HIV-positive. Most of the study participants (67%) had previous history of anti-TB treatment, the literacy rate was 64% and 51% were urban dwellers. In addition, 18% had history of contact with MDR/RR-TB patients, 18% had a family history of TB and 91% were smear positive at the time of diagnosis. Geographically, 42% of MDR-TB patients were recruited from the University of Gondar Referral Hospital and 40% of MDR-TB patients were living in North Gondar zone (Table 1).
Population structure of M. tuberculosis
Analysis by MIRU-VNTR
A total of 45 MDR/RR-TB isolates were submitted for MIRU-VNTR 24-loci testing. Two isolates were excluded from the final MIRU-VNTR analysis. One isolate did not have a PCR amplicon for more than 4 loci and the other isolate was contaminated and was not identified as M. tuberculosis. Isolates with no PCR amplicon at only one locus were included into the analysis and categorized under missing data. Three isolates showed double alleles in a single locus.
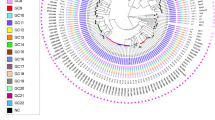
The most prevalent M. tuberculosis lineages were TUR (23/43; 53.5%) and Delhi/CAS (16/43; 37.2%). Haarlem and Endo-Oceanic both accounted for 4.6% (2/43) (Fig. 1, Supporting file: Fig S1).

Radial UPGMA tree based on the MIRU-VNTR 24-loci copy numbers. The tree was calculated using MIRU-VNTRplus website. EAI: East African Indian/ Indo-Oceanic
Twenty-four of the 43 (55.8%) M. tuberculosis isolates were clustered into four different MIRU-VNTR typing groups containing 2, 3, 5 and 14 strains in each. Nineteen isolates had unique MIRU-VNTR patterns. The recent transmission index using MIRU-VNTR pattern analysis was 46.5%.
Out of the 24 loci, the least (low) allelic diversity (h < 0.3) was seen in 9 loci (MIRU 02, MIRU 04, MIRU 20, Mtub29, ETRB, MIRU 23, MIRU 24, MIRU 27 and Mtub34). Moderate allelic diversity (h = 0.6 − 0.3) was seen in 14 loci (Mtub04, ETRC, MIRU 40, MIRU 10, MIRU 16, Mtub21, QUB-11b, ETRA, MIRU 26, MIRU 31, Mtub39, QUB-26, QUB-4156 and MIRU 39). The highest allelic diversity indices (h > 0.6) was seen only for Mtub30.
Phylogenetic relationship with representative branch lengths demonstrates 4 clusters of MDR/RR-TB isolates based on the 24-loci VNTR pattern (Fig. 2).

Phylogenetic tree based on 24-loci MIRU-VNTR pattern of 4 clustered MDR/RR-TB isolates. The VNTR loci copy number for each isolates with sub-lineage of M. tuberculosis
Analysis by WGS
Four of the 45 samples (A126, A132, A143 and A191) had poor coverage and/or contaminated or one isolate was not identified as M. tuberculosis, and excluded from the final analysis. Of the 41 remaining isolates, the statistics used to summarize the data showed that the isolates exhibited read-quality scores between 32 and 33 Q, over 99% genome coverage, average sequencing read-length of 142 bp, and an average coverage of 97.1 depth.
A total of 4456 SNP sites were identified. The maximum likelihood tree identified 3 distinct clades based on M. tuberculosis lineage classification in accordance with bioinformatics pipeline vSNP or SNP based barcode nomenclature [11] (Fig. 3). Overall, three major M. tuberculosis lineages were identified; the predominant lineage was Euro-American (lineage 4) accounted for 58.5% (24/41), followed by 36.6% (15/41) lineage 3 (Delhi/CAS) and 5% (2/41) Endo-Oceanic lineage (lineage 1).

Maximum likelihood of M. tuberculosis phylogeny. Three clades were identified and colored to represent the M. tuberculosis lineages: H37Rv-04/lineage 4 (n = 24, blue branches), H37Rv-03/lineage 3 (n = 15, magenta branches), and H37Rv-01/lineage 1 (n = 2, red branches)
The predominant sub-lineages of Euro-American lineage was TUR, 92% (22/24) (Coll sub-lineage 4.2.2). Haarlem (Coll sub-lineage 4.1.2) and X-type (Coll sub-lineage 4.1.1) each accounted 4% (1/24). Isolates belonging to lineage 3 harbored the CAS specific RD750 deletion and its sub-lineage was identified as CAS for 53% (8/15) and CAS1-kili for 47% (7/15) spoligotypes. Lineage 1 isolates had RD239 deletion and contain two spoligotype families (EAI3 and EAI5).
41% (17/41) of the isolates were clustered into four WGS groups (Fig. 4). The remaining isolates were more than 12 SNPs apart, known as unique strains. Compared to the lineages identified by vSNP, cluster 1 is composed by nine isolates belonging to lineage 4 with two recent transmission links (A76-A194 and A110-A114) denoted as red circles in Fig. 4. Clusters 2 and cluster 3 has 3 isolates each belonging to lineages 4 and 3, respectively. Cluster 4 is composed of two isolates belonging to lineage 4.

Cluster analysis based on pairwise SNP difference between clustered isolates. The number indicates the SNP differences that exist between two isolates. Red circle indicates recent transmission links between isolates and the blue circle shows isolates within a transmission cluster
Four different cgMLST clusters were identified within the 41 WGS-genotyped isolates; 18 isolates were clustered and 23 isolates were unique. The upper limit of cluster threshold was defined as a maximum of 12 allele difference (Fig. 5, Supporting file: Fig S2).

Minimum spanning tree for 41 M. tuberculosis isolates based upon cgMLST analysis. M. tuberculosis cgMLST complex type / cluster-alert distance were 12 alleles. The numbers displayed on lines between circles represent the numbers of different alleles between different samples. Samples were grouped by color according to their cgMLST complex type (right side)
Cluster 1 had 10 isolates (A184, A73, A110, A114, A29, A161, A101, A194, A76 and A85); cluster 2 had 3 isolates (A158, A74, and A88); cluster 3 had 3 isolates (A117, A19, and A170) and cluster 4 had 2 isolates (A173 and A162). Two recent transmission events were identified in cluster 1 (A76-A194, and A110-A114-A73-A184).
In total, 4 WGS transmission clusters in both SNP and cgMLST based analysis corresponding to a clustering proportion of 44% (18/41) and resulting in a clustering rate or recent transmission index of 34% (calculated as (18 − 4)/41)).
45% of TUR lineage was resistance to all first line drugs and 73% had previous history of anti-TB treatment (Table 2).
Discussion
We analyzed 45 MDR/RR-TB isolates collected from 2016 to 2018 from the Amhara region health institutions. However, only 41 isolates were eligible for further analysis and the remaining four isolates were excluded from WGS analysis.
Transmission clusters analysis is the basis for tracing outbreak of M. tuberculosis clones and to improve TB control policies and strategies. Clustering is an indicator of recent transmission [48]. Recent transmissions association with MDR-TB strains has been reported in various geographical settings [26, 28, 49,50,51].
The present study suggests possible high transmission of MDR/RR-TB in the Amhara region, as shown by 44% (18/41) and 55.8% of MDR-TB isolates being clustered based on SNPs/alleles difference and VNTR genotype, respectively. This variation is due to the fact that WGS generates smaller clusters than MIRU-VNTR typing. As a result, MIRU-VNTR typing is less predicative of close genetic relatedness compared with WGS and has led to overestimation of TB transmission in the community [52].
Although an extended contact evaluation or epidemiological link data were not collected to determine direct transmission links, the majority of the patient characteristic within the transmission clusters obtained from SNPs or allele difference matched with variable degree the patients’ admission/ treatment initiation dates within one year apart and/or proximity of location within hospitals and/or zones or Districts.
A recent MDR-TB transmission index can be used to assess the country’s efficacy of TB prevention and care programs. Looking specifically the four recent transmissions grouped isolates using SNPs (A76-A194 and A110-A114), the linked patients live in the same district area (two were in Achefere and the other two were in Tegedie district). It is unknown if the patients may have had direct contact history and/or social interaction with each other but the geographical proximity supports epidemiological transmission links. Interestingly, cgMLST also identified the same two recent transmission clusters (A76-A194 and A110-A114-A73-A184). The first one is same as SNPs analysis but the second has a total of 4 patients compared to two by SNPs. Patients, labelled as A73 and A184, were not living in the same geographic location like patients labelled as A110 and A114, but one of the patient was admitted at the same hospital with others and lives in the same zone but not in the same district like others.
In our finding, the MDR-TB clustering rate (34%) is in concordance with other countries such as China (33.7%) and Saudi-Arabia (32%), indicating high rate of drug resistance TB transmission [53, 54]. Compared to our finding, previous study have reported higher cluster rate of MDR-TB transmission in Ethiopia (86-86.5%), Turkey (79%), Saudi-Arabia (67.6%), Tunisia (65%) and Europe (43%) [19, 20, 26, 49, 51, 55]. MDR-TB cluster rates lower than our studies have also been reported in the US (20%) and in the UK (15%) [56, 57]. The disparity between studies in the transmission cluster rate might be due to differences in genotyping methods, types of TB patients (DS- vs. MDR or XDR-TB), geographical location, variation in TB intensive contact evaluation and social interaction or network and population movement. In our study, the TUR strains are more clustered compared to the other strains.
In Ethiopia, the health extension program is functioning well at the community level that results for early detection and treatment of TB suspected patients within few weeks of onset of TB symptoms. But the relatively higher clustering rate observed in our study might be linked to insufficient TB prevention and care programs in Ethiopia or the region. Given the new genotyping cluster data, a recommendation to the regional TB program to increase active tracing of contacts of patients are needed in conjunction with improving the diagnostic facilities for MDR-TB. Lastly, early treatment of infected patients is essential to block the transmission chain. Amhara region has only two TB culture laboratories. It is very important to expand the capabilities for TB culture and DST to combat the spread of drug resistant TB in Ethiopia and in the Amhara region.
Our finding revealed that Euro-American lineage (lineage 4) was the most dominant among MDR/RR-TB strains in the Amhara region, followed by Delhi/Central Asia (lineage 3) and Endo-oceanic lineage (lineage 1) using WGS analysis. The main lineage classifications are the same in the MIRU-VNTR analysis except one sub-lineage classification variation: One isolate was Haarlem by MIRU-VNTR but X-type by WGS analysis. Our finding also revealed the dominance of lineage 4 that is already explained previously that Euro-American lineage is mainly associated with drug resistance [58].
A recent meta-analysis study of Ethiopia shown that Euro-American lineage was predominant in Ethiopia, followed by lineage 3/Delhi/CAS and lineage 1 in all TB forms [59]. Other studies reported that Euro-American and EAI lineage were predominant among MDR-TB strains in Ethiopia, Euro-American strains accounted 52% and 36% using sploligotyping alone [19, 20] and 83% from 12 strains in Ethiopia [28] and other countries reported, 55%, 54% and 100% in Saudi Arabia, Lebanon and Tunisia, respectively [49, 51, 60]. No Beijing strain was present in our study although it is commonly associated with drug resistance. This is in contrast to studies in Belarus, Turkey and Central Asia, which reported the dominance of Beijing strains and elucidated its association with drug resistance [26, 61, 62]. This might be attributed to that different lineages are prevalent in specific regions/countries and adapted to a particular human populations/ host-pathogen compatibility/ and more likely to transmit and cause disease of the same ethnicity [12].
The most prevalent genotypes within the Euro-American main lineage were sub-lineage TUR (92%; 22/24) and Haarlem (4%) and X-type (4%) strains. From lineage 3, we found CAS (53%) and CAS1-kili (47%) spoligotype families and two EAI3 and EAI5 spoligotype families were found under lineage 1 category. Although Haarlem is one of the widespread sub-lineage globally, the TUR sub-lineage is geographically restricted mainly in Asia and Africa [63]. Previous studies in Ethiopia reported the presence of Haarlem, CAS and EAI strains in MDR-TB isolates characterized by limited sample size or use of spoligotyping technique alone. TUR strains (designated previously as LAM7-TUR) have not been reported yet among MDR-TB strains in Ethiopia. Large cluster of TUR strains have been identified in Turkey and Saudi Arabia [49, 50]. The presence of high percentage of TUR strains in Ethiopia may be explained by the introduction of these strains from Turkey/Saudi Arabia to Ethiopia due to the growing economic partnership between Ethiopia and Turkish and/or Saudi Arabia or due to the return of the Ethiopian immigrants from Saudi Arabia to Ethiopia. Another theory proposed by Comas et al. that proposed an African origin for M. tuberculosis complex with some genotypes already present on the African continent before migration to European and Asia [27], and so this lineage could be emerged in Ethiopia.
In addition, two of the pre-XDR-TB strains were lineage 1 and one isolate in each represented the TUR and Delhi/CAS strains. From 3 non-MDR-TB isolates, two of them were grouped in lineage 3 and sub-lineage CAS and the other one isolate was TUR strain. The Pre-XDR-TB strains are grouped under the three lineages. The three unique strains were not found in a cluster that might be due to de novo evolution of resistance. This hypothesis might be in agreement with the hypothesis that XDR-TB acquisition appeared mainly by de novo evolution of resistance in addition to patient to-patient transmission [62].
One of the limitations of this study is the lack of extended epidemiological links data between the clustered cases and thus unable to confirm the genotype clusters and small number of MDR-TB isolates were genotyped (due to limited funding) that may not be representative of the region and may affect the cluster analysis. Genotyping of 200 MDR/RR-TB isolates may identify additional clusters. Plan is underway to genotype all of the remaining isolates.
Conclusions
Our finding showed that Amhara region appears to have ongoing MDR/RR-TB transmission dynamics indicated by high cluster proportion of MDR-TB isolates. Three major lineages were found among MDR-TB isolates in the Amhara region with three dominant sub-lineages. Improved TB infection control with expanded contact investigation, early diagnosis and treatment with appropriate regimen will be crucial to stop the growing spread and interrupt the transmission chain of MDR-TB strains. Improved TB surveillance, diagnosis and treatment of DS-TB will decrease treatment failure and re-infection of community.
Data availability
Raw sequence data are available under the project accession number PRJNA935744 (https://www.ncbi.nlm.nih.gov/sra/PRJNA935744).
Abbreviations
- TB:
-
Tuberculosis
- MDR-TB:
-
Multi-drug resistant tuberculosis
- WGS:
-
Whole genome sequence
- SNP:
-
Single nucleotide polymorphisms
- MIRU-VNTR:
-
Mycobacterial interspersed repetitive unit variable number of tandem repeats
References
Jabbar A, Phelan JE, de Sessions PF, Khan T, Rahman H, Khan SN, et al. Whole genome sequencing of drug resistant Mycobacterium tuberculosis isolates from a high burden tuberculosis region of North West Pakistan. Sci Rep. 2019;9:14996. https://doi.org/10.1038/s41598-019-51562-6.
World Health Organization. Global tuberculosis report 2021. 2021.
World Health Organization. WHO announces updated definitions of extensively drug-resistant tuberculosis. 2021:https://www.who.int/news/item/27-01-2021-who-announces-updated-definitions-of-extensively-drug-resistant-tuberculosis. Access on August, 20, 2022.
Menzies D, Benedetti A, Paydar A, Royce S, Pai M, Burman W, et al. Standardized treatment of active tuberculosis in patients with previous treatment and/or with monoresistance to isoniazid: a systematic review and meta-analysis. PLoS Med. 2009;6:e1000150.
da Garrido S, Bührer-Sékula M, Souza S, de Oliveira AB, Antunes GP, Mendes A. Multidrug-resistant tuberculosis in the Amazonas State, Brazil, 2000–2011. Int J Tuberc Lung Dis. 2015;19:531–6.
Li Y, Ehiri J, Oren E, Hu D, Luo X, Liu Y, et al. Are we doing enough to Stem the Tide of Acquired MDR-TB in Countries with High TB Burden? Results of a mixed method study in Chongqing, China. PLoS ONE. 2014;9(2):e88330.
Lukoye D, Adatu F, Musisi K, Kasule GW, Were W, Odeke R, et al. Anti-Tuberculosis Drug Resistance among New and previously treated Sputum SmearPositive Tuberculosis Patients in Uganda: results of the First National Survey. PLoS ONE. 2013;8(8):e70763.
Kendall EA, Fofana MO, Dowdy DW. The burden of transmitted multi-drug resistance among epidemics of tuberculosis: a transmission model. Lancet Respir Med. 2015;3(12):963–72.
Suen S-c, Goldhaber-Fiebert BE. Disease Control Implications of India’s changing Multi-Drug resistant tuberculosis epidemic. PLoS ONE. 2014;9(3):e89822.
Kodama C, Lange B, Olaru ID, Khan P, Lipman M, Seddon JA, et al. Mycobacterium tuberculosis transmission from patients with drug-resistant compared to drug-susceptible TB: a systematic review and meta-analysis. Eur Respir J. 2017;50:1701044.
Coll F, McNerney R, Guerra-Assunc¸a˜o JA, Glynn JR, Perdiga˜o J, Viveiros M, et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 2014;5:4812.
Gagneux S. Host-pathogen coevolution in human tuberculosis. Philos Trans R Soc Lond B Biol Sci. 2012;367:850–9.
Nebenzahl-Guimaraes H, Yimer SA, Holm-Hansen C, de Beer J, Brosch R, Soolingen D. Genomic characterization of Mycobacterium tuberculosis lineage 7 and a proposed name: ‘Aethiops vetus’. Microb Genom. 2016;2(6):e000063.
Lo´pez B, et al. A marked difference in pathogenesis and immune response induced by different Mycobacterium tuberculosis genotypes. Clin Exp Immunol. 2003;133:30–7.
Ford CB, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 2013;45:784–90.
Kato-Maeda M, Kim E. Differences among sublineages of the East-Asian lineage of Mycobacterium tuberculosis in genotypic clustering. Int J Tuberc Lung Dis. 2010;14:538–44.
Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, et al. Evolution and transmission of drug-resistant tuberculosis in a russian population. Nat Genet. 2014;46:279–86.
Niemann S, Supply P. Diversity and evolution of Mycobacterium tuberculosis: moving to whole-genome-based approaches. Cold Spring Harb Perspect Med. 2014;4:a021188.
Agonafir M, Lemma E, Wolde-Meskel D, Goshu S, Santhanam A, Girmachew F, et al. Phenotypic and genotypic analysis of multidrug-resistant tuberculosis in Ethiopia. Int J Tuberc Lung Dis. 2010;14(10):1259–65.
Diriba B, Berkessa T, Mamo G, Tedla Y, Ameni G. Spoligotyping of multidrug-resistant Mycobacterium tuberculosis isolates in Ethiopia. Int J Tuberc Lung Dis. 2013;17(2):246–50.
Supply P, Mazars E, Lesjean S, Vincent V, Gicquel B, Locht C. Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol Microbiol. 2000;36:762–71.
Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103:2869–73.
Jones RC, Harris LG, Morgan S, Ruddy MC, Perry M, Williams R, et al. Phylogenetic analysis of Mycobacterium tuberculosis strains in Wales by Use of Core Genome Multilocus sequence typing to analyze whole-genome sequencing data. J Clin Microbiol. 2019;57(6):e02025–18. https://doi.org/10.1128/JCM.-18.
Kohl TA, Diel R, Harmsen D, Rothgänger J, Walter KM, Merker M, et al. Whole-genome-based Mycobacterium tuberculosis surveillance: a standardized, portable, and expandable approach. J Clin Microbiol. 2014;52(7):2479–86. https://doi.org/10.1128/JCM.00567-14.
Zozio T, Allix C, Gunal S, Saribas Z, Alp A, et al. Genotyping of Mycobacterium tuberculosis clinical isolates in two cities of Turkey: description of a new family of genotypes that is phylogeographically specific for Asia Minor. BMC Microbiol. 2005;5:44.
Kisa O, Tarhan G, Gunal S, Albay A, Durmaz R, Saribas Z, et al. Distribution of Spoligotyping defined genotypic lineages among drug-resistant Mycobacterium tuberculosis Complex Clinical isolates in Ankara, Turkey. PLoS ONE. 2012;7(1):e30331.
Comas I, Hailu E, Kiros T, Bekele S, Mekonnen W, Gumi B, et al. Population Genomics of Mycobacterium tuberculosis in Ethiopia contradicts the virgin Soil Hypothesis for Human Tuberculosis in Sub-Saharan Africa. Curr Biol. 2015;25:3260–6.
Tessema B, Beer J, Merker M, Emmrich F, Sack U, Rodloff AC, et al. Molecular epidemiology and transmission dynamics of Mycobacterium tuberculosis in Northwest Ethiopia: new phylogenetic lineages found in Northwest Ethiopia. BMC Infect Dis. 2013;13:131.
Shibabaw A, Gelaw B, Gebreyes W, Robinson R, Wang S-H, Tessema B. The burden of pre-extensively and extensively drugresistant tuberculosis among MDR-TB patients in the Amhara region, Ethiopia. PLoS ONE. 2020;15(2):e0229040.
van Embden IDA, Cave MD, Crawford JT, Dale JW, Eisenach KD, Gicoquel B, et al. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J Clin Microbiol. 1993;31:406–9.
de Beer JL, Kremer K, Ködmön C, Supply P, van Soolingen D, Global Network for the Molecular Surveillance of Tuberculosis. 2009. First worldwide proficiency study on variablenumber tandem-repeat typing of Mycobacterium tuberculosis complex strains. J Clin Microbiol. 2012;50:662-9.
Allix-Béguec C, Harmsen D, Weniger T, Supply P, Niemann S. Evaluation and user-strategy of MIRU-VNTRplus, a multifunctional database for online analysis of genotyping data and phylogenetic identification of Mycobacterium tuberculosis complex isolates. J Clin Microbiol. 2008;46(8):2692–9.
Glynn JR, Vynnycky E, Fine PEM. Influence of sampling on estimates of clustering and recent transmission of Mycobacterium tuberculosis derived from DNA fingerprinting techniques. Am J Epidemiol. 1999;149:366–71.
Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of numerical index of the discriminatory ability of typing systems : an application of Simpson’s index of diversity. J Clin Microbiol. 1988;26:2465–6.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform Bioinformatics (Oxford, England). 2009;25(14):1754–60. doi:10.093/bioinformatics/btp324.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis Toolkit: a Map reduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. https://doi.org/10.101/gr.107524.110.
Van der Auwera GA, Carneiro MC, Hartl C, Poplin R, DelAngel G, Levy-Moonshine A, et al. From FastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protocols Bioinf. 2013;43(11):1–33.
Brennan M. The enigmatic PE/PPE Multigene Family of Mycobacteria and Tuberculosis Vaccination. Infect Immun. 2017;85(6):1–8.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv preprint arXiv:1207.3907 [q-bio.GN] 2012.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6. https://doi.org/10.1038/nbt.754.
Stamatakis A. Bioinf (Oxford England). 2014;30(9):1312–3. 10.093/bioinformatics/btu033. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies.
Kohl TA, Harmsen D, Rothgänger J, Walker T, Diel R, Niemann S. Harmonized genome wide typing of Tubercle Bacilli using a web-based Gene-By-Gene nomenclature system. EBioMedicine. 2018;34:131–8. https://doi.org/10.1016/j.ebiom.2018.07.030.
Hamming RW. Error detecting and error correcting codes. Bell Syst Technol J. 1950;29(2).
Walker TM, CIp CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–46.
Coll F, McNerney R, Preston MD, Guerra-Assunção JA, Warry A, Hill-Cawthorne G, et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015;7:51.
Feuerriegel S, Schleusener V, Beckert P, Kohl TA, Miotto P, Cirillo DM, et al. PhyResSE: web tool delineating Mycobacterium tuberculosis antibiotic resistance and lineage from whole-genome sequencing data. J Clin Microbiol. 2015;53(6):1908–14.
Barnes PF, Yang Z, Preston-Martin S, Pogoda JM, Jones BE. Patterns of tuberculosis transmission in Central Los Angeles. JAMA. 1997;278:1159–63.
Al-Ghafli H, Kohl TA, Merker M, Varghese B, Halees A, Niemann S, et al. Drug-resistance profiling and transmission dynamics of multidrug-resistant Mycobacterium tuberculosis in Saudi Arabia revealed by whole genome sequencing. Infect Drug Resist. 2018;11:2219–29.
Durmaz R, Zozio T, Gunal S, Yaman A, Cavusoglu C, et al. Genetic diversity and major spoligotype families of drug-resistant Mycobacterium tuberculosis clinical isolates from different regions of Turkey. Infect Genet Evol. 2007;7:513–9.
Bouzouita I, Cabibbe AM, Trovato A, Daroui H, Ghariani A, Midouni B, et al. Whole-genome sequencing of drug-resistant Mycobacterium tuberculosis strains, Tunisia, 2012–2016. Emerg Infect Dis. 2019;25(3):547–50.
Wyllie DH, Davidson JA, Smith EG, Rathod P, Crook DW, Peto TEA, et al. A quantitative evaluation of MIRU-VNTR typing against whole-genome sequencing for identifying Mycobacterium tuberculosis transmission: a prospective Observational Cohort Study. EBioMedicine. 2018;34:122–30.
Varghese B, Shoukri M, Memish Z, Abuljadaye N, Alhakeem R, Alrabiah F, et al. Occurrence of diverse mutations in Isoniazidand Rifampicin-Resistant Mycobacterium tuberculosis isolates from Autochthonous and immigrant populations of Saudi Arabia. Microb drug Resist. 2014;20:6DOI. https://doi.org/10.1089/mdr.2014.0065.
Wang F, Shao L, Fan X, Shen Y, Diao N, Jin J, et al. Evolution and transmission patterns of extensively drug-resistant tuberculosis in China. Antimicrob Agents Chemother. 2015;59:818–25. https://doi.org/10.1128/AAC.03504-14.
Devaux I, Kremer K, Heersma H, van Soolingen D. Clusters of Multidrug-Resistant Mycobacterium tuberculosis cases, Europe. Emerg Infect Dis. 2009;15(7):1052–62.
Anderson LF, Tamne S, Brown T, Watson JP, Mullarkey C, Zenner D, et al. Transmission of multidrug-resistant tuberculosis in the UK: a cross-sectional molecular and epidemiological study of clustering and contact tracing. Lancet Infect Dis. 2014;14(5):406–15.
Metcalfe JZ, Kim EY, Lin SG, Cattamanchi A, Oh P, Flood J, et al. Determinants of Multidrug-Resistant tuberculosis clusters, California, USA, 2004–2007. Emerg Infect Dis. 2010;16(9):1403–09.
Oppong YEA, Phelan J, Perdigão J, Machado D, Miranda A, Portugal I, et al. Genome-wide analysis of Mycobacterium tuberculosis polymorphisms reveals lineage-specific associations with drug resistance. BMC Genomics. 2019;20:252.
Mekonnen D, Derbie A, Chanie A, Shumete A, Biadglegne F, Kassahun Y, et al. Molecular epidemiology of M. tuberculosis in Ethiopia: a systematic review and meta-analysis. Tuberculosis. 2018;118:101858.
Panossian B, Salloum T, Araj GF, Khazen G, Tokajian S. First insights on the genetic diversity of MDR Mycobacterium tuberculosis in Lebanon. BMC Infect Dis. 2018;18:710.
Engström A, Antonenka U, Kadyrov A, Kalmambetova G, Kranzer K, Merker M, et al. Population structure of drug-resistant Mycobacterium tuberculosis in Central Asia. BMC Infect Dis. 2019;19:908.
Wollenberg KR, Desjardins CA, Zalutskaya A, Slodovnikova V, Oler AJ, Quiñones M, et al. Whole-genome sequencing of Mycobacterium tuberculosis provides insight into the evolution and genetic composition of drug-resistant tuberculosis in Belarus. J Clin Microbiol. 2017;55(2):457–69.
Stucki D, Brites D, Jeljeli L, Coscolla M, Liu Q, Trauner A, et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet. 2016;48(12):1535–43.
Acknowledgements
We extended our gratitude to the University of Gondar for awarding the Mega Project Grant. We also appreciate the scholarship funding of The Ohio State University, Global One Health initiative to Agumas Shibabaw. A special thanks to Michigan Department of Health and Human Services for genotyping support and educating Agumas on advanced molecular genotyping methods. Last but not least, we thank all of the study participants for participation and staffs at the MDR/RR-TB treatment center hospitals and TB culture laboratories in the Amhara region for their assistance in collecting the clinical and laboratory data from MDR-TB patients.
Funding
This research was funded by University of Gondar (Ethiopia) and Global One Health Initiative, The Ohio State University (USA) for data collection and genotyping works. The funders had no role in the design, data collection, analysis and interpretation, decision to publish and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
AS conceived and designed the experiment, collected data and experimental samples, analyzed the sequenced and overall data, and wrote the original manuscript; BG, BT, SHW and WG designed, supervised, analyzed the data, and revised the manuscript; LCMS, NL and MG analyzed the sequenced data and revised the manuscript; AMS and MKS revised the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was obtained from University of Gondar Ethical Review Board (Ref. No: O/V/P/RCS/05/19/2016) and all experiments in human participants were performed according to the Declaration of Helsinki guidelines and regulations. All data were collected from each study participant after obtaining written informed consent (written form) to participate in the study. Material transfer agreement was made between The Ohio State University, USA and University of Gondar, Ethiopia for shipment of research materials (extracted DNA samples) for sole purpose of genotyping research to Columbus, Ohio. All left over DNA samples were discarded accordingly. Data confidentiality was maintained using study codes for each participant.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material
: Fig S1: Minimum spinning tree based on the 24-loci MIRU-VNTR typing data of 43 M. tuberculosis isolates in the Amhara region, generated by MIRU-VNTRplus website (minimum size: two VNTR types). The size of each circle is proportional to the number of MIRU-VNTR types belonging to a particular complex. Classification of the isolates into the different phylogenetic lineages is visualized by color coding. Fig S2: A neighbor joining tree showing relatedness between 41 isolates based on the M. tuberculosis complex cgMLST v2.1; 2891 targets. M. tuberculosis H37Rv genome was used to root the tree. Samples were grouped by color according to their cgMLST complex type.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shibabaw, A., Gelaw, B., Ghanem, M. et al. Molecular epidemiology and transmission dynamics of multi-drug resistant tuberculosis strains using whole genome sequencing in the Amhara region, Ethiopia. BMC Genomics 24, 400 (2023). https://doi.org/10.1186/s12864-023-09502-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09502-2