
Abstract
Background
Early-onset renal cell carcinoma (eoRCC) is typically associated with pathogenic germline variants (PGVs) in RCC familial syndrome genes. However, most eoRCC patients lack PGVs in familial RCC genes and their genetic risk remains undefined.
Methods
Here, we analyzed biospecimens from 22 eoRCC patients that were seen at our institution for genetic counseling and tested negative for PGVs in RCC familial syndrome genes.
Results
Analysis of whole-exome sequencing (WES) data found enrichment of candidate pathogenic germline variants in DNA repair and replication genes, including multiple DNA polymerases. Induction of DNA damage in peripheral blood monocytes (PBMCs) significantly elevated numbers of \(\gamma\)H2AX foci, a marker of double-stranded breaks, in PBMCs from eoRCC patients versus PBMCs from matched cancer-free controls. Knockdown of candidate variant genes in Caki RCC cells increased \(\gamma\)H2AX foci. Immortalized patient-derived B cell lines bearing the candidate variants in DNA polymerase genes (POLD1, POLH, POLE, POLK) had DNA replication defects compared to control cells. Renal tumors carrying these DNA polymerase variants were microsatellite stable but had a high mutational burden. Direct biochemical analysis of the variant Pol δ and Pol η polymerases revealed defective enzymatic activities.
Conclusions
Together, these results suggest that constitutional defects in DNA repair underlie a subset of eoRCC cases. Screening patient lymphocytes to identify these defects may provide insight into mechanisms of carcinogenesis in a subset of genetically undefined eoRCCs. Evaluation of DNA repair defects may also provide insight into the cancer initiation mechanisms for subsets of eoRCCs and lay the foundation for targeting DNA repair vulnerabilities in eoRCC.
Similar content being viewed by others


Background
Early onset renal cell carcinoma (eoRCC) in patients under the age of 60 has been increasing in frequency over the past decade [1]. In the United States alone, the most recent analyses report a range of 3.0% annual increase in RCC incidence among individuals aged 45–49 years to as high as a 6.2% increase in incidence among those aged 25–29 years [1]. EoRCC is in some cases linked to pathogenic germline variants (PGVs) in genes associated with RCC familial syndromes (VHL, MET, FLCN, TSC1, TSC2, FH, SDHx, PTEN, BAP1) [1,2,3]; these genes are also often somatically mutated in sporadic RCC cases [2,3,4,5]. Identification of a PGV in defined RCC familial syndrome genes guides clinical recommendations for surveillance, often improving survival due to early diagnosis of eoRCC. However, in recent work we found that only ~ 3.7% of eoRCC patients undergoing cancer risk assessment report a PGV in the currently defined RCC familial syndrome genes [6], reflecting the fact that the majority of eoRCC cases remain genetically not well characterized. Currently, there are no National Comprehensive Cancer Network (NCCN) guidelines for detection, prevention, or risk reduction in individuals who present with an eoRCC but lack a PGV in a familial RCC gene [7].
Recently, we reported that a significant subset of eoRCC patients undergoing cancer risk assessment carry PGVs in DNA damage response and repair genes (~ 8.55% vs. 3.7% in familial RCC genes) [6]. Similarly, Carlo et al. reported an increased prevalence of PGVs in DNA repair genes in advanced clear cell and non-clear cell renal cancer patients [3, 8]. Although PGVs in DNA repair genes are not currently defined by clinical testing guidelines as increasing risk of RCC, these recent studies suggest a potential role of defective DNA repair pathways in eoRCC carcinogenesis that could also lead to novel therapeutic options for RCC patients. Owing to the rising incidence of eoRCC and limited genetic data in younger RCC patients, we performed germline and tumor whole exome sequencing (WES) and functional assays on biospecimens from high-risk eoRCC patients diagnosed before 60 years of age, who were negative for PGVs in familial RCC syndrome genes and had a family history of RCC and/or other familial cancers. Our results suggest that constitutional defects in DNA repair underlie at least a subset of eoRCC cases. Screening patient lymphocytes to identify genotype–phenotype associations via functional assays may provide insight into the mechanism of carcinogenesis for a subset of genetically undiagnosed eoRCCs.
Results
eoRCC patients at the Fox Chase Cancer Center (FCCC) and family history of cancer
We analyzed the personal and family history of the probands in a cohort of 22 eoRCC patients. Multiple probands (6/22, 27%) had a second primary cancer, with breast cancer diagnosed in 3 probands (3/22, 14%) prior to diagnosis of RCC (Fig. 1A-E and Table 1). Here, 73% (n = 16/22) of probands had a family history of RCC, with 50% (n = 11/22) of probands having a first-degree relative with RCC. Intriguingly, 64% (n = 14/22) of probands had a family history of cancers of the prostate, bladder, and thyroid, and melanoma, which have been associated with an RCC diagnosis [9].

Select pedigrees from the eoRCC patient cohort and enrichment of predicted pathogenic variants in DNA repair genes in the cohort. A-E. Pedigrees of eoRCC patients with variants in: A— POLD1 and POLH; B— POLE; C— ATM; D— RRM2B and BCL2L1; E— OGG1, NEIL3 and UBR5. F. Summary of variants in genes and pathways, identified in the cohort. In color—number of variants identified for each gene. For detailed information, see Supplementary tables 1 and 2
Analysis of whole-exome sequencing data reveals enrichment of germline variation in DNA repair and replication genes in eoRCC patients
We performed WES on lymphocyte DNA from the 22 eoRCC probands, which we analyzed to detect candidate variants in genes included in a candidate gene list (n = 613) that was developed in our prior studies ([10]; see (Supplementary Table 1) and Supplementary Materials and Methods). Here, our intention was to analyze whole exomes of the probands with an expanded list of genes beyond the targeted set of genes on clinical germline panels with the following justification:
-
a)
genes involved in genome stability (using Gene Ontology terms such as DNA repair, DNA replication, DNA damage checkpoints, cell cycle, mitotic machinery, replication stress, DNA damage response, chromatin remodeling) would be important for hereditary cancer risk. This is in line with recent work from our group and others in renal cancer [3, 6, 11, 12]. b) an expanded network of genes relevant to renal cell biology (such as cellular metabolism) and genes somatically mutated in RCC that might be relevant for eoRCC-predisposition [5, 13, 14]. For example, there are several genes that are relevant to RCC biology (e.g., PBRM1 [15, 16] SETD2 [15, 17] that are not tested as hereditary risk genes as there is currently no evidence to suggest they impact cancer risk.
Novel candidate variants were stringently defined as those that are predicted to disrupt protein function by the consensus of at least 4 protein predictor algorithms; are rare (gnomAD allele frequency < 0.01); and are nonsynonymous variants (frameshifts, stop gains, and splicing) (see Supplementary Methods for variant prioritization). After applying American College of Medical Genetics (ACMG) criteria [18], we identified candidate variants in 17/22 eoRCC patients in the study, yielding a total of 41 variants in 38 genes (Table 1, and Supplementary Table 2). Gene Ontology analysis confirmed that the candidate variants were enriched in DNA repair and replication pathway genes (Fig. 1F and Supplementary Fig. 1, WebGestalt) [19]. Here, 10 patients (46%; 10/22) had 17 candidate variants in 14 genes currently associated with hereditary cancers across major organ systems (ATM, BRCA2, POLD1, POLE, FH, MITF, MSH3, MUTYH, PDGFRA, RET, SDHB, SMARCA4, SMARCE1, TSC2). Only 4 patients had candidate variants in RCC familial syndrome genes (4/22 – FH, MITF, SDHB, TSC2). Finally, a total of 14 patients (64%; 14/22) had candidate variants from our expanded candidate gene list, from genes not currently defined as RCC-predisposing.
Among the DNA repair-associated genes, candidate variants were found in BRCA2 (Pt #2, Table 1, Supplementary Table 2) and in ATM (2 variants in Pt #14, Table 1, Supplementary Table 2). In addition, 5 candidate variants in DNA replication-repair genes (4/22 patients; 18%, Table 1, Supplementary Table 2, POLD1 and POLH (Pt #1), POLE (Pt #2), POLK (Pt #16), and RRM2B (Pt #4)). Pt #1 had candidate missense variants in PolD1, a catalytic subunit of the replicative DNA polymerase, Pol δ, and in the translesion synthesis DNA polymerase, Pol η. POLD1 G2275A p.V759I is in a highly conserved region of PolD1 subunit of the Pol δ (coded by POLD1 gene) [20] and occurs at a high allele frequency in the Ashkenazi Jewish population (0.0213) reported in gnomAD (versus 0.0018 in the complete gnomAD dataset [21]). POLH G626T (p.G209V) is in the Pol η catalytic core [22, 23]. Pt #2 has a candidate stop-gain G4872A (p.W1624X) variant in the POLE gene, coding replicative DNA polymerase Pol ε, in the conserved C-terminal domain [24]. Pt #4 had a splice site (intronic) variant in RRM2B, coding a subunit of p53-inducible ribonucleotide reductase, which performs de novo conversion of ribonucleotide diphosphates into the corresponding deoxyribonucleotide diphosphates for DNA synthesis [25], in genome position #103,237,248 (chromosome 8q23). Pt #16 had a missense variant in the highly conserved N-terminus domain of another translesion synthesis polymerase, POLK G85A (p.E29K); this variant has been described previously as compromising enzyme activity [26]. ACMG classification and ClinVar evidence are presented in Supplementary Table 2, with most variants currently classified as variants of uncertain significance (VUS).
Primary lymphocytes from eoRCC patients have reduced capacity to suppress DNA double strand breaks (DSBs)
To begin to assess the functional effect of candidate variants in genes linked to DNA replication and repair, we assessed the numbers of γ (phospho)-H2AX foci (a marker of DSBs, [27]) in patient peripheral blood monocytes (PBMCs) at baseline and after treatment with the DNA polymerase inhibitor aphidicolin (Fig. 2A). In PBMCs from both matched cancer-free controls (by age and gender) and eoRCC patients, aphidicolin significantly elevated the number of γH2AX foci; however, aphidicolin-treated cells from eoRCC patients had markedly higher numbers of γH2AX foci than those from similarly treated controls on treatment, indicating reduced DSB repair mechanism in eoRCC patient cells (Fig. 2A, P < 0.001). In complementary work, we tested whether the genes bearing candidate variants were specifically needed to suppress DNA DSBs in RCC cells. For this, we used siRNA to deplete the POLD1, POLE, POLH, POLK, RRM2B, and ATM genes in the Caki RCC cell line. For each gene, knockdown significantly increased γH2AX foci relative to control (Supplementary Fig. 2) further supporting a role for these proteins in DSB repair in renal cells.

Cell-based functional analysis revealed defects in DNA repair and DNA replication in lymphocytes from eoRCC patients. A. γH2AX foci immune fluorescence staining in primary PBMCs from eoRCC patients versus matched controls, at baseline or post treatment with aphidicolin (2 h). PBMCs from patients showed statistically significant elevation of γH2AX foci post treatment with aphidicolin. Data were normalized and are presented as percent of positive γH2AX foci. B. Representative Western blots showing expression of PolD1, Pol η, Pol ε and Pol κ in EBV-transformed cell lines carrying variants versus matched controls (without the variants). Data quantification was performed based on 3 independent biological repeats, technical repeats are presented on gels. C-E. Relative viability of EBV-transformed cell lines was assessed by CTB assay at baseline or after treatment with aphidicolin or UV. Data were normalized to CTB values for controls and are presented as percent cellular viability for POLD1/POLH (C), POLE (D), and POLK (E) cell lines. Data from 3 independent biological repeats are presented. F-G. Difference in DNA replication fork elongation/restoration in EBV-transformed cell lines (F— POLD1, POLH; G— POLE lines) at the baseline and post replications stress was assessed using DNA fiber assay. At baseline the EBV-transformed cells were labeled with IdU for 20 min, for fork restoration cells then were treated with 100 µM aphidicolin or 1 uM MNNG for 2 h, and then labeled with CldU for 40 min. For all conditions, post labeling, cells were lysed, and DNA fibers stretched onto glass-slides, fixed, denatured, blocked, and stained with corresponding antibodies. Fiber images were captured using the Nikon TS2R Inverted Microscope and analyzed in ImageJ software. Data for 3 independent repeats are presented as IdU tract length or CldU/IdU tract length ratio. For all graphs: *** for p < 0.001, ** for p < 0.01, * for p < 0.05 and NS for p > 0.05, unpaired, non-parametric t-test, Mann–Whitney criteria
Patient-derived cell lines with candidate PGVs in DNA polymerases exhibit DNA replication defects
We prepared EBV-transformed cell lines from the primary lymphocytes of 3 patients bearing candidate variants (henceforth referred to as the POLD1/POLH cell line, POLE cell line, and POLK cell line) and from several age-and gender-matched cancer-free controls. The POLD1/POLH cell line had significantly reduced levels of the Pol η; for the other candidate variants, the level of the polymerase bearing the candidate variant was not affected (Fig. 2B). Cell Titer Blue (CTB) cellular assays showed significantly better viability than control-derived cell lines when treated with aphidicolin, or with ultraviolet light (which causes bulky adducts in DNA), suggesting that cell lines from patients had better ability to tolerate DNA damage (Figs. 2C-E). Such increased viability in the context of alterations in polymerases has been reported in a number of studies [28,29,30].
Analysis of cell cycle did not show any significant differences in patients and matched control cell lines (Supplementary Fig. 3A).
To further expand on these cell-based findings, we used a DNA fiber assay (see Supplementary Methods) and directly compared DNA replication in patient-derived versus control cells, either untreated or following treatment with aphidicolin or with a DNA-alkylating agent, 1-Methyl-3-nitro-1-nitrosoguanidine (MNNG) (Fig. 2F-G Supplementary Fig. 4A-D). The POLD1/POLH cell line and the POLE cell line exhibited a significantly lower rate of DNA replication in untreated cells (~ 1.4-fold decrease, p < 0.001 for the POLD1/POLH cell line, and ~ 1.9-fold decrease, p < 0.05 for the POLE cell line versus controls). We also observed a significantly lower replication fork recovery after 2 h treatment with aphidicolin (~ 1.44-fold decrease, p < 0.001 for the POLD1/POLH cell line, and ~ 1.88-fold decrease, p < 0.01 for the POLE cell line versus controls) (Fig. 2G-H). Intriguingly, the POLD1/POLH cell line showed defective replication fork restoration (~ 1.2-fold decrease, p < 0.001 versus control line) 2 h post-treatment with MNNG (Fig. 2G). The POLK cell line did not show any defects in DNA replication and replication recovery under the conditions tested (Supplementary Fig. 4A). A complete summary of results for the DNA polymerase variants is provided in Supplementary Table 3.
Altered enzymatic activity of Pol δ and Pol η variant proteins
Among the candidate variants detected in polymerases, the Pol κ variant E29K has previously been biochemically shown to possess not only a significantly reduced catalytic efficiency but also reduced replication fidelity [26]. E29K is in a conserved region of the Pol κ N-terminus, the N-clasp subdomain (1–32 aa), which is essential to maintaining the stability of the open conformation of the Pol κ active site [31]. Intriguingly, a previous study showed that deletion of the first 67 amino acids reduces Pol κ activity during translesion synthesis (TLS, i.e., replication by efficient bypass of bulky lesions in DNA) [32].
To directly test effects of the other candidate variants on polymerase activity, we first purified the polymerase delta (Pol δ) protein complex, with the PolD1 (POLD1), PolD2 (POLD2), PolD3 (POLD3), and PolD4 (POLD4) subunits from recombinant protein co-expressed in E. coli, and with preparations containing either wild type (wt) PolD1 or PolD1 V759I variant (Fig. 3). Both the wt and the variant-containing Pol δ complexes extended a Cy3-labeled DNA primer-template; however, the V579I variant complex had significantly less robust polymerase activity than the wt complex (Fig. 3A, p < 0.001). Furthermore, when Pol δ complexes containing PolD1 wt or PolD1 V759I proteins were mixed in a ratio of 1:1, the appearance of the extended primer-template was significantly more robust than the variant alone but significantly less robust than the wt alone. This result suggests that the variant is not only impaired for function but has a partial dominance over the wt in this assay (Supplementary Fig. 5, p < 0.001).

Structural and biochemical assays revealed altered enzymatic activities of the PolD1 and Pol η variants. A. Pol δ complex and primer extension assay. On the left—representative gel image of purified wt and variant Pol δ protein complexes, containing 4 subunits: PolD1 (125 kDa), PolD2 (50 kDa), PolD3 (66 kDa) and PolD4 (12.5 kDa). Center and right – Pol δ complex primer extension assay with quantification. Representative gel image showing reactions performed with 20 nM Cy-3 labeled DNA-duplex template (SA#1), 20 nM of indicated proteins and 500 uM dNTPs. PolD1 V759I complex extended DNA-template less efficiently comparing to wt protein complex. Data for 3 independent repeats are presented. B. Pol η and primer extension assay. On the left—representative gel image of purified wt and variant Pol η catalytic cores (432 amino acids), molecular weight ~ 56 kDa. Center and right – Pol η primer extension assay with quantification. Representative gel image showing reactions performed with 20 nM Cy-3 labeled DNA-duplex template (SA#1), 20 nM of indicated proteins and 500 uM dNTPs. Data for 3 independent repeats is presented. C. Pol η lesion (8-oxoG) bypass assay with quantification. Representative gel image showing reactions performed with 20 nM Cy-3 labeled DNA-duplex template with 8-oxoG in the position, opposite to 3`-OH group (SA#4), and template of the same sequence without lesion (SA#3), 20 nM of indicated proteins and 500 uM dNTPs. Data for 3 independent repeats are presented. For A-C: *** for p < 0.001, ** for p < 0.01, * for p < 0.05 and NS for p > 0.05, unpaired, non-parametric t-test, Mann–Whitney criteria. All template sequences may be found in Supplementary Table 4. D-E. Homology modeling structures using yeast protein templates for human PolD1 V759I (D) and for human Pol η G209V (E)
Pol η is a low fidelity polymerase, which contributes to its ability to perform TLS [33]. Hence, G209V variant and wt Pol η prepared in E. coli were assessed for their ability to extend labeled DNA primer-template duplexes (Figs. 3B-C). In the absence of DNA damage (e.g., in a normally base-paired template), the wt and variant proteins both extended the template (Fig. 3B, p > 0.05), but the observed bands suggest higher processivity for the variant on template without lesions compared to wt (Fig. 3B). To evaluate repair of DNA damage, TLS activity was also tested using a template containing an 8-oxoGuanine (8-oxoG) DNA lesion [33]. Pol η wt bypassed the 8-oxoG lesion robustly compared to the Pol η variant (p < 0.05, Fig. 3C), suggesting better processivity for the wt protein on template with DNA lesion [33].
Finally, biochemical analysis of a purified Pol ε variant, W1624X, was not performed as it is a stopgain variant in the C-terminal domain or CTD, truncating 662 amino acids of the protein. The CTD region is not well-studied, but is thought to be essential for stability of the Pol ε holoenzyme [34].
Structural modeling of DNA polymerase variants in eoRCC suggests impact on polymerase function
The PolD1 V759I variant is located two amino acids away from residue D757 (Fig. 3D, in dark green). In the PolD1 active site, D757 coordinates the Mg2+ ions (neon green spheres) required for DNA synthesis and plays a direct role in the catalytic mechanism and binding of DNA [20]. Structural modeling indicates that a substitution of the valine (V) 759 to isoleucine (I) could plausibly alter the position of D757 and disrupt the efficiency of DNA polymerization. To further understand the structural changes that each polymerase variant might induce, we calculated the change in stability of each amino acid substitution as described in Supplemental Methods and Supplementary Table 4. Interestingly, the PolD1 I759 yielded a mild stabilization (ΔΔG of -1.36 kcal/mole) relative to the wildtype V759. The I759 residue makes twice the number of hydrophobic contacts as the wildtype V759 with the long helix below. The variant could lock this strand in an overly rigid position that compromises DNA polymerization steps that involve flexibility [35, 36], consistent with the biochemical results observed in Fig. 3A.
The Pol η G209 residue is in the catalytic core of the polymerase (Fig. 3E, residues 1–432, colored green) at a position often called the C-cap, i.e. the residue in this position is proximal to the C-terminal end of the α-helix (in orange) [37]. Typically, valine, threonine, and isoleucine residues are not preferred in the C-cap, due to poor solvation at the C-terminus of the helix when the side chains are bulky [37]. Structural modeling of the G209V substitution showed that the valine with a bulkier side chain could not only alter the stability of the α-helix, but also the nearby β-strands of the catalytic active site (in pink). Rosetta modeling shows the variant G209V is capable of making 5 hydrophobic contacts across the cleft with R24, which is just downstream from the key residue D13, which coordinates active site metals that are central to the catalytic mechanism and the binding of the incoming NTP. The predicted change in stability of the Pol η G209V variant revealed a significant destabilization (ΔΔG of 5.67 kcal/mole) relative to the wildtype G209, consistent with lower levels observed in protein (Fig. 2B, Fig. 3B (left gel), and Supplementary Table 4). Thus, a significant destabilization in a relatively rigid region may impact enzyme function, as supported by [35, 36]. Interestingly, the activity of the G209V compares well with wt for a normal primer template and might even be more processive (Fig. 3B, middle gel). However, the variant appears defective for TLS when the template contains an 8oxoG (Fig. 3C). Consideration of these data together shows that changes in stability and conformation may be subtle and even result in an alteration of substrate preference. Thus, a careful combination of both computational and biochemical methods is required to gain a clear understanding of the role a given polymerase variant might have in DNA replication, repair, and possibly cancer initiation and/or progression.
EoRCCs carrying candidate PGVs in DNA polymerases are hypermutant and microsatellite stable (MSS)
To extend these functional tests, we next explored tumor mutation burden (TMB) in tumors from RCC patients in TCGA and from the FCCC eoRCC patients. Previous studies have shown that colorectal and endometrial tumors carrying mutations in POLE exonuclease domain (ExoD) and in POLD1 exhibit a high burden of mutations, are typically MSS but few cases with microsatellite instability (MSI) have been reported, and do not exhibit loss of heterozygosity (LOH) [20, 38,39,40,41,42,43,44,45,46]. RCCs are typically non-hypermutated, with an average TMB of ~ 1 mut/Mb [47]; however, rare hypermutated (≥ 10 mut/Mb) and rarer ultra-hypermutated (> 100 mut/Mb) RCCs carrying polymerase mutations, with or without MSI, have been reported [47]. Analysis of TCGA renal tumor data found that several genes that were mutated in our study (Fig. 4A), including DNA polymerase genes, were somatically mutated in TCGA hypermutant clear cell RCCs (ccRCCs) (Fig. 4A). We analyzed the MSI/MSS and TMB status of tumors from the FCCC eoRCC patients. Both tumors were reported as MSS from clinical testing and were hypermutated (POLD1/POLH tumor- 12.85 mut/Mb, POLE tumor- 14.44 mut/Mb) (Fig. 4B). We analyzed mutational signatures from the POLD1/POLH tumor and the POLE tumor. In single-base substitutions (SBS), signatures SBS5 (clock-like aging signature) was the dominant signature in both tumors; this signature is typically observed in all tissues in the body, normal and tumor, and not associated with polymerase exonuclease defects. Notably, we did not observe SBS signatures (SBS10 subtypes) associated with exonuclease domain mutations, which was expected as the candidate variants in POLE and POLD1 are not within the exonuclease domain. Intriguingly, analysis of doublet-base substitutions (DBS) revealed presence of signature DBS3; DBS3 is typically associated with SBS10 signature but is not always observed in tumors with SBS10 signatures and/or exonuclease domain mutations [48, 49]. We also confirmed that the tumors from the FCCC eoRCC patients did not exhibit LOH of the polymerase genes, as has been observed in other polymerase-mutated tumors [47, 50] (Fig. 4C).

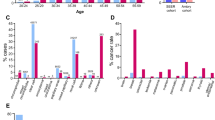
Renal tumors carrying polymerase variants showed high TMB, MSS, and no LOH. A. Percent alteration frequency in 897 tumors from TCGA in different histological types of RCC: chromophobe (n = 66), ccRCC—clear cell renal cell carcinoma (n = 538), ccRCC (hyper)—hypermutated samples (n = 12), papillary (n = 293). B. TMB and MSS data are presented for Pt #1 (POLD1 V759I, POLH G209V) and Pt #2 (POLE W1624X). C. Tumor and normal Sanger sequencing for variants in Pt #1 (POLD1 V759I, POLH G209V) and Pt #2 (POLE W1624X) showing no LOH. Arrows show variants of interest on sequencing tracks
To expand the analysis of DNA polymerases in RCC, we next modeled the structural consequences of somatic PolD1 and Pol ε variants in hypermutated ccRCCs from TCGA. Supplementary Table 5 shows the predicted changes in stability for 20 different PolD1 variants and 23 Pol ε variants from hypermutated ccRCCs in TCGA. A broad range of both stabilizing and destabilizing variants was found in all domains of each of these polymerases. Figure 5 shows these variants in relation to known pathogenic variants in POLD1 (Fig. 5A-E) and POLE (Fig. 5F-I). PolD1 R823G/L is in a β-sheet of the polymerase domain close to the DNA (Fig. 5C). The substitution of a positively charged arginine (R) to a hydrophobic glycine (G) or leucine (L) could destabilize the β-sheet and impact DNA binding. PolD1 D893N is positioned close to the DNA and a variant in this region may destabilize DNA binding or position for DNA–protein interactions (Fig. 5D). PolD1 P151S is in a β-sheet in the ExoD and the change from proline (P) to serine (S) could destabilize the β-sheet geometry (Fig. 5E). P151 is close to a known cancer driver mutation [47, 51], E245K, in the unfolded region of the ExoD (Fig. 5A).

Structure mapping of the novel PolD1 and Pol ε variants from hypermutated ccRCCs in TCGA. A-E. DNA-bound PolD1 3D-model was refined from PDB:3IAY. The colored functional domains are exonuclease (light blue, residues 131–477) and polymerase (green, residues 550–978). A. Red spheres represent known cancer drivers. B. Blue spheres represent variants of uncertain significance in ccRCC. C-E. Fragments of PolD1 model showing variants: A810S and R823G (C), D893N, A810S, and R978C (D), P151S and E245K (E). F. DNA-bound N-terminal domain of Pol ε was refined from PDB: 4M8O. The colored functional domains are N-terminal subdomain (dark grey, residues 31–281), exonuclease (wheat, residues 282–527), polymerase (light pink, palm: 528–950; cyan, fingers: 769–833; lime, thumb: 951–1186). Red spheres represent known cancer drivers (structure above). Blue spheres represent variants of uncertain significance in ccRCC (structure below). G. A fragment of Pol ε model showing variant P696L. H. 3D-model of whole-length Pol ε (without DNA). Structure was refined as described in Methods based on [34]. The colored functional domains are N-terminal subdomain (dark grey, residues 31–281), exonuclease (wheat, residues 282–527), polymerase (light pink, palm: 528–950; cyan, fingers: 769–833; lime, thumb: 951–1186), C-terminal domain (light grey, residues 1308–2222). Blue spheres represent variants of uncertain significance in ccRCC. I. Fragments of Pol ε model showing variants S803L and F753
Pol ε P696R is in the palm region of the polymerase domain, which is highly conserved among replicative polymerases (Fig. 5G). Arginine (R) has a very large positively charged side chain when compared to smaller proline (P), suggesting this variant may disrupt the polymerase structure and impact DNA synthesis. Pol ε S803L and F753L are in the flexible region of the polymerase domain or the fingers (in cyan, Fig. 5I). This finger region shifts (27° tilt) on DNA binding [52], and thus plays an essential role in polymerase function. S803 is close to the positively charged lysine (K) 431 in the ExoD, and serine (S) is polar and a smaller residue than the hydrophobic leucine (L). Pol ε S803L is near a site of known cancer driving mutations, C810 [47], suggesting that specific alterations in this α-helix could impact polymerase functioning. F753L is on the border of the fingers and the polymerase domain, close to the ExoD and could be important in the coordination of these regions with or without DNA binding. Finally, several variants were found in the C-terminal domain (Fig. 5H, in light grey), which is currently not well-studied, but is known for stabilizing the Pol2 (human Pol ε) complex in yeast [34].
Discussion
In this study, we focus on analysis of candidate variants in DNA repair and replication genes in probands with RCC diagnosed prior to 60 years of age who were undergoing cancer risk assessment at our cancer center and who tested negative for RCC familial syndrome genes. We applied a well-curated pipeline of candidate genes in genome stability, metabolism, metabolic stress, normal renal function, RCC biology, and chromatin remodeling to germline WES data from eoRCC patients. Gene Ontology analysis confirmed that the identified candidate variants were enriched in DNA repair and replication genes. Intriguingly, we found that many eoRCC patients exhibit defects in suppression of DSBs in their primary PBMCs, with PBMCs from eoRCC patients exhibiting higher γH2AX foci than matched cancer-free controls in response to DNA damage. Direct knockdown of some of these candidate variant genes in Caki RCC cell line also led to increased γH2AX foci. Genes with candidate variants were found to be mutated in sporadic RCCs, with specific enrichment of alterations in DNA polymerases (POLE, POLD1, POLH1, and POLK) and BRCA2 in hypermutant RCCs in TCGA. Importantly, detailed analysis of candidate variants in DNA polymerase genes from the FCCC eoRCC patients confirmed the damaging nature of the candidate variants and suggested a mechanistic basis for association of these variants with observed defects in DNA repair and replication. Several PolD1 and Pol ε variants from the hypermutant RCCs in TCGA were proximal to the catalytic center or substrate binding regions and will benefit from similar future biochemistry experiments shown in this study.
This work complements a number of recent studies indicating inherited defects in DNA replication machinery may increase cancer risk. Candidate variants in the ExoD of POLE and POLD1 predispose to cancer and exhibit a strong mutagenic effect, however, the role of non-ExoD variants in mutagenesis and cancer risk has been controversial [39, 41, 53]. A recent study reported that the POLD1 candidate variant (p.V759I, in Pt #1) is frequently present in the Ashkenazi Jewish population and proposed this gene as a founder mutation [2]. Mertz et. al. have demonstrated a strong mutator effect of the PolD1 polymerase domain variant R689W in human cells [54]. Barbari et. al have recently shown that defective proofreading is not the only important determinant of variant pathogenicity; polymerase fitness is also a key factor [55]. Several TLS polymerases including Pol η and Pol κ, are important for preventing accumulation of single strand DNA gaps, and the replication of DNA fragile sites. Owing to their high error-propensity, TLS polymerases are likely to contribute to oncogene-induced mutagenesis [56, 57].
It is likely that the candidate variants in DNA replication and repair genes detected here interact with other germline variants to impact eoRCC risk. In this study, Pt #1 also harbored candidate variants in MTOR, PARP1, FH, MCM2, suggesting the POLD1 and POLH variants assessed here may act together with other variants to augment the DNA repair defects observed. Pt #2 with a POLE variant also harbored candidate variants in BRCA2, PDGFRA. In other studies, hypermutant ccRCCs also carried mutations in TP53, PTEN, VHL, and UNC5C [58, 59]. POLE and POLD1 are currently not considered classical tumor suppressor genes, and LOH is typically not observed in tumors. Increased mutation frequency is observed with a heterozygous mutation in these replicative DNA polymerases, but only the homozygous mice have increased susceptibility to cancer; suggesting that there are other additional factors important for carcinogenesis [38, 39, 41, 60]. It is possible that defects in polymerase genes impact cancer risk by affecting biological processes beyond DNA replication and repair. Conversely, some familial RCC genes (such as FH, VHL, PBMR1, and SDHx) have also been implicated in suppression of DSBs and in replication stress [16, 61,62,63], based on mechanisms that are not well understood.
It is important to note that while the family history of the high-risk probands in this study is suggestive of underlying genetics [64], clinical testing for RCC familial syndrome genes did not yield any actionable PGVs according to current NCCN recommended guidelines. Our results suggest that in the absence of PGVs in RCC familial syndrome genes or phenotypic features of familial RCC or family history of RCC, a comprehensive assessment of general cancer predisposition genes, including DNA repair genes, may be beneficial. RCC may be one type of cancer induced by mutations, rather than the sole type of cancer. This is supported by the pedigree data in this study as multiple probands (27%) had at least one additional primary cancer with breast cancer being the most common additional primary cancer (14%). Here, 64% of probands had an extensive family history of cancers of the prostate, bladder, and thyroid, and melanoma, all of which have previously been associated with RCC diagnosis [9]. In fact, PGVs in DNA repair genes have been reported as risk factors for bladder, skin, thyroid, and prostate cancers [65,66,67,68,69,70]. A recent retrospective analysis of the Swedish Cancer Registry showed that ~ 10% of RCC patients develop another second primary cancer, and this is currently thought to be independent of the primary RCC, suggesting broader cancer predisposition [71], compatible with PGVs in genes affecting DNA repair.
Besides genetic screening, these data suggest the value of functional assessment for the families of individuals with eoRCC. In this study, the majority of eoRCC PBMC biospecimens samples exhibited elevated γH2AX levels and candidate variants in DNA repair genes. Our data supports a potential role of germline variation in DNA repair/replication leading to suboptimal encoding protein activity, and genome instability. Overall, these data suggest that assays of γH2AX foci in normal cells, supporting germline variation in DNA repair/replication genes, could be a potential tool for the identification of individuals with genetically unexplained eoRCC. As these defects could be detected in normal cells, it could lead to the identification of individuals in need of cancer risk assessment. This is especially relevant because case–control studies suggest that an elevated familial RCC risk may be multifactorial, and or due to an interaction of the heritable genetics and the shared environment [64]. It is possible that defective DNA repair in the heterozygous state could be a recessive heritable factor that when combined with other RCC risk factors may jointly increase the risk of eoRCC.
Currently, the therapeutic significance of DNA repair genes is not clinically defined for RCC. Evidence is emerging that PARP inhibitors could be therapeutics of choice in RCCs that may not carry mutations in the classical BRCA genes, but which have other defects in DNA repair, with recent clinical trials assessing the use of PARP inhibitors in RCC [72, 73]. Hence, there is a critical need to not only understand the biological impact of defective DNA repair in renal tissue but to also define risk of RCC due to a germline defect in DNA repair genes. A limitation of this study is a relatively small cohort which is not representative of all individuals with eoRCC (i.e. as the cohort subjects had clinical characteristics and/or family history to support germline testing). The candidate variants identified, and the functional assays suggest a mechanistic basis, but more studies are needed to conclude impact on RCC risk. Further work in a larger and more diverse (by race/ethnicity) patient population is clearly of interest for the future.
Conclusions
The results presented here suggest that constitutional defects in DNA repair such as DNA replication repair underlie a subset of eoRCC cases. Screening patient lymphocytes to identify these defects may provide insight into mechanisms of carcinogenesis in a subset of genetically undefined eoRCCs. Evaluation of DNA repair defects may also provide insight into the cancer initiation mechanisms for subsets of eoRCCs and lay the foundation for targeting DNA repair vulnerabilities in eoRCC.
Materials and methods
eoRCC patient population, and peripheral blood DNA analysis
Case-only eoRCC probands that underwent clinical germline genetic testing between 2010–2016 were included in this study (n = 22). Patients were followed by the Genitourinary Program at the Fox Chase Cancer Center and had undergone evaluation for inherited cancer risk at the FCCC Family Risk Assessment Program (RAP). Blood samples were banked in the FCCC Biosample Repository Facility under broad informed consent for research and deidentified. Each participant had a strong family cancer history as shown in Table 1, with either multiple first-degree or second-degree relatives with RC, RC-associated cancers, or other cancers. The mean age at eoRCC diagnosis was 48 years (range 36–59 years). No pathogenic mutations were identified from sequencing the following RC-specific genes: VHL, MET, FLCN, TSC1, TSC2, FH, SDHx, PTEN and BAP1. The patients reported here were self-reported white, non-Hispanic. Family histories were obtained by trained licensed genetic counselors and verified by attending physicians. All patient data obtained were de-identified and included family history of cancer, genetic test results, personal history of cancer(s), presence of multifocal tumors, cancer subtype/stage. Any de-identified personal or family history information including sex, ethnicity/race, age of cancer diagnosis, tumor histology, history of additional personal cancer, and history of family cancer and types was reported first as summarized data and later as de-identified individual case reports. See Supplementary Methods for more methods.
Availability of data and materials
The data supporting the conclusions of this article are included within the article and its additional files. The informed consent was not obtained for data sharing of raw sequencing reads in publicly accessible databases.
Change history
10 July 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12864-023-09486-z
Abbreviations
- 8-oxoG:
-
8-OxoGuanine
- BWA:
-
Burrows-Wheeler aligner
- ccRCC:
-
Clear cell renal cell carcinoma
- CTB:
-
Cell Titer Blue
- CldU:
-
5-Chloro-2'-deoxyuridine
- DSBs:
-
Double-strand breaks
- DSB:
-
Doublet-base substitutions
- eoRCC:
-
Early-onset renal cell carcinoma
- DDR:
-
DNA damage response
- EBV:
-
Epstein-Barr virus
- ExAC:
-
Exome Aggregation Consortium
- ExoD:
-
Exonuclease domain
- FCCC:
-
Fox Chase Cancer Center
- FDR:
-
False discovery rate
- GATK:
-
Genome analysis toolkit
- GnomAD:
-
Genome Aggregation Database
- GO:
-
Gene ontology
- IdU:
-
5-Iodo-2′-deoxyuridine
- LOH:
-
Loss of heterozygosity
- MMR:
-
Mismatch repair
- MNNG:
-
1-Methyl-3-nitro-1-nitrosoguanidine
- MSI:
-
Microsatellite instability
- MSS:
-
Microsatellite stability
- NCCN:
-
National Comprehensive Cancer Network
- PBMCs:
-
Peripheral blood monocytes
- PGVs:
-
Pathogenic germline variants
- Pt:
-
Patient
- RAP:
-
Risk assessment program
- RCC:
-
Renal cell carcinoma
- SNPs:
-
Single nucleotide polymorphisms
- SBS:
-
Single-base substitutions
- TLS:
-
Translesion synthesis
- TMB:
-
Tumor mutation burden
- VUS:
-
Variant of uncertain significance
- WebGestalt:
-
WEB-based GEne SeT AnaLysis Toolkit
- WES:
-
Whole-exome sequencing
- wt:
-
Wild type
References
Sung H, Siegel RL, Rosenberg PS, Jemal A. Emerging cancer trends among young adults in the USA: analysis of a population-based cancer registry. The Lancet Public Health. 2019;4(3):e137–47.
Rosner G, Gluck N, Carmi S, Bercovich D, Fliss-Issakov N, Ben-Yehoyada M, Aharon-Caspi S, Kellerman E, Strul H, Shibolet O, et al. POLD1 and POLE gene mutations in Jewish cohorts of early-onset colorectal cancer and of multiple colorectal adenomas. Dis Colon Rectum. 2018;61(9):1073–9.
Carlo MI, Mukherjee S, Mandelker D, Vijai J, Kemel Y, Zhang L, Knezevic A, Patil S, Ceyhan-Birsoy O, Huang KC, et al. Prevalence of germline mutations in cancer susceptibility genes in patients with advanced renal cell carcinoma. JAMA Oncol. 2018;4(9):1228–35.
Linehan WM, Bratslavsky G, Pinto PA, Schmidt LS, Neckers L, Bottaro DP, Srinivasan R. Molecular diagnosis and therapy of kidney cancer. Annu Rev Med. 2010;61:329–43.
Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7(5):277–85.
Hartman TR, Demidova EV, Lesh RW, Hoang L, Richardson M, Forman A, Kessler L, Speare V, Golemis EA, Hall MJ, et al. Prevalence of pathogenic variants in DNA damage response and repair genes in patients undergoing cancer risk assessment and reporting a personal history of early-onset renal cancer. Sci Rep. 2020;10(1):13518.
Motzer RJ, Jonasch E, Michaelson MD, Nandagopal L, Gore JL, George S, Alva A, Haas N, Harrison MR, Plimack ER, et al. NCCN Guidelines Insights: Kidney Cancer, Version 2.2020: Featured Updates to the NCCN Guidelines. J Natl Compr Canc Netw J Natl Compr Canc Netw. 2019;17(11):1278–85.
Truong H, Sheikh R, Kotecha R, Kemel Y, Reisz PA, Lenis AT, Mehta NN, Khurram A, Joseph V, Mandelker D, et al. Germline variants identified in patients with early-onset renal cell carcinoma referred for germline genetic testing. Eur Urol Oncol. 2021;4:993.
Liu H, Sundquist J, Hemminki K. Familial renal cell carcinoma from the Swedish family-cancer database. Eur Urol. 2011;60(5):987–93.
Nicolas E, Demidova EV, Iqbal W, Serebriiskii IG, Vlasenkova R, Ghatalia P, Zhou Y, Rainey K, Forman AF, Dunbrack RL Jr, et al. Interaction of germline variants in a family with a history of early-onset clear cell renal cell carcinoma. Mol Genet Genomic Med. 2019;7(3): e556.
Carlo MI, Mukherjee S, Offit K. CHEK2 Alleles predispose to renal cancer in Poland-in reply. JAMA Oncol. 2019;5(4):576–7.
Truong H, Sheikh R, Kotecha R, Kemel Y, Reisz PA, Lenis AT, Mehta NN, Khurram A, Joseph V, Mandelker D, et al. Germline variants identified in patients with early-onset renal cell carcinoma referred for germline genetic testing. Eur Urol Oncol. 2021;4(6):993–1000.
Henegan JC Jr, Gomez CR. Heritable cancer syndromes related to the hypoxia pathway. Front Oncol. 2016;6:68.
Berti M, Vindigni A. Replication stress: getting back on track. Nat Struct Mol Biol. 2016;23(2):103–9.
D’Avella C, Abbosh P, Pal SK, Geynisman DM. Mutations in renal cell carcinoma. Urol Oncol. 2020;38(10):763–73.
Chabanon RM, Morel D, Eychenne T, Colmet-Daage L, Bajrami I, Dorvault N, Garrido M, Meisenberg C, Lamb A, Ngo C, et al. PBRM1 deficiency confers synthetic lethality to DNA repair inhibitors in cancer. Cancer Res. 2021;81(11):2888–902.
Kanu N, Grönroos E, Martinez P, Burrell RA, Yi Goh X, Bartkova J, Maya-Mendoza A, Mistrík M, Rowan AJ, Patel H, et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene. 2015;34(46):5699–708.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–24.
Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47(W1):W199-w205.
Nicolas E, Golemis EA, Arora S. POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene. 2016;590(1):128–41.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Tomczyk P, Synowiec E, Wysokiński D, Woźniak K. Eukaryotic TLS polymerases. Postepy Hig Med Dosw (Online). 2016;70:522–33.
Acharya N, Manohar K, Peroumal D, Khandagale P, Patel SK, Sahu SR, Kumari P. Multifaceted activities of DNA polymerase η: beyond translesion DNA synthesis. Curr Genet. 2019;65(3):649–56.
Burgers PMJ, Kunkel TA. Eukaryotic DNA replication fork. Annu Rev Biochem. 2017;86:417–38.
Iqbal W, Demidova EV, Serrao S, ValizadehAslani T, Rosen G, Arora S. RRM2B is frequently amplified across multiple tumor types: implications for DNA repair, cellular survival, and cancer therapy. Front Genet. 2021;12: 628758.
Yadav S, Mukhopadhyay S, Anbalagan M, Makridakis N. Somatic mutations in catalytic core of POLK reported in prostate cancer alter translesion DNA synthesis. Hum Mutat. 2015;36(9):873–80.
Arora S, Yan H, Cho I, Fan HY, Luo B, Gai X, Bodian DL, Vockley JG, Zhou Y, Handorf EA, et al. Genetic variants that predispose to DNA double-strand breaks in lymphocytes from a subset of patients with familial colorectal carcinomas. Gastroenterology. 2015;149(7):1872-1883 e1879.
Sviderskiy VO, Blumenberg L, Gorodetsky E, Karakousi TR, Hirsh N, Alvarez SW, Terzi EM, Kaparos E, Whiten GC, Ssebyala S, et al. Hyperactive CDK2 activity in basal-like breast cancer imposes a genome integrity liability that can be exploited by targeting DNA polymerase ε. Mol Cell. 2020;80(4):682-698.e687.
Uphoff S. Real-time dynamics of mutagenesis reveal the chronology of DNA repair and damage tolerance responses in single cells. Proc Natl Acad Sci U S A. 2018;115(28):E6516-e6525.
Nik-Zainal S, Hall BA. Cellular survival over genomic perfection. Science. 2019;366(6467):802–3.
Jha V, Ling H. 2.0 Å resolution crystal structure of human polκ reveals a new catalytic function of N-clasp in DNA replication. Sci Rep. 2018;8(1):15125.
Lone S, Townson SA, Uljon SN, Johnson RE, Brahma A, Nair DT, Prakash S, Prakash L, Aggarwal AK. Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Mol Cell. 2007;25(4):601–14.
Suarez SC, Beardslee RA, Toffton SM, McCulloch SD. Biochemical analysis of active site mutations of human polymerase η. Mutat Res. 2013;745–746:46–54.
Yuan Z, Georgescu R, Schauer GD, O’Donnell ME, Li H. Structure of the polymerase ε holoenzyme and atomic model of the leading strand replisome. Nat Commun. 2020;11(1):3156.
Caswell RC, Gunning AC, Owens MM, Ellard S, Wright CF. Assessing the clinical utility of protein structural analysis in genomic variant classification: experiences from a diagnostic laboratory. Genome Med. 2022;14(1):77.
Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins. 2011;79(3):830–8.
Doig AJ, Baldwin RL. N- and C-capping preferences for all 20 amino acids in alpha-helical peptides. Protein Sci. 1995;4(7):1325–36.
Barbari SR, Shcherbakova PV. Replicative DNA polymerase defects in human cancers: Consequences, mechanisms, and implications for therapy. DNA Repair (Amst). 2017;56:16–25.
Church DN, Briggs SE, Palles C, Domingo E, Kearsey SJ, Grimes JM, Gorman M, Martin L, Howarth KM, Hodgson SV, et al. DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013;22(14):2820–8.
Hodel KP, de Borja R, Henninger EE, Campbell BB, Ungerleider N, Light N, Wu T, LeCompte KG, Goksenin AY, Bunnell BA, et al. Explosive mutation accumulation triggered by heterozygous human Pol ε proofreading-deficiency is driven by suppression of mismatch repair. Elife. 2018;7:e32692.
Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45(2):136–44.
Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A. 2013;110(8):2916–21.
Henninger EE, Pursell ZF. DNA polymerase ε and its roles in genome stability. IUBMB Life. 2014;66(5):339–51.
Shinbrot E, Henninger EE, Weinhold N, Covington KR, Göksenin AY, Schultz N, Chao H, Doddapaneni H, Muzny DM, Gibbs RA, et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014;24(11):1740–50.
Shlien A, Campbell BB, de Borja R, Alexandrov LB, Merico D, Wedge D, Van Loo P, Tarpey PS, Coupland P, Behjati S, et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet. 2015;47(3):257–62.
Arora S, Xiu J, Sohal D, Lou E, Goldberg RM, Weinberg BA, Grothey A, Korn WM, Khushman M, Shields AF, et al. The landscape of POLE variants in colorectal and endometrial tumors: Correlation with microsatellite instability (MSI) and tumor mutation burden (TMB). J Clin Oncol. 2020;38(15_suppl):e13538–e13538.
Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, Davidson S, Edwards M, Elvin JA, Hodel KP, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171(5):1042-1056.e1010.
Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91(2):355–8.
Park VS, Pursell ZF. POLE proofreading defects: Contributions to mutagenesis and cancer. DNA Repair (Amst). 2019;76:50–9.
He J, Ouyang W, Zhao W, Shao L, Li B, Liu B, Wang D, Han-Zhang H, Zhang Z, Shao L, et al. Distinctive genomic characteristics in POLE/POLD1-mutant cancers can potentially predict beneficial clinical outcomes in patients who receive immune checkpoint inhibitor. Ann Transl Med. 2021;9(2):129–129.
Lancey C, Tehseen M, Raducanu VS, Rashid F, Merino N, Ragan TJ, Savva CG, Zaher MS, Shirbini A, Blanco FJ, et al. Structure of the processive human Pol δ holoenzyme. Nat Commun. 2020;11(1):1109.
Mur P, García-Mulero S, del Valle J, Magraner-Pardo L, Vidal A, Pineda M, Cinnirella G, Martín-Ramos E, Pons T, López-Doriga A, et al. Role of POLE and POLD1 in familial cancer. Genet Med. 2020;22(12):2089–100.
Mertz TM, Baranovskiy AG, Wang J, Tahirov TH, Shcherbakova PV. Nucleotide selectivity defect and mutator phenotype conferred by a colon cancer-associated DNA polymerase δ mutation in human cells. Oncogene. 2017;36(31):4427–33.
Barbari SR, Beach AK, Markgren JG, Parkash V, Moore EA, Johansson E, Shcherbakova PV. Enhanced polymerase activity permits efficient synthesis by cancer-associated DNA polymerase ϵ variants at low dNTP levels. Nucleic Acids Res. 2022;50(14):8023–40.
Vaziri C, Rogozin IB, Gu Q, Wu D, Day TA. Unravelling roles of error-prone DNA polymerases in shaping cancer genomes. Oncogene. 2021;40(48):6549–65.
Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer. 2011;11(2):96–110.
Park VS, Sun MJS, Frey WD, Williams LG, Hodel Karl P, Strauss Juliet D, Wellens Sydney J, Jackson JG, Pursell ZF. Mouse model and human patient data reveal critical roles for Pten and p53 in suppressing POLE mutant tumor development. NAR Cancer. 2022;4(1):zcac004.
Serebriiskii IG, Pavlov V, Tricarico R, Andrianov G, Nicolas E, Parker MI, Newberg J, Frampton G, Meyer JE, Golemis EA. Comprehensive characterization of PTEN mutational profile in a series of 34,129 colorectal cancers. Nat Commun. 2022;13(1):1618.
Albertson TM, Ogawa M, Bugni JM, Hays LE, Chen Y, Wang Y, Treuting PM, Heddle JA, Goldsby RE, Preston BD. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A. 2009;106(40):17101–4.
Demidova EV, Ghatalia P, Arora S. Combination Strategies for Immune Checkpoint Inhibitors in PBRM1-mutant Renal Cell Carcinoma: To PARP or Not To PARP?. Eur Urol. 2022;81(2):149–50. https://doi.org/10.1016/j.eururo.2021.10.028.
Sulkowski PL, Sundaram RK, Oeck S, Corso CD, Liu Y, Noorbakhsh S, Niger M, Boeke M, Ueno D, Kalathil AN, et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet. 2018;50(8):1086–92.
Scanlon SE, Hegan DC, Sulkowski PL, Glazer PM. Suppression of homology-dependent DNA double-strand break repair induces PARP inhibitor sensitivity in VHL-deficient human renal cell carcinoma. Oncotarget. 2018;9(4):4647–60.
Hung RJ, Moore L, Boffetta P, Feng BJ, Toro JR, Rothman N, Zaridze D, Navratilova M, Bencko V, Janout V, et al. Family history and the risk of kidney cancer: a multicenter case-control study in Central Europe. Cancer Epidemiol Biomarkers Prev. 2007;16(6):1287–90.
Nicolas E, Arora S, Zhou Y, Serebriiskii IG, Andrake MD, Handorf ED, Bodian DL, Vockley JG, Dunbrack RL, Ross EA, et al. Systematic evaluation of underlying defects in DNA repair as an approach to case-only assessment of familial prostate cancer. Oncotarget. 2015;6(37):39614–33.
Shah SM, Demidova EV, Lesh RW, Hall MJ, Daly MB, Meyer JE, Edelman MJ, Arora S. Therapeutic implications of germline vulnerabilities in DNA repair for precision oncology. Cancer Treat Rev. 2022;104:102337.
Allen EMV, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–11.
Pemov A, Wegman-Ostrosky T, Kim J, Koutros S, Douthitt B, Jones K, Zhu B, Baris D, Schwenn M, Johnson A, et al. Identification of genetic risk factors for familial urinary bladder cancer: an exome sequencing study. JCO Precis Oncol. 2021;5:1830–9.
Sandler JE, Huang H, Zhao N, Wu W, Liu F, Ma S, Udelsman R, Zhang Y. Germline Variants in DNA Repair Genes, Diagnostic Radiation, and Risk of Thyroid Cancer. Canc Epidemiol Biomark Prev : Publ Am Assoc Canc Res Cosponsored Am Soc Prev Oncol. 2018;27(3):285–94.
Vlachostergios PJ, Faltas BM, Carlo MI, Nassar AH, Alaiwi SA, Sonpavde G. The emerging landscape of germline variants in urothelial carcinoma: Implications for genetic testing. Canc Treat Res Commun. 2020;23:100165.
Czene K, Hemminki K. Kidney cancer in the Swedish family cancer database: familial risks and second primary malignancies. Kidney Int. 2002;61(5):1806–13.
Study of Olaparib in Metastatic Renal Cell Carcinoma Patients With DNA Repair Gene Mutations (ORCHID). [https://clinicaltrials.gov/ct2/show/NCT03786796?term=olaparib&cond=kidney+cancer&draw=2&rank=1].
Talazoparib and Axitinib for People With Previously Treated Advanced Kidney Cancer [https://clinicaltrials.gov/ct2/show/NCT04337970?term=talazoparib&cond=kidney+cancer&draw=2&rank=1].
Acknowledgements
The work in this manuscript was supported by the resources and expertise provided by the FCCC Cell Culture, Biosample Repository, Genomics, Biostatistics, Population Sciences, Molecular Modelling, and High Throughput Screening Facilities. We acknowledge Nina Shah (summer undergraduate at FCCC) for assistance with knockdown experiments. We acknowledge Emmanuelle Nicolas (Genomics Facility, FCCC), Waleed Iqbal (Drexel University, PA) and Yan Zhou (Biostatistics Facility, FCCC) for assistance with genomic analysis. We express gratitude to Lisa Bealin, Andrea F. Forman, and Kim Rainey (Department of Clinical Genetics, FCCC) for valuable assistance on the study.
Funding
All Fox Chase Cancer Center affiliated authors are in part supported by the NCI Core Grant, P30 CA006927, to the Fox Chase Cancer Center. S.A. was supported by the DOD W81XWH-18–1-0148, CEP Award from the Yale Head and Neck Cancer SPORE, and NIH (NCI) 1UH2CA271230-01 grant. M.J.H. was supported by funding from the American Cancer Society. M.B.D was supported by the NIH U01 CA164920, R01 CA207365 grants. E.A.G. was supported by NIH R01 DK108195. E.V.D. and I.G.S. were partially supported by the Kazan Federal University Strategic Academic Leadership Program (PRIORITY-2030). R.J.D. was supported by NIH R35 GM122517. R.T.P. was supported by NIH grants 1R01GM130889 and 1R01GM137124. G.L.R. was supported by NSF grants #1919691, #1936782, and #2107108.
Author information
Authors and Affiliations
Contributions
E.V.D. performed most studies, data analysis and interpretation, all figure and table generation, writing & editing; T.R.H., I.G.S. performed data analysis, and contributed to discussion; R.V. performed TCGA analysis; S.K. & M.A. & R.L.D. performed structure modelling, discussion; T.K., R.T.P. assisted with Pol delta purification & methods; J.V., G.L.R. mutation signatures; E.A.G. assisted with discussion; M.J.H. & D.Y.T.C. & M.B.D. patients, discussion; M.B.D. & S.A., worked on discussion, designing, planning studies, writing & editing. All authors reviewed the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The FCCC Institutional Review Board Committee provided study oversight and approval (protocol number 14–831). The FCCC IRB determined the criteria for the approval of research outlined in 45 CFR 46.111. All experiments using human biospecimens were performed in accordance with relevant guidelines and regulations. Blood samples were banked in the FCCC Biosample Repository Facility under broad informed consent for research and deidentified.
Consent for publication
Not applicable.
Competing interests
All authors declare that there is no competing interest regarding this manuscript. M.J.H. performs collaborative research (with no funding) with the following: Myriad Genetics, Invitae Corporation, Ambry Genetics, Foundation Medicine, Inc. He also performs collaborative research (with no funding) and is part of a Precision Oncology Alliance funded by Caris Life Sciences (cover travel and meals at meetings). S.A. performs collaborative research (with no funding) with Caris Life Sciences, Foundation Medicine, Inc., Ambry Genetics, and Invitae Corporation. S.A.’s spouse is employed by Akoya Biosciences and has stocks in Akoya Biosciences, HTG Molecular Diagnostics, Abcam Plc., and Senzo Health. S.A., M.J.H., E.A.G., I.G.S. have patents and/or pending patents related to cancer diagnostics/treatment.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
In the original version supplementary tables 1, 2 and 8 were missing from the published article and the incorrect versions of Fig. 1 and Fig. 4 were published.
Supplementary Information
Additional file 1:
Peripheral blood lymphocyte DNA analysis: whole exome sequencing, gene variants annotation and prioritization. Supplementary Figure 1. Pathway enrichment for the genes with identified DDR germline variants according to over-representation analysis (ORA). FDR is provided inside the boxes of the heat map. Supplementary Figure 2. A. siRNA depletion of POLD1, POLE, POLH, POLK, RRM2B and ATM genes in Caki RCC cell line. A. Cells were transfected with the designated siRNAs (two per gene), or GL2 control or WRN positive control. Cells were fixed, permeabilized, blocked and stained for γH2AX antibody. Cells were scored for γH2AX foci and the data are plotted as relative induction of γH2AX to GL2 control from 2 independent experiments. *** for p<0.001, ** for p<0.01, * for p<0.05 and NS for p>0.05, Wilcoxon signed rank test. Supplementary Figure 3. Representative cell cycle data from immortalized B-cell lines from Pt #1 (POLD1, POLH), Pt#2 (POLE), Pt#16 (POLK) PBMCs and corresponding immortalized B-cell lines from matched control. A. Percent positive gated cells is presented for each cell cycle phase for each cell line (controls on the left in blue, cases on the right in red). Data for 3 independent repeats are presented. *** for p<0.001, ** for p<0.01, * for p<0.05, NS for p>0.05, Wilcoxon signed rank test. Supplementary Figure 4. Difference in DNA replication fork elongation/restoration in EBV-transformed cell line with POLK (Pt #16) variant, and representative pictures of DNA fibers for the main figures 2E and 2F. For all graphs: *** for p<0.001, ** for p<0.01, * for p<0.05 and NS for p>0.05, unpaired, non-parametric t-test, Mann-Whitney criteria. Means with SD are plotted. Data for 3 independent repeats are presented as IdU tract length or CldU/IdU tract length ratio. A. Difference in DNA replication fork elongation/restoration in EBV-transformed cell line with POLK E29K variant at the baseline and replication stress was assessed using DNA fiber assay. At baseline the EBV-transformed cells were labeled with IdU for 20 min, for fork restoration cells then were treated with 100 μM aphidicolin and then labeled with CldU for 40 min. For all conditions, after labeling, cells were lysed, and DNA fibers stretched onto glass-slides, fixed, denatured, blocked, and stained with corresponding antibodies. Fiber images were captured using the Nikon TS2R Inverted Microscope and analyzed in ImageJ software. B-D. Additional representative pictures for each DNA fiber experiment: B for IdU 20 min labeling (main figures 2E, 2F, current figure A); C for short aphidicolin treatment (main figures 2E, 2F, current figure A); D for treatment with MNNG (main figure 2E). Scale bar = 5 μm. Supplementary Figure 5. Pol δ complex primer extension competition assay with quantification. Representative gel image showing reactions performed with 20 nM Cy-3 labeled DNA-duplex template (SA#1), 20 nM of indicated proteins (wild type complex, V759I complex and both wild type + V759 variant complexes in ratio 1:1) and 500 uM dNTPs. Under presence of the both wild type and PolD1 V759I Pol δ complexes, DNA-template was extended less efficiently compared to wild type and more efficiently compared to V759I complexes alone. Data for 3 independent repeats are presented. *** for p<0.001, ** for p<0.01, * for p<0.05 and NS for p>0.05, unpaired, non-parametric t-test, Mann-Whitney criteria. Supplementary Figure 6. Raw uncropped gel images, corresponding to sub-figures in Figure 2B: A, B and C for PolD1, Pol η and loading control exposures used to generate the main figure, D for Pol ε, E for Pol κ. Supplementary Figure 7. Raw uncropped gel images, corresponding to sub-figures in Figure 3 and Supplementary Figure 5. A for Figure 3A for Pol δ complex purification, B for Figure 3B Pol η purification, C for Figure 3A on right- Pol δ complex primer extension assay, D for 3B on right- Pol η primer extension assay, E for 3C - Pol η lesion bypass assay, F for Supplementary Figure 5. Supplementary Figure 8. Mutational signature analysis in tumors from Pts 1 and 2. A. SBS signatures are reported in tumors from Pts 1 and 2. B. DBS signatures are reported in tumors from Pts 1 and 2. Supplementary table 1. List of candidate genes for WES analysis (Excel file). Supplementary table 2. Annotation of candidate variants identified in the 22 eoRCC patients (Excel file). Supplementary table 3. Complete summary of results for the DNA polymerase variants identified in the eoRCC patients. Supplementary table 4. Protein stability or ddG values for the PolD1 V759I and Pol η G209V variant proteins that were assessed biochemically in the study. Supplementary table 5. POLE and POLD1 variants in hypermutated ccRCC in TCGA. The table shows the variants from TCGA with allele counts typically observed in the GnomAD database, TMBs observed in association with these variants in other studies [60], and finally protein stability or ddG values for the variant proteins. Supplementary table 6. DNA substrates used in biochemical assays. The following are the references for the DNA substrates [47, 49, 50]. Supplementary table 7. ddG application file. Supplementary Table 8. Results from analysis of CNVs, SNVs and Indels using tumor sequencing data from Pt #1 (POLD1, POLH), and Pt #2 (POLE) (Excel file).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Demidova, E.V., Serebriiskii, I.G., Vlasenkova, R. et al. Candidate variants in DNA replication and repair genes in early-onset renal cell carcinoma patients referred for germline testing. BMC Genomics 24, 212 (2023). https://doi.org/10.1186/s12864-023-09310-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09310-8