
Abstract
Background
Pueraria montana var. lobata (kudzu) is an important food and medicinal crop in Asia. However, the phylogenetic relationships between Pueraria montana var. lobata and the other two varieties (P. montana var. thomsonii and P. montana var. montana) remain debated. Although there is increasing evidence showing that P. montana var. lobata adapts to various environments and is an invasive species in America, few studies have systematically investigated the role of the phylogenetic relationships and evolutionary patterns of plastomes between P. montana var. lobata and its closely related taxa.
Results
26 newly sequenced chloroplast genomes of Pueraria accessions resulted in assembled plastomes with sizes ranging from 153,360 bp to 153,551 bp. Each chloroplast genome contained 130 genes, including eight rRNA genes, 37 tRNA genes, and 85 protein-coding genes. For 24 newly sequenced accessions of these three varieties of P. montana, we detected three genes and ten noncoding regions with higher nucleotide diversity (π). After incorporated publically available chloroplast genomes of Pueraria and other legumes, 47 chloroplast genomes were used to construct phylogenetic trees, including seven P. montana var. lobata, 14 P. montana var. thomsonii and six P. montana var. montana. Phylogenetic analysis revealed that P. montana var. lobata and P. montana var. thomsonii formed a clade, while all sampled P. montana var. montana formed another cluster based on cp genomes, LSC, SSC and protein-coding genes. Twenty-six amino acid residues were identified under positive selection with the site model. We also detected six genes (accD, ndhB, ndhC, rpl2, rpoC2, and rps2) that account for among-site variation in selective constraint under the clade model between accessions of the Pueraria montana var. lobata clade and the Pueraria montana var. montana clade.
Conclusion
Our data provide novel comparative plastid genomic insights into conservative gene content and structure of cp genomes pertaining to P. montana var. lobata and the other two varieties, and reveal an important phylogenetic clue and plastid divergence among related taxa of P. montana come from loci that own moderate variation and underwent modest selection.
Similar content being viewed by others
Background
Pueraria montana var. lobata (Willd.) Sanjappa & Pradeep (2n = 2x = 22) is a semi-woody, perennial liana, which belongs to the Leguminosae family. It is native to Southeast Asia and introduced in Africa, America, and Europe [1]. As an economic crop, it contains puerarin and other functional components and is used in the production of both pharmaceuticals and health foods [2]. Its dried rhizome is listed in the Chinese Pharmacopeia as kudzu [3]. Kudzu was introduced from Asia to America about 150 years ago for soil erosion control and cattle feed, but later became a highly invasive species [4].
Due to considerable morphological diversity, complex domestication history, and similar health and cosmetic benefits, Pueraria montana var. lobata and the other two varieties faced a protracted period of taxonomic uncertainty. The dried rhizome of Pueraria montana var. thomsonii (Benth.) Wiersema ex D.B. Ward (common name “Fenge” in Chinese) is usually used as a vegetable and for extracting starch, while Pueraria montana var. montana (Willdenow) Maesen & S. M. Almeida ex Sanjappa & Predeep (common name “Gemamu” in Chinese) is confusedly used medicinally and sometimes as a cover crop and fodder [5]. Some taxonomic questions on P. montana var. lobata and the other two varieties are up for debate because of the phenomena of homonym, unscientific classification, genetic relation confusion, and transitional states of highly variable traits. The synonyms or some combinations of P. montana var. lobata in the different studies complicated the species delimitation on account of variation in leaf shape, inflorescence and flower size, and geography by different authors (Table S1), which may have further hindered the classification of P. montana var. lobata and the other two varieties. Several researchers consider that these taxa of P. montana were one species with three varieties [5,6,7,8], while Ohashi et al. [9] considered that they should be treated as two species. Pueraria montana var. lobata and the other two varieties share the following diagnostic characteristics [5]: robust climbers with tuberous roots, dorsifixed stipules entire to fringed at the base, trifoliolate, and flowers clustered at each node of the rachis (Fig. 1, Fig. S1, Table S2). The previous study focusing on the samples located in Taiwan showed that P. lobata (= P. montana var. lobata) and P. montana (= P. montana var. montana) are single species, while P. lobata subsp. thomsonii (Benth.) Ohashi and Tateishi is considered the subspecies of P. lobata (= P. montana var. lobata) based on the leaflets, the relative length between inflorescence and leaves, and the relative length between the wing and keel-petal [9]. The morphology of leaflets from entire to trilobed had no quantitative standard among these three varieties we observed in the field (Fig. S1), as well as the width and the length. Furthermore, the range of the length of the pedicel, bracteole, flower, and calyx overlapped among these taxa [6, 9]. For instance, the length of the pedicel of P. lobata subsp. lobata (= P. montana var. lobata), P. lobata subsp. thomsonii (= P. montana var. thomsonii) and P. montana (= P. montana var. montana) are 2–6 mm, 3–7 mm, and 1.5-4 mm, respectively [9]. Especially, the intermediate morphology of quantitative traits (i.e. the width and length of apex leaflets, the length of pedicel) between P. montana var. thomsonii and P. montana var. lobata reveal that it may be questionable to classification and rank of P. montana merely based on the morphological characteristics (Table S2) [7, 9]. For example, P. montana var. lobata and P. montana var. thomsonii shared the pedicel with white short spreading hairy [9].

Morphological diversity of Pueraria montana var. lobata and the other two varieties in this study. (A)-(B)Pueraria montana var. lobata (YG_127); (C)-(D)Pueraria montana var. thomsonii (FG_43); (E)-(H)Pueraria montana var. montana (GMM_96 and GMM_94). The code of the individual is consistent with Table 1
Molecular phylogenies are often incongruent with morphologically-based classification schemes, leading to difficulty in understanding the evolution of morphological traits in many crops involving domestication. Several molecular markers have been used to reconstruct a phylogenetic frame of Pueraria or legume species, but the phylogenetic relationships between P. montana and closely related species are still disputed due to morphological intermediacy and varying molecular markers [7, 10,11,12,13,14,15,16,17,18,19,20]. In previously published works, Pueraria montana var. lobata and the other two varieties formed a clade and nested within Pueraria sensu stricto based on two cpDNA markers, while they formed a polytomy based on one nuclear marker (AS2) [16]. Pueraria montana var. montana should be considered as a single species, and P. montana var. thomsonii sister to P. montana var. lobata based on nrITS sequences involving four accessions of P. montana and its varieties [11]. Pueraria montana var. thomsonii, P. montana var. lobata, and P. montana var. montana clustered within a monophyletic clade but relationships among these taxa are less resolved based on nrITS sequences of 11 accessions of Pueraria [15]. Therefore, insufficiently informative characters and sparse representative accessions in sampling had been the problems for investigating the phylogeny between P. montana var. lobata and closely related taxa.
Full plastome sequencing is useful in resolving complex evolutionary relationships in closely related species due to more informative sites and lower nucleotide substitution rates [21,22,23,24,25]. Moreover, accumulating studies showed that positive selection may be critical for the evolution of genes in plastomes, even if the structure and sequence of plastomes among closely related taxa are conservative [26,27,28,29,30,31,32,33]. Pueraria montana var. lobata is distributed widely and become an invasive plant due to the breeding system, favorable climatic conditions, and human-mediated movement [34, 35]. Given the many important biological processes in plastids, including photosynthesis, the assimilation of sulfur and nitrogen, and phytohormones, it is to be expected that the plastome would be diversified or adapted to a specific environment due to natural selection [36].
Here, we sequenced twenty-six chloroplast genomes for P. montana var. lobata and its allies to explore plastid diversity in the genetic background of kudzu vine that originated from different geographical regions. We aim to (1) establish a phylogenetic framework for P. montana var. lobata and closely related taxa; and (2) compare the structural variation and positive selection of the chloroplast genomes of P. montana var. lobata and closely related taxa.
Results
General features of Plastomes
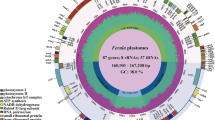
We sequenced and assembled twenty-six plastomes from P. montana and its allies (Table 1, Table S3). All newly sequenced genomes showed typical quadripartite structure, ranging from 153,360 bp (P. montana var. montana_GMM_112) to 153,551 bp (P. edulis_SYG_78) (Fig. 2), which was similar to previously published complete chloroplast genomes of P. montana var. lobata and P. montana var. thomsonii [18,19,20]. The GC content of these plastomes was 35.4%, except for P. mirifica A. Shaw and Suvat that was 35.5%. We identified 112 genes comprising four ribosomal RNAs, 30 transfer RNAs, and 78 protein-coding genes. Seven protein-coding genes, four rRNAs, and seven tRNAs are duplicated in the inverted repeat regions, and thus each chloroplast genome harbored 130 genes in total (Table S4). The size of the inverted repeat (IR), large single copy (LSC) and small single copy (SSC) regions ranged from 25,632 bp (P. montana var. montana_GMM_112) to 25,684 bp (P. mirifica_TG_79), 84,077 bp (P. mirifica_TG_79) to 84,248 bp (P. edulis_SYG_78), and 17,971 bp (P. mirifica_TG_79) to 18,005 bp (P. montana var. montana_GMM_112), respectively.

Gene maps of chloroplast genomes of Pueraria accessions in the present study. Genes outside the circles are transcribed in counter clockwise direction and those inside in counterclockwise direction. The colored bars indicate known protein-coding genes, transfer RNA genes, and ribosomal RNA genes. The dashed, dark grey area in the inner circle denotes GC content, and the light grey area indicates genome AT content. LSC indicates large single copy; SSC, indicates small single copy and IR, indicates inverted repeat
The mVISTA analysis revealed that the chloroplast genomes of P. montana var. lobata and closely related taxa were conserved generally with a few variable regions (Fig. S2). The proportion of variable sites was higher in the LSC regions than the SSC and IR regions, and higher in the intergenic spacers than the coding regions. We also found there was low variability of adjacent genes in the IRb/SSC and SSC/IRa boundaries of P. montana var. lobata and the other two varieties (Fig. S3). We did not detect any inversions and large indels either within the varieties of P. montana or in comparison to the P. edulis and P. mirifica (Fig. S4). The range of nucleotide diversity was 0-0.00191, with an average of 0.07839. Three genes had a higher π: ndhJ, ycf1 and rps15, and the corresponding noncoding regions trnK-rbcL, trnG-psbZ, psbD-trnT, trnD-psbM, petN-trnC, psbK-trnQ, rps16-accD, psbB-psbT, rps3-rps19, and ccsA-ndhA (Fig. S5).
Plastome phylogenomics
The multiple sequence alignment of all 47 plastomes (including both newly generated and previously published plastomes) was 154,176 bp after excluding one of the inverted repeat regions. The best-fit model for the chloroplast genomes, SSC, IR, LSC, and coding sequences was listed in Table 2. The aligned sequences of the five sequence datasets were little saturated and were thus useful for phylogenetic analyses (Table S5). The topologies estimated from the 47 complete chloroplast sequences (Fig. 3) were broadly similar to those estimated using the other four datasets (Fig. S6-S9), where two major clades of the accessions of P. montana var. lobata and closely related taxa were identified. The monophyly of the Pueraria montana var. lobata clade including all the individuals of P. montana var. lobata and P. montana var. thomsonii was strongly supported in all phylogenetic analyses. All sampled individuals of P. montana var. montana were monophyletic in LSC, SSC and protein-coding sequences dataset and paraphyletic in IR dataset with low support. Thus, we limited the discussion to phylogenetic trees estimated using chloroplast genomes (Fig. 3).

Phylogenetic tree reconstructed based on the 47 complete plastome sequences. Tree topology was based on BI analysis. Numbers at the nodes represent BI posterior probability (PP) / ML bootstrap (MLB) values. PP or MLB values lower than 0.5 or 50% were indicated by hyphens
Pueraria mirifica (PP/MLB = 1.00/80) and P. edulis (PP/MLB = 1.00/100) was sister to the varieties of P. montana with strong support, respectively. Pueraria montana var. lobata and the other two varieties are divided into two clades. The Pueraria montana var. montana clade, containing all accessions of P. montana var. montana, showed high support (PP/MLB = 1.00/100). The Pueraria montana var. lobata clade, which includes all accessions of P. montana var. lobata and P. montana var thomsonii, is a monophyletic lineage with strong support (PP/MLB = 1.00/100), but the resolution among the accessions within this clade was relatively low.
Topology testing
Since the tree topologies of the major clades based on the taxon removal experiments showed no changes when outgroups were removed (Fig. S10), we concluded that the tree topology was not affected by long-branch attraction. The incompletely resolved relationships in the network suggested substantial conflicting signals (Fig. 4). All accessions were sorted into respective clades, and the clades were almost distinct. However, the relationships within the Pueraria montana var. montana clade were less clear.

Neighbor-net analyses of Pueraria accessions based on 27 plastome sequences. Clades revealed are labeled with different colors
Positive selection
Pueraria montana var. lobata and the other two varieties from different productive areas were divided into two clades. The branch model detected only one gene (ycf2) with a significantly decelerated rate of evolution (ω = 0.62093, p < 0.001). We detected one gene (ndhB, one site) under positive selection at one amino-acid site (p < 0.001, BEB value > 0.95) in the analyses of the branch-site models, taking the Pueraria montana var. lobata clade as the foreground clade. However, no signals of positive selection were detected in the Pueraria montana var. montana clade in either model. Because chloroplast genes tend to be conservative, we compared the fit of the Clade model C (CmC) with Likelihood Ratio Tests (LRT) to detect subtle differences in specific selective constraints between the Pueraria montana var. lobata clade and Pueraria montana var. montana clade. Our CmC analysis showed that accessions of the Pueraria montana var. lobata clade exhibited significant relaxation in six genes (accD, ndhB, ndhC, rpl2, rpoC2, rps2), compared to accessions of the Pueraria montana var. montana clade (Table S6). 26 amino acid residues were identified as having a sign of positive selection in the analyses of the Site model, which predicted that 36 sites greater than 0.95 and 33 sites greater than 0.99 (Table S7) of all legume taxa.
Discussion
Phylogenetic inference
Previous studies revealed that Pueraria s.l. is a polyphyletic group since several lineages could be treated as five distinct genera, namely Pueraria s.s., Teyleria stricta (Kurz) (A) N. Egan & (B) Pan, Neustanthus Bentham, Haymondia A.N. Egan & B. Pan, and Toxicopueraria A.N. Egan & B. Pan based on several key morphological and phylogenetic studies. The remaining species include Pueraria tuberosa (Roxb. ex Willd.) DC. grouped into Pueraria s.s. [7, 10,11,12,13,14,15,16]. The infrageneric classification of the Pueraria s.s. is still largely in dispute, and it is an intricate challenge for taxonomic species, subspecies, or varieties as many synonyms are reported for a single species. For example, Pueraria edulis is treated as the kudzu vine in the southwestern region of China such as Sichuan and Yunnan provinces [37]. In previous works, the phylogenetic tree based on two cpDNA markers or one nuclear maker (AS2) showed that P. montana var. lobata and the other two varieties formed a subclade and nested within Pueraria s.s. with other closely related Pueraria species [16]. Our plastomic data support the previous findings that Haymondia wallichii diverges from Pueraria taxa and demonstrate P. edulis and P. mirifica are separated from three varieties of P. montana, then further revealing that P. montana var. lobata and the other two varieties were divided into two clades, of which one was comprised of P. montana var. lobata and P. montana var. thomsonii, and the other clade included all sampled P. montana var. montana.
For the most part, our phylogenetic results revealed that the Pueraria montana var. montana clade is resolved as a monophyletic group with strong support except for phylogenetic analysis based on IR regions. The phylogenetic analysis based on nrITS sequences showed that P. montana var. montana formed a monophyletic clade sister to the other two varieties, which suggested that P. montana var. montana should be treated as a single species [11, 15, 17]. Pueraria montana var. lobata and P. montana var. thomsonii were different from P. montana var. montana in aspects of the morphology of leaflets, the relative length between inflorescence and leaves, the length and hairiness of the pedicel, the relative length between the wing and keel-petal (Table S2) [9, 10]. Furthermore, the composition and abundance of secondary metabolites also showed significant differences in P. montana var. montana compared to P. montana var. lobata and P. montana var. thomsonii [38].
All our phylogenetic analyses support the sister relationship of P. montana var. lobata and P. montana var. thomsonii with strong support but the relationship among accessions are less resolved. The uncertain association between P. montana var. lobata and P. montana var. thomsonii was also revealed in previous studies [9, 10]. Although these two varieties were having high medicinal values, there are considerable differences in terms of the metabolites, material basis, and efficacy [38,39,40,41]. Morphological intermediacy, insufficiently informative characters, artificial domestication, species complex and putative hybridization/introgression contribute to the lack of agreement between morphological-based classification schemes and recent molecular phylogenies among P. montana var. lobata and closely related taxa [11, 15,16,17]. Further studies of Pueraria species should focus on other markers to achieve better phylogenetic resolution, such as RAD-seq or transcriptome.
Conservation and adaptation of kudzu plastomes
Our results reveal no obvious expansion or contraction of IR region, indels and inversion in the chloroplast genome between P. montana var. lobata and closely related taxa, even compared to the outgroup species of P. mirifica. The results of the mVISTA analysis showed variable intergenic regions in the LSC region. Lower sequence diversity existed in the SSC and IR regions of 24 newly sequenced three varieties of P. montana. The nine noncoding regions (trnK-rbcL, trnG-psbZ, psbD-trnT, trnD-psbM, petN-trnC, psbK-trnQ, rps16-accD, psbB-psbT, and rps3-rps19) and one gene (ndhJ) were detected as relatively divergent in the LSC region, while two genes (rps15, ycf1) and one noncoding region (ccsA-ndhA) were detected in SSC region. These variable regions identified in the present study could be potential markers for phylogenetic or population genetics studies in P. montana var. lobata and its related taxa.
Pueraria species are delineated as climbing vines. Pueraria montana var. lobata (kudzu) inhabits temperate environments or higher altitudes in the tropics [7] and prefers to occur in high-light forest edge areas, even though it can live under both shade and sun [34]. Thus, Pueraria montana var. lobata has been widely found in its native area, and probably introduced elsewhere [7]. The ecology, climatic considerations, growth and establishment pattern, cultivation and genetic divergence may influence the distribution of P. montana var. lobata, even other Pueraria plants [42]. Interestingly, neither similar environment nor genetics seems to be a major factor in the distribution of kudzu, as is the case in the United States and Switzerland [42]. The ecology and genetics of P. montana var. lobata in Southern Switzerland is similar to that in the South-Eastern USA [42]. Pueraria montana var. lobata is not expected to spread on a large scale in Southern Switzerland [42, 43], while it was officially listed as a Federal Noxious Weed due to widespread ecological and economic impacts in the southeastern USA [34, 44]. For example, its invasion has the potential to raise ozone pollution in the southeastern United States [45]. To clarify the adaptation or differentiation of P. montana var. lobata and its closely related taxa, our data provide novel evidence of positive selection of genes under four models from the perspective of the chloroplast genome, as well as in Panax [27], and Rhodiola [31].
The clade model detected the variation in accD, ndhB, ndhC, rpl2, rpoC2, rps2 as being significantly different from the accessions of the Pueraria montana var. montana clade. The plastid accD gene encodes a key enzyme for fatty acid biosynthesis, namely a beta subunit of acetyl-CoA carboxylase, which is important for leaf development [46,47,48]. The ndh genes in plastomes encode a protein act by adjusting the electron transfer from NADH to photosystem I [31, 48]. The gene rpl2 encodes the ribosomal protein L2 of 80 S ribosomes, which is critical for the peptidyl transferase activity [49]. The gene rpoC2 encodes RNA polymerase subunits and the gene rps2 encodes the ribosomal protein, but it is unclear their specific function in the evolution of the plastome of Pueraria.
The site model detected 26 amino acid residues across all sampled species. Using the Pueraria montana var. lobata clade as the foreground, the branch model revealed only ycf2 may have undergone negative selection. ycf2, as the largest coding sequence in plastome and conserved open reading frames, may exhibit similarities with fstH, such as activity associated with cell division, ATPase, and chaperone function [50, 51]. We also detected one possible positively selected gene (ndhB) using the branch-site model, which may be involved in photosynthesis and temperature sensitivity [52, 53]. However, taking the Pueraria montana var. montana clade as the foreground, both the branch and branch-site models failed to detect the positive genes.
The combination of key traits such as relatively high photosynthetic rates, rapid leaf movements, and the ability to fix N2 make P. montana var. lobata an aggressive competitor [34]. Thus, our results propose that the positive selection of these genes, involving leaf development or photosynthesis, may play an important role in adaptation to different habitats in kudzu or other kudzu species, which may influence their plastid differentiation pattern. This adaptation of kudzu and its related taxa occurred when their common ancestor diverged from other Pueraria species.
Conclusions
In this study, we reported the chloroplast genomes for 26 Pueraria accessions. Through the comparisons of these newly sequenced chloroplast genomes to additional sequences from species of legume species in NCBI, we found that gene content and order of P. montana var. lobata and its closely related taxa are highly conserved. The phylogenetic results based on 47 plastomes, LSC, SSC, IR, and protein-coding sequences supported P. edulis and P. mirifica was sister to remaining Pueraria accessions respectively. The remaining Pueraria accessions were divided into two clades but could not resolve the further division within these two clades. We also detected several genes that may play the important role in the evolution of these two clades. Overall, these results demonstrated the power of chloroplast genomes in solving the phylogenetic relationship, as well as the variation of chloroplast genomes between P. montana var. lobata and its closely related species. Future work with more taxa sampling and markers will help to illustrate gene introgression, ILS, and other biological factors that may play various roles among lineages of P. montana var. lobata and P. montana var. montana.
Methods
Taxon sampling, DNA extraction and sequencing
Three to five fresh leaf samples from each individual were collected in their native habitats and dried in silica gel and stored at room temperature for DNA extraction. Then we selected these accessions from representative productive areas. Twenty-six accessions, including P. montana var. lobata and the other two varieties, were selected for Illumina sequencing. Pueraria mirifica Airy Shaw & Suvat. and Pueraria edulis Pampan. were also sequenced as outgroups. All vouchers were deposited in the Guangxi Academy of Agricultural Sciences (GAAS, Nanning, China) (Table 1). The samples of Pueraria accessions were identified by Hua-Bing Yan and Xiao-Hong Shang in the field. Silica-dried leaf material was sent to the Novogene Bioinformatics Technology Co. Ltd. (Tianjin, China) for library preparation and Illumina sequencing on NovaSeq 6000. A paired-end library (150 × 2) was constructed with an insert size of 350 base pairs (bp). Total genomic DNA was isolated using a DNeasy Plant Mini Kit (Qiagen, CA, United State).
Sequence assembly and annotation
The raw data were first filtered using fastp to remove those paired reads that contained more than 10% of N bases, and more than 50% of bases with low Phred quality (Q ≤ 5), and adapter sequences. The clean data were mapping assembly to create initial reference sequences using MIRA v4.0.2 [54]. These newly created reference sequences were assembled as the complete plastome sequences using MITObim v1.8 [55]. The previously published complete plastome sequences of P. montana var. lobata (GenBank nos., MT818508 and MN180247) and P. montana var. thomsonii (GenBank no., MN515038) were used as seed references. These assemblies were manually remapped and inspected using Geneious v. R9.0.2 [56] under default parameters to check for misassemblies. The tRNAscan-SE ver. 1.21 software [57] was applied to verify the tRNA genes with default parameters. Gene annotations were assigned using DOGMA [58]. Inverted repeat boundaries were determined using the BLAST method [59] (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and verified using Geneious v. R9.0.2. The circular maps of newly sequenced chloroplast plastomes were drawn using the online program OGDRAW v. 1.2 [60].
Identification of the hypervariable regions
To better identify possible rearrangements, five newly sequenced chloroplast genomes of P. montana var. lobata and closely related taxa were compared using the global alignment algorithm Shuffle-LAGAN [61] with mVISTA (https://genome.lbl.gov/vista/mvista/submit.shtml), with plastome of P. mirifica was chosen as the reference genomes to detect the gene content and structural variation between P. montana var. lobata and the other two varieties. To further identify specific patterns of structural variation at the species level, five chloroplast genomes were used as the representatives to align with default parameters using MAUVE plugin [62] in Geneious v R9.0.2. We also detected the contraction and expansion of the boundaries between SC and IR regions using IRscope [63]. The nucleotide diversity (π) analysis of 24 newly sequenced plastomes of three varieties of P. montana was implemented using DnaSP v. 6.12.03 [64] with window length set to 500 bp, and the step size set as 200 bp.
Phylogenetic analyses
To better determine the phylogenetic position of P. montana var. lobata and other varieties, we reconstructed a phylogeny using 47 chloroplast genomes, including the representatives of the major subclades of legume species referring to the previous studies [16, 18,19,20]. Twenty-one previously published plastomes were downloaded from the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/) (Table S8). The phylogenetic analyses were conducted based on the following five datasets: (1) the complete chloroplast genome sequences that excluded one copy of the inverted repeat (IR) region; (2) the large single-copy region (LSC); (3) the small single-copy region (SSC); (4) one inverted repeats region (IR), and (5) 73 protein-coding genes excluding genes infA, rpl33, rps16, ycf1, ycf4 that were lost in several outgroups (Table S8). These regions were aligned in Geneious v R9.0.2 using Mauve progressive using default options. DAMBE v6.4.29 was used to assess substitution saturation [65, 66]. Phylogenetic analyses were inferred using both maximum likelihood (ML) and Bayesian inference (BI) via the CIPRES Science Gateway server (http://www.phylo.org/) [67]. ML analysis using rapid bootstrapping and 1000 replications was performed in RAxML [68] with the GTR-GAMMA model. The appropriate model for each dataset was determined using jModelTest v2.1.4 [69]. For the BI analysis, two independent Markov chain Monte Carlo (MCMC) runs with three heated and one cold chain were executed using MrBayes v3.2.7 [70]. Trees were sampled every 1000 generations. The first 25% of trees were discarded as burn-in and the remaining samples were then summarized and a majority-rule consensus tree was constructed. Two million generations were run for the IR dataset and data-complete in the taxon removal experiments dataset, whereas three million generations were run for the other four datasets (Table 2). The stationarity was considered to be reached when the average standard deviation of split frequencies fell below 0.01. We also determined the convergence of runs using Tracer v 1.6 [71] to assess the effective sample size (ESS) > 200 for all parameters.
Topology hypothesis testing
Taxon removal experiments can aid in the investigation of whether distantly related outgroups have a biased attraction to long branches within the ingroups [72,73,74]. To test if outgroup selection affected the topology of the ingroups, we recovered phylogenetic trees by removing outgroups to include only 30 accessions of P. montana var. lobata and its closely related taxa. Haymondia wallichii (DC.) (A) N. Egan & (B) Pan bis was chosen as the outgroup based on similar morphology and relatedly phylogenetic relationship with Pueraria taxa in our analysis. To better visualize and evaluate the phylogenetic signal conflicts between ingroup taxa, the neighbor-net algorithm based on uncorrected P-distances was implemented in SplitsTree v4.14.4 [75], excluding all outgroup taxa to leave P. montana var. lobata and the other two varieties.
Positive selection analysis
We chose the ML tree based on 47 chloroplast genomes to identify positively selected genes in the EasyCodeML [76] with the likelihood ratio test (LRT) since the topology of ML trees and BI trees were congruent. Because the genes infA, rpl33, rps16, ycf1 and ycf4 were lost in several legume species (Table S8), we selected 73 protein-coding genes for further analysis. The amino acid sequences of the 73 protein-coding genes were aligned using MAFFT and converted nucleotide alignments into PAML format in EasyCodeML according to the manual [76]. Four different models (i.e. site model, branch model, branch-site model, and clade model) were used to test the selection pressure. We set two clades to detect positive evolution at the clade level: one is the Pueraria montana var. lobata clade and the other is the Pueraria montana var. montana clade using the other three model selections, namely branch model, branch-site model and clade model. Firstly, we used a branch model to identify rapidly evolving genes. Genes with p < 0.05 and ω > 1 are considered under positive selection, while others with p < 0.05 and 0 < ω < 1 are considered under purifying selection. The branch-site models were combined with heterogeneous ω across sites and branches to find instances of neutral evolution and negative selection. The clade model was performed to identify subtler differences in site-specific selective constraint among partitions of a phylogeny [77].
Data Availability
The assembled sequences described in this study have been deposited in the National Center for Biotechnology and Information (NCBI) under the accessions as summarized in Table 1. The phylogenetic genome datasets used and analyzed in this study were retrieved from the National Center for Biotechnology Information Organelle Genome Resource Database.
Abbreviations
- BI:
-
Bayesian interference
- IR:
-
Inverted repeat
- LRT:
-
Likelihood ratio test
- LSC:
-
Long single copy
- LSR:
-
Long sequence repeat
- MCMC:
-
Markov chain Monte Carlo
- ML:
-
Maximum likelihood
- PP:
-
Posterior probability
- NCBI:
-
National Center for Biotechnology Information
- π:
-
nucleotide diversity
- rRNA:
-
ribosomal RNA
- SSC:
-
Short single copy
- tRNA:
-
transfer RNA
References
Haynsen MS, Vatanparast M, Mahadwar G, Zhu D, Moger-Reischer RZ, Doyle JJ, et al. De novo transcriptome assembly of Pueraria montana var. lobata and Neustanthus phaseoloides for the development of eSSR and SNP markers: narrowing the US origin(s) of the invasive kudzu. BMC Genomics. 2019;19:439. https://doi.org/10.1186/s12864-018-4798-3.
Wang S, Zhang S, Wang S, Gao P, Dai L. A comprehensive review on Pueraria: insights on its chemistry and medicinal value. Biomed Pharmacother. 2020;131:110734. https://doi.org/10.1016/j.biopha.2020.110734.
China Pharmacopoeia Committee. Pharmacopoeia of the People’s Republic of China Part 1. Beijing: China Medical Science and Technology Press; 2020. p. 347.
ISSG. 100 of the world’s worst invasive alien species. Pueraria montana var. lobata. http://www.issg.org/database/species/search.asp?st=100ss&fr=1&sts= 2013.
Wu TL, Pueraria DC. In: Li SK ed. Flora Reipublicae Popularis Sinicae. Beijing: Science Press. 1995; 41: 219–229.
Wu DL, Thulin M. Pueraria. Flora of China. 2020;10:244–8.
van der Maesen LJG. Revision of the genus Pueraria DC. with some notes on Teyleria Backer (Leguminiosae). Ph.D. Dissertation. Wageningen: Agricultural University. 1985; pp. 5–13.
van der Maesen LJG. Pueraria: botanical characteristics. In: Keung WM, editor. The genus Pueraria. London: Taylor & Francis; 2002. pp. 1–30.
Ohashi H, Tateishi Y, Nemoto T, Endo Y. Taxonomic studies on the Leguminosae of Taiwan III. Sci Rep Tohoku Univer 4th Ser (Biol). 1988;39:191–248.
Lackey JA. A synopsis of the Phaseoleae (Leguminosae, Papilionoideae). Ph.D. Dissertation. Iowa: Iowa State University. 1977; pp. 293.
Zeng M, Ma YJ, Zheng SQ, Xu JF, Qiu XH. Studies on ribosomal DNA sequence analyses of Radix Puerariae and its sibling species. Chin Pharmacol J. 2003;38(3):173–5.
Lee J, Hymowitz T. A molecular phylogenetic study of the subtribe Glycininae (Leguminosae) derived from the chloroplast DNA rps16 intron sequences. Am J Bot. 2001;88:2064–73.
Stefanovic S, Pfeil BE, Palmer JD, Doyle JJ. Relationships among phaseoloid legumes based on sequences from eight chloroplast regions. Syst Biol. 2009;34:115–28. https://doi.org/10.1600/036364409787602221.
Cagle W. Parsing polyphyletic Pueraria: delimiting distinct evolutionary lineages through phylogeny. Ph.D. Dissertation. North Carolina: East Carolina University. 2013.
Jiang XH, Liu LK, She CW. Genetic analysis of 11 Pueraria species based on nrITS sequence. Jiangsu Agr Sci. 2015;43(7):46–9.
Egan AN, Vatanparast M, Cagle W. Parsing polyphyletic Pueraria: delimiting distinct evolutionary lineages through phylogeny. Mol Phylogenet Evol. 2016;104:44–59. https://doi.org/10.1016/j.ympev.2016.08.001.
Jiang XH, Liu LK, She CW. Study on classification consistency based on morphology and rDNA ITS sequences of Pueraria species. Hubei Agr Sci. 2016;55(4):939–42.
Miao XR, Niu JQ, Wang AQ, Wang DB, Fan J. Complete chloroplast genome sequence of Pueraria thomsonii, an important traditional chinese medicine plant. Mitochondrial DNA B. 2019;4(2):4163–5. https://doi.org/10.1080/23802359.2019.1693301.
Cao TX, Ma XL, Zhang Y, Su WZ, Li BC, Zhou QH, et al. The complete chloroplast genome sequence of the Pueraria lobata (Willd.) Ohwi (Leguminosae). Mitochondrial DNA B. 2020;5(3):3754–6. https://doi.org/10.1080/23802359.2020.1835576.
Sun JH, Wang L, Yuan Y, Zhou RR, Zhao YP. Complete chloroplast genome sequence of Pueraria lobata (Willd.) Ohwi (Fabaceae): a traditional chinese medicinal herb. Mitochondrial DNA B. 2020;5(1):25–6. https://doi.org/10.1080/23802359.2019.1694850.
Ye JF, Niu YT, Liu B, Hai LS, Wen J, Chen ZD. Taxonomy and biogeography of Diapensia (Diapensiaceae) based on chloroplast genome data. J Syst Evol. 2020;58(5):696–709. https://doi.org/10.1111/jse.12597.
Chang H, Zhang L, Xie HH, Liu JQ, Xi ZX, Xu XT. The conservation of chloroplast genome structure and improved resolution of infrafamilial relationships of Crassulaceae. Front Plant Sci. 2021;12:631884. https://doi.org/10.3389/fpls.2021.631884.
Guo MY, Pang XH, Xu YQ, Jiang WJ, Liao BS, Yu JS, et al. Plastid genome data provide new insights into the phylogeny and evolution of the genus Epimedium. J Adv Res. 2021;36:175–85. https://doi.org/10.1016/j.jare.2021.06.020.
Nguyen HQ, Nguyen TNL, Doan TN, Nguyen TTN, Pham MH, Le TL, et al. Complete chloroplast genome of novel Adrinandra megaphylla Hu species: molecular structure, comparative and phylogenetic analysis. Sci Rep. 2021;11:11731. https://doi.org/10.1038/s41598-021-91071-z.
Yan LJ, Zhu ZG, Wang P, Fu CN, Guan XJ, Kear P et al. Comparative analysis of 343 plastid genomes of Solanum section Petota: insights into potato diversity, phylogeny and species discrimination. J Syst Evol. 2022.
Bock DG, Andrew RL, Rieseberg LH. On the adaptive value of cytoplasmic genomes in plants. Mol Ecol. 2014;23:4899–911. https://doi.org/10.1111/mec.12920.
Jiang P, Shi FX, Li MR, Liu B, Wen J, Xiao HX, et al. Positive selection driving cytoplasmic genome evolution of the medicinally important ginseng plant genus Panax. Front Plant Sci. 2018;9:359. https://doi.org/10.3389/fpls.2018.00359.
Ye WQ, Yap ZY, Li P, Comes HP, Qiu YX. Plastome organization, genome-based phylogeny and evolution of plastid genes in Podophylloideae (Berberidaceae). Mol Phylogenet Evol. 2018;127:978–87. https://doi.org/10.1016/j.ympev.2018.07.001.
Gao LZ, Liu YL, Zhang D, Li W, Gao J, Liu Y, et al. Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun Biol. 2019;2:278. https://doi.org/10.1038/s42003-019-0531-2.
Shrestha B, Weng ML, Theriot EC, Gilbert JE, Ruhlman TA, Krosnick SE, et al. Highly accelerated rates of genomic rearrangements and nucleotide substitutions in plastid genomes of Passiflora subgenus Decaloba. Mol Phylogenet Evol. 2019;138:53–64. https://doi.org/10.1016/j.ympev.2019.05.030.
Zhao DN, Ren Y, Zhang JQ. Conservation and innovation: Plastome evolution during rapid radiation of Rhodiola on the Qinghai-Tibetan Plateau. Mol Phylogenet Evol. 2020;144:106713. https://doi.org/10.1016/j.ympev.2019.106713.
Wang J, Fu GF, Tembrock LR, Liao XZ, Ge S, Wu ZQ. Mutational meltdown or controlled chain reaction: the dynamics of rapid plastome evolution in the hyperdiversity of Poaceae. J Syst Evol. 2022. https://doi.org/10.1111/jse.12854.
Xia MQ, Liao RY, Zhou JT, Lin HY, Li JH, Li P, et al. Phylogenomics and biogeography of Wisteria: implications on plastome evolution among inverted repeat-lacking clade (IRLC) legumes. J Syst Evol. 2022;60(2):253–65. https://doi.org/10.1111/jse.12733.
Forseth IN, Innis AF. 2004. Kudzu (Pueraria montana): history, physiology, and ecology combine to make a major ecosystem threat. Criti Rev Plant Sci. 2004; 23(5): 401–413. doi: https://doi.org/10.1080/07352680490505150.
Follak S. Potential distribution and environmental threat of Pueraria lobata. Cent Eur J Biol. 2011;6(3):457–69. https://doi.org/10.2478/s11535-010-0120-3.
Körner C. Functional plant ecology of high mountain ecosystems. Alpine plant life. Berlin:Springer. 2003.
Wu ZG, Deng KZ, Ge F, Wu B, Zhu YY, Hu SF, et al. Herbal textual research of kudzu vine root on original plants, traditional functions, and resources distribution. J Jiangxi Univer TCM. 2020;32(1):1–4.
Shang XH, Huang D, Wang Y, Xiao L, Ming RH, Zeng WD, et al. Identification of nutritional ingredients and medicinal components of Pueraria lobata and its varieties using UPLC-MS/MS-Based metabolomics. Molecules. 2021;26:6587. https://doi.org/10.3390/molecules26216587.
Chen SB, Liu HP, Tian RT, Yang DJ, Chen SL, Xu HX, et al. High-performance thin-layer chromatographic fingerprints of isoflavonoids for distinguishing between Radix Puerariae Lobate and Radix Puerariae Thomsonii. J Chromatogr A. 2006;1121:114–9. https://doi.org/10.1016/j.chroma.2006.04.082.
Wong KH, Razmovski-Naumovski V, Li KM, Li GQ, Chan K. Differentiating Puerariae Lobatae Radix and Puerariae Thomsonii Radix using HPTLC coupled with multivariate classification analyses. J Pharmaceut Biomed. 2014;95:11–9. https://doi.org/10.1016/j.jpba.2014.02.007.
Wong KH, Razmovski-Naumovski V, Li KM, Li GQ, Chan K. Comparing morphological, chemical and anti-diabetic characteristics of Puerariae Lobatae Radix and Puerariae Thomsonii Radix. J Ethnopharmacol. 2015;164:53–63. https://doi.org/10.1016/j.jep.2014.12.050.
Gigon A, Pron S, Buholzer S. Ecology and distribution of the southeast asian invasive liana kudzu, Pueraria lobata (Fabaceae), in southern Switzerland. EPPO Bull. 2014;44(3):490–501. https://doi.org/10.1111/epp.12172.
Sun JH, Liu Z, Britton KO, Cai P, Orr D, Goldstein JH. Survey of phytophagous insects and foliar pathogens in China for a biocontrol perspective on kudzu, Pueraria montana var. lobata (Willd.) Maesen and S. Almeida (Fabaceae). Biol Control. 2006; 36: 22–31.
Blaustein RJ. Kudzu’s invasion into Southern United States life and culture. In: McNeeley JA, editor. The great reshuffling: human dimensions of invasive species. UK: Switzerland and Cambridge, The World Conservation Union, UK.; 2001. pp. 55–62.
Hickman JE, Wu SL, Mickley LJ, Lerdau MT. Kudzu (Pueraria montana) invasion doubles emissions of nitric oxide and increases ozone pollution. P Natl Acad Sci USA. 2010;107(22):10115–9. https://doi.org/10.1073/pnas.0912279107.
Kode V, Mudd EA, Iamtham S, Day A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005;44(2):237–44. https://doi.org/10.1111/j.1365-313X.2005.02533.x.
Bock R. Structure, function, and inheritance of plastid genomes. In: Bock R ed. Cell and Molecular Biology of Plastids. Berlin: Springer 2007; 19: 29–63.
Li X, Yang JB, Wang H, Song Y, Corlett RT, Yao X, et al. Plastid ndh pseudogenization and gene loss in a recently derived lineage from the largest hemiparasitic plant genus Pedicularis (Orobanchaceae). Plant Cell Physiol. 2021;62(6):971–84. https://doi.org/10.1093/pcp/pcab074.
Nagano Y, Ishikawa H, Matsuno R, Sasaki Y. Nucleotide sequence and expression of the ribosomal protein L2 gene in pea chloroplasts. Plant Mol Biol. 1991;17(3):541–5. https://doi.org/10.1007/BF00040653.
Wolfe KH. Similarity between putative ATP-binding sites in land plant plastid ORF2280 proteins and the FtsH/CDC48 family of ATPases. Curr Genet. 1994;25:379–83. https://doi.org/10.1007/BF00351493.
Machado LDO, Vieira LDN, Stefenon VM, Faoro H, Pedrosa FO, Guerra MP, et al. Molecular relationships of Campomanesia xanthocarpa within Myrtaceae based on the complete plastome sequence and on the plastid ycf2 gene. Genet Mol Biol. 2020;43(2):e20180377. https://doi.org/10.1590/1678-4685-GMB-2018-0377.
Joët T, Cournac L, Horvath EM, Medgyesy Peltier G. Increased sensitivity of photosynthesis to antimycin a induced by inactivation of the chloroplast ndhB gene. Evidence for a participation of the NADH-Dehydrogenase complex to cyclic electron flow around photosystem I. Plant Physiol. 2001;125(4):1919–29. https://doi.org/10.1104/pp.125.4.1919.
Karcher D, Bock R. Temperature sensitivity of RNA editing and intron splicing reactions in the plastid ndhB transcript. Curr Genet. 2002;41:48–52. https://doi.org/10.1007/s00294-002-0278-y.
Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced SETs. Genome Res. 2004;14:1147–59. https://doi.org/10.1101/gr.1917404.
Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 2013;41:e129. https://doi.org/10.1093/nar/gkt371.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9. https://doi.org/10.1093/bioinformatics/bts199.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. https://doi.org/10.1093/nar/25.5.955.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–5. https://doi.org/10.1093/bioinformatics/bth352.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. https://doi.org/10.1016/S0022-2836(05)80360-2.
Lohse M, Drechsel O, Kahlau S, Bock R. Organellar Genome DRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41:W575–81. https://doi.org/10.1093/nar/gkt289.
Brudno M, Malde S, Poliakov A, Do CB, Couronne O, Dubchak I, et al. Glocal alignment: finding rearrangements during alignment. Bioinformatics. 2003;19S1:i54–i62.
Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5(6):e11147.
Amiryousefi A, Hyvönen J, Poczai P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 2018;34:3030–1. https://doi.org/10.1093/bioinformatics/bty220.
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol. 2017;34:3299–302. https://doi.org/10.1093/molbev/msx248.
Xia X, Xie Z, Salemi M, Chen L, Wang Y. An index of substitution saturation and its application. Mol Phylogenet Evol. 2003;26:1–7. https://doi.org/10.1016/S1055-7903(02)00326-3.
Xia X, Lemey P. Assessing substitution saturation with DAMBE. In: Philippe L, editor. The phylogenetic handbook: a practical approach to DNA and protein phylogeny. London: Cambridge University Press; 2009. pp. 615–30.
Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE). LA: New Orleans, 2010; pp. 1–8.
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 2008;57(5):758–71. https://doi.org/10.1080/10635150802429642.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat methods. 2012;9:772. https://doi.org/10.1038/nmeth.2109.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42. https://doi.org/10.1093/sysbio/sys029.
Rambaut A, Suchard MA, Xie D, Drummond AJ. Tracer version 1.6. Available at http://beast.bio.ed.ac.uk/Tracer. Accessed December 11, 2014.
Bergsten J. A review of long-branch attraction. Cladistics. 2005;21:163–93. https://doi.org/10.1111/j.1096-0031.2005.00059.x.
Attigala L, Wysocki WP, Duvall MR, Clark LG. Phylogenetic estimation and morphological evolution of Arundinarieae (Bambusoideae: Poaceae) based on plastome phylogenomic analysis. Mol Phylogenet Evol. 2016;101:111–21. https://doi.org/10.1016/j.ympev.2016.05.008.
Zhou Y, Zhang YQ, Xing XC, Zhang JQ, Ren Y. Straight from the plastome: molecular phylogeny and morphological evolution of Fargesia (Bambusoideae: Poaceae). Front Plant Sci. 2019; 10: 981. doi: https://doi.org/10.3389/fpls.2019.00981.
Huson DH, Kloepper T, Bryant D. SplitsTree 4.0-Computation of phylogenetic trees and networks. Bioinformatics. 2008;14:68–73.
Gao FL, Chen CJ, Arab DA, Du ZG, He YH, Ho SYW. EasyCodeML: a visual tool for analysis of selection using CodeML. Ecol Evol. 2019;9(7):3891–8. https://doi.org/10.1002/ece3.5015.
Weadick CJ, Chang BS. An improved likelihood ratio test for detecting site-specific functional divergence among clades of protein-coding genes. Mol Biol Evol. 2012;29(5):1297–300. https://doi.org/10.1093/molbev/msr311.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (31960420 to X.H. Shang and 32200179 to Y. Zhou), Guangxi Natural Science Foundation Project (2021JJB130122 to Y. Zhou and 2021GXNSFBA220026 to S. Cao).
Funding
This study was supported by the National Natural Science Foundation of China (31960420 to X.H. Shang and 32200179 to Y. Zhou), Guangxi Natural Science Foundation Project (2021JJB130122 to Y. Zhou and 2021GXNSFBA220026 to S. Cao).
Author information
Authors and Affiliations
Contributions
HBY conceived and designed the experiments. LX, ZDW and SC collected materials and conducted field investigations; XHS and HBY identified the Pueraria species. YZ and XHS analyzed the data; HBY and YZ together discussed the results and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We comply with relevant institutional, national, and international guidelines and regulations for plant studies.
Consent for publication
Not applicable.
Competing interests
The authors have declared that no competing interests exist.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, Y., Shang, XH., Xiao, L. et al. Comparative plastomes of Pueraria montana var. lobata (Leguminosae: Phaseoleae) and closely related taxa: insights into phylogenomic implications and evolutionary divergence. BMC Genomics 24, 299 (2023). https://doi.org/10.1186/s12864-023-09356-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09356-8