
Abstract
Background
The serum is rich in nutrients and plays an essential role in electrolyte and acid–base balance, maintaining cellular homeostasis. In addition, serum parameters have been commonly used as essential biomarkers for clinical diagnosis. However, little is known about the genetic mechanism of the serum parameters in ducks.
Results
This study measured 18 serum parameters in 320 samples of the F2 segregating population generated by Mallard × Pekin duck. The phenotypic correlations showed a high correlation between LDH, HBDH, AST, and ALT (0.59–0.99), and higher coefficients were also observed among TP, ALB, HDL-C, and CHO (0.46–0.87). And then, we performed the GWAS to reveal the genetic basis of the 18 serum biochemical parameters in ducks. Fourteen candidate protein-coding genes were identified with enzyme traits (AST, ALP, LDH, HBDH), and 3 protein-coding genes were associated with metabolism and protein-related serum parameters (UA, TG). Moreover, the expression levels of the above candidate protein-coding genes in different stages of breast muscle and different tissues were analyzed. Furthermore, the genes located within the high-LD region (r2 > 0.4 and − log10(P) < 4) neighboring the significant locus also remained. Finally, 86 putative protein-coding genes were used for GO and KEGG enrichment analysis, the enzyme-linked receptor protein signaling pathway and ErbB signaling pathway deserve further focus.
Conclusions
The obtained results can contribute to new insights into blood metabolism and provide new genetic biomarkers for application in duck breeding programs.
Similar content being viewed by others

Background
In the animal organism, blood components reflect immune activity and nutrient metabolism. The serum is the fluid and solute component of blood that does not contain leukocytes, erythrocytes, platelets, or clotting factors [1, 2]. Furthermore, the serum is rich in nutrients, including all proteins, electrolytes, antigens, antibodies, hormones, and exogenous substances not used in the clotting process. In addition, serum plays an essential role in electrolyte and acid–base balance, maintaining the homeostasis of the intracellular environment, and transporting nutrients to the body [3, 4]. Therefore, measuring the content of various serum components is helpful in many applications, such as medical diagnostics and animal husbandry.
Serum parameters are commonly used as essential biomarkers for clinical diagnosis in the medical field [5, 6]. For instance, serum Ca, phosphorus, and alkaline phosphatase (ALP) are essential indicators of bone metabolism [7]. The level imbalance of triglyceride (TG), cholesterol (CHO), high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) in serum were usually accompanied by lipid metabolism disease [8]. In animal breeding, serum biochemical parameters indirectly reflect animal health status and economic traits. Studies have found that high levels of Lactate Dehydrogenase (LDH), Creatine Kinase (CK), Blood glucose (GLU), and aspartate transaminase (AST) were associated with pale, soft, and exudative (PSE) meat. It provides a new and effective method for detecting PSE meat by measuring the blood biochemical parameters [9]. Dong compared serum biochemical parameters between two broiler chicken lines and identified serum HDL-C and LDL-C levels as potential biomarkers for selecting of lean birds [10]. It has been reported that comparing serum metabolite compositions between obese and lean-growing pigs based on the metabonomic approach provides a useful model for childhood obesity research [11]. In Shanma duck, it was reported that the possibility of early breeding of duck by using serum biomarkers [12]. In addition, the GWAS analysis of 42 days old Pekin duck found that 54 significant QTLs associated with 23 candidate genes may contribute to 12 serum parameters [13]. Therefore, serum biomarkers have been developed as indicators for clinical in humans and breeding purposes in animals, and elucidating the genetic basis of these serum biomarkers is critical to the livestock breeding process.
With the development of genome re-sequencing technology, more and more genome-wide association (GWAS) analyses have been performed on serum biochemical indicators of livestock and poultry in recent years. In different livestock and poultry, quantitative trait loci (QTL) for serum biochemical indicators have been identified [14,15,16]. Although studies have conducted GWAS analysis on serum parameters of poultry, there is a lack of GWAS focused on the early growth period of ducks. The study of serum parameters in the early stage of poultry growth can improve the efficiency of seed selection, conduct early assessment of poultry growth and development, and improve the economic benefits. At present, many studies are mainly limited to association analysis of serum parameters and phenotypic traits, the study of genetic variability in blood parameters could contribute to design new strategies to overcome the limited effectiveness of the traditional selection programs to improve disease resistance, tolerance and resilience [17]. In addition, serum parameters have been studied by GWAS in livestock and poultry [14, 18], but few studies have been conducted on their genetic basis in ducks. Hence, in this study, total 320 samples of 3-week-old ducks were used as experimental animals to perform the GWAS of serum parameters and identify the candidate regions and genes to facilitate early breeding.
Results
Phenotypic correlation between serum biochemical indicators
Eighteen serum biochemical parameters were detected in this study, including CHO, TG, ALP, UA, HDL-C, LDL-C, etc. By calculating the coefficient variation of all serum biochemical indicators in ducks, the TP (0.11), ALB (0.10), TBIL (0.16), GLU (0.11), P (0.11), CHO (0.13), and HDL-C (0.14) showed a lower variation coefficient. In contrast, some biochemical indicators have higher variation coefficients, mainly AST (0.59), ALP (1.22), (LP(a)) (0.55), LDH (0.54), and HBDH (0.76), the variation coefficients of which exceed more than 0.60 (Table S1). Through the correlation analysis of all 18 blood biochemical indicators, the Pearson correlation coefficient ranged from 0.46–0.87 among the serum indicators, including TP, ALB, HDL-C and CHO, and the higher correlation coefficients were observed among LDH, HBDH, AST, and ALT (r = 0.59–0.99) (Fig. 1).

Pearson’s correlation coefficients among the 18 analyzed serum biochemical parameters. The value in the box represents the Pearson correlation coefficient between the two serum parameters
GWAS analysis
In this study, 18 serum biochemical indicators of 3-week-old F2 ducks, were selected as phenotypes for GWAS analysis (Correction threshold = 8.916). Significant signals in Manhattan plots were only observed among AST, ALP, LDH, HBDH, UA, and TG. The correlation QQ plots (Figure S1) showed that the model we used was reasonable, most of the observed P values were consistent with the expected values, and significant SNPs were found, indicating that the above association analysis results for serum parameter traits are reliable. The Manhattan and QQ plots for the other 14 serum biochemical indicators were indicated in Figure S2 and Figure S3, GWAS analysis revealed that these SNP were not significant.
The six biochemical indicators that showed significant signals above were divided into two categories: The enzyme traits (AST, ALP, LDH, and HBDH) and the metabolism and protein-related traits (UA and TG).
I. GWAS for enzyme traits
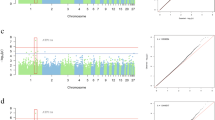
The Manhattan plot of AST showed obvious signals on chromosome 1 (Fig. 2a). A total of 20 SNPs reached the significant threshold level, of which 11 SNPs are distributed on chromosome 1. The genes annotated by Top10 SNPs are shown in Table 1, including SHANK3, SPOP, SLC30A7, ABL1, and SLC26A5. The results of ALP showed that 18 SNPs reached the significance threshold level (Fig. 2b), of which potential SNPs were mainly distributed in the PCDH11X, LDLRAD4, ABCB7, CAPZA2, MALRD1, and PRSS12 (Table 1). Only 5 SNPs reached the significant threshold level regarding the results of LDH and were distributed in the ABL1, NUP214, and KAT7 (Fig. 2c, Table 1). Regarding HBDH, 7 SNPs reached the significant threshold level in the Manhattan plot (Fig. 2d), the genes annotated by these SNPs included ABL1, KAT7, CAB2, and TRAP1 (Table 1).

The Manhattan plots of the enzyme traits. a Manhattan plot of AST. b Manhattan plot of the ALP. c Manhattan plot of the LDH. d Manhattan plot of the HBDH. Abscissa numbers represent different chromosomes. The dotted line in the Figure represents the threshold level (Correction threshold = 8.916)
II. GWAS for metabolism and protein-related traits
The Manhattan plot of UA only showed obvious signals on chromosome 2 (Fig. 3a). A total of 3 SNPs reached the significant threshold level. The significant SNPs were distributed in the 44113911 – 45103209 bp on chromosome 2. The regions harbored the candidate genes, including ATP2C1 and TMEM108 (Table 2). Then, we examined the leader SNP (Chr2: 44113911 bp) closely by calculating correlations between the SNPs within the QTL (Chr2:43.61–44.61 Mbp) surrounding the leader SNP on chromosome 2, and 56 SNPs were highly correlated (pairwise r2 > 0.6; Table S2 and Fig. 3b). In this range, we identified three candidate genes, including ATP2C1, ASTE1, NEK11 (Fig. 3c). The results of TG only showed that 1 SNP (Chr20: 2228453 bp) reached the significance threshold level (Fig. 4a), and was distributed in the CTXN1 on chromosome 20 (Table 2). Similarly, we calculated correlations between the SNPs within the QTL (Chr20: 1.73–2.73 Mbp) surrounding the leader SNP on chromosome 20, and 6 SNPs were highly correlated (pairwise r2 > 0.6; Table S3 and Fig. 4b). Only CTXN1 (Fig. 4c) is near the leader SNP.

GWAS analysis of the UA. a Manhattan plot of UA. The gray horizontal dashed lines indicate the Bonferroni significance threshold of the GWAS (Correction threshold = 8.916). b Regional plots for the loci ranging from 43.6 to 44.6 Mbp associated with UA. c there are three genes (ATP2C1, ASTE1, NEK11) in the candidate region

GWAS analysis of the TG. a Manhattan plot of TG. The gray horizontal dashed lines indicate the Bonferroni significance threshold of the GWAS (Correction threshold = 8.916). b Regional plots for the loci ranging from 2.13 to 2.33 Mbp associated with TG. c, there is only CTXN1 in the candidate region
Candidate genes expression analysis
I. Candidate genes expression analysis for enzyme traits
In animal breeding, serum biochemical parameters indirectly indicate health status and economic traits. To further determine the candidate genes for serum biochemical indicators, we compared the mRNA expression of candidate genes based on the transcriptome data in breast muscle tissues between Pekin ducks and Mallards during 2, 4, 6, and 8 weeks. Among the enzyme traits, 14 candidate protein-coding genes under the SNPs reached the significant threshold. Interestingly, ABL1 was annotated in AST, LDH, and HBDH (Table 1) and had high expression in the breast muscle of Mallard and Pekin duck. At 2, 4, and 6 weeks, the expression level in the breast muscle of the Mallard was higher than that of the Pekin ducks, but at 8 weeks, the expression level in the breast muscle of the Pekin duck was higher than that of the Mallard. The CAPZA2 gene was annotated in ALP and highly expressed in the breast muscle, and the expression level was higher in Mallard. Besides, SHANK3, SPOP, LDLRAD4, and ABCB7 also have a relatively high expression level in the breast muscle (Fig. 5a and c).

The expression level of candidate protein-coding genes related to enzyme traits was analyzed. a, b, Expression levels of putative candidate protein-coding genes in different stages of breast muscle and different tissues (Pekin duck). c, d, Expression levels of putative candidate protein-coding genes in different stages of breast muscle and different tissues (Mallard)
In addition, we also analyzed the expression of candidate genes in different tissues (breast muscle, skin fat, liver, belly fat, brain, heart, kidney, lung, spleen). ABL1 is widely and highly expressed in all tissues, especially in skin fat and lung, with the highest expression in the skin fat of Pekin duck and the highest expression in Mallard’s lung. SHANK3, PCDH11X, and PRSS12 have the highest expression levels in the brain, SPOP expression level was highest in the brain, LDLRAD4 was highly expressed in the liver and spleen, and the expression level in Mallard was higher than Pekin duck. CAPZA2 has the highest expression level in belly fat (Fig. 5b and d).
II. Candidate genes expression analysis for metabolism and protein-related traits
The genes annotated by metabolism and protein-related traits only include ATP2C1, TMEM108, and CTXN1. Same as above, we analyzed the expression levels of candidate genes in the breast muscles of Pekin duck and Mallard at different growth stages (2, 4, and 6 weeks). ATP2C1 was highly expressed in breast muscle, and CTXN1 was less expressed in breast muscle. The expression level of TMEM108 in the breast muscle of Pekin duck and Mallard at different stages showed a decreasing trend (Fig. 6a and c). Besides, the expression analysis of candidate genes in different tissues showed that CTXN1 was especially highly expressed in the brain. ATP2C1 was expressed in all tissues, with the highest in bally fat and the lowest in the liver. TMEM108 was mainly expressed in the brain, kidney, and lung, and hardly expressed in other tissues (Fig. 6b and d).

The expression level of candidate protein-coding genes related to metabolism and protein-related traits was analyzed. a, b, Expression levels of putative candidate protein-coding genes in different stages of breast muscle and different tissues (Pekin duck). c, d, Expression levels of putative candidate protein-coding genes in different stages of breast muscle and different tissues (Mallard)
Candidate genes and functional annotation
The annotation of each important locus is considered as candidate gene. Moreover, the genes located within the high-LD region (r2 > 0.4 and − log10(P) < 4) neighboring the significant locus also remained. Finally, 86 putative protein-coding genes were used for GO and KEGG enrichment analysis (Table S4). Enzyme-linked receptor protein signaling pathway (GO:0,007,167) was the most significant GO term in the biological process. Within molecular function, ATP binding (GO:0,005,524) was the most dominant GO subcategory (Fig. 7b). Besides, another 10 GO terms also were significantly enriched, including adenyl ribonucleotide binding, adenyl nucleotide binding, purine ribonucleoside, etc. (Table S5). Five KEGG pathways were significantly enriched, including the ras signaling pathway, axon guidance, focal adhesion, proteoglycans in cancer, and the ErbB signaling pathway (Fig. 7b and Table S6).

GO and KEGG enrichment analysis for 86 candidate genes. a GO enrichment analysis for 86 candidate genes. The x-axis indicates the number of genes for each GO term; the y-axis corresponds to the GO terms. The color of the bar represents the P value. b KEGG enrichment analysis for 86 candidate genes. The x-axis shows the gene ratio; the y-axis represents KEGG pathways. The dot color represents the P value, and the dot size represents the number of genes enriched in the reference pathway
Discussion
Serum biochemical indicators can be used for the clinical detection of poultry's nutritional metabolism and growth performance. In this study, we measured the 18 serum parameters of ducks. The phenotypic correlation analysis showed that some serum biochemical indicators have high correlations. Like LDH and HBDH have high correlation, HBDH is an indirect reflection of LDH activity, and its activity changes parallel to the total LDH activity, the increase or decrease of LDH and HBDH remained concomitant, and serum parameters are mostly determined by biological genetic material [19], so we can analyze the genetic mechanism of these serum parameters at the genome level. This has aroused our interest in revealing the genetic determinants of duck serum biochemical indicators through genome-wide association study.
AST mainly exists in the mitochondria and cytoplasm of hepatocytes and is an essential enzyme for protein synthesis in hepatocytes. This enzyme is released into the blood when liver cells degenerate and necrosis or increase cell membrane permeability and are usually used to detect liver health as a clinical biomarker [20]. LDH and HBDH are usually used as one liver function indicators, and they are also used as cardiomyocyte markers in the clinical. Interestingly, our results showed that ABL1, SPOP, and KAT7 were all annotated by these three enzymes (AST, LDH, HBDH) as candidate genes.
These genes can be proved to directly or indirectly affect the normal progress of liver or body life activities. It was reported that hepatocellular carcinoma (HCC) samples have increased levels of ABL1 compared with nontumor liver tissues, and overexpress ABL1 correlates with shorter survival times for patients. Knocking out ABL1 or inhibiting its expression reduced HCC cells and slowed liver tumor growth in mice [21]. In addition, recent research has found that ABL1 was associated with immune infiltration and the prognosis of HCC [22]. Gene expression analysis showed that the expression of the ABL1 gene was most expressed in skin fat and lung, decreased in breast muscle at different stages, but increased at 8 W of Pekin duck. Studies have identified the critical role of SPOP in regulating proliferation and migration in liver cancer [23, 24]. Expression analysis showed that there was no differential expression of this gene. Bai found that miR-639 inhibits the proliferation and migration of human hepatocellular carcinoma cells through the KAT7/Wnt/β-Catenin Pathway, KAT7 expression promotes cell proliferation and migration of human HCC cells in vitro [25]. Duck viral hepatitis (DVH) is one of the most serious infectious diseases in Pekin ducks [26]. Therefore, genetic analysis of serum biochemical indicators for evaluating liver function and screening candidate genes are highly important for duck quality breeding and liver performance determination.
ALP has essential physiological functions in the body, as a marker of osteoblasts maturation and an important indicator of bone metabolism [27, 28]. Our results showed that the candidate genes of ALP mainly include ATRX, ALG13, CHRDL1, and AMMECR1. Hypomorphic mutations of the ATRX could lead to skeletal deformities, and individuals with ATRX mutations show delayed bone age [29, 30]. A study has identified ALG13 as a potential osteoporosis marker gene related to osteoclast activity and hypogonadal bone loss. CHRDL1 is a secreted glycoprotein, which can bind to BMPs family ligands and promote osteoblast differentiation in vivo [31]. Research in humans found that AMMECR1 is potentially involved in cell cycle control and linked to a new syndrome with bone alterations [32].
The metabolism and protein-related traits included UA and TG. UA, the end product of purine metabolism is excreted predominantly by the proximal tubules. UA is a marker of kidney disease and is also associated with hypertension, gout, hyperuricemia, and some cardiovascular diseases [33, 34]. PIK3R4 is the candidate gene for UA, which can cause ciliopathies and affect kidney function [35]. In human, TMEM108 is a candidate gene associated with stroke by GWAS [36]. TG is an important indicator of heart health. In this study, there is only CTXN1 in the candidate QTL region. However, there are few studies on this gene. Interestingly, this gene is highly expressed in the brain, then the details of the gene may require further study.
To gain insight into the function of 86 candidate genes, we performed GO and KEGG enrichment analyses. GO terms with major enrichment of candidate genes, including the enzyme linked-receptor protein signaling pathway, ATP binding, and enzymatic activity. Therefore, we speculate that candidate genes related to blood biochemical indicators play an essential role in biological processes such as energy metabolism. ALP in the body plays a crucial role in cell cycle, growth, apoptosis and signal transduction pathways, also is a marker of osteoblast maturation and an important indicator of bone metabolism [27, 37]. As a candidate gene related to ALP, CAPZA2 highly expressed in the breast muscle and belly fat, which enriched in enzyme-linked receptor protein signaling pathway, regulation of cellular process and biological process. Researchers found that a de novo inframe deletion variant in CAPZA2 tentacle domain with global developmental delay and skeletal malformation of head [38]. Therefore, CAPZA2 could be further studied as an essential gene. In addition, ErbB signaling pathway focused by us, the candidate gene ABL1 of AST, LDH and HBDH is enriched in this pathway, and the pathway plays a key role in the development of many cancers and the immune response [39], ABL1 also is also involved in the carcinogenesis and immune process.
Conclusion
In summary, we detected 18 serum biochemical indicators and analyzed them by GWAS in this study. We found 6 serum parameter phenotypic indicators showing significant signals by GWAS analysis. Expression analysis of 14 putative candidate protein-coding genes related to enzyme traits and 3 candidate protein-coding genes related to metabolism and protein-related traits were performed. The candidate genes and SNPs found in this study may contribute to the future research of serum biomarkers and provide a reference for the early breeding of ducks.
Methods
Experimental population and sample preparation
The Mallard × Pekin F2 resource population used in this study was established by the Key Laboratory of Animal (Poultry) Genetics Breeding and Reproduction. The F2 segregating population description refers to Zhou et al. [40]. In the orthogonal cross, 10 ♂ Pekin ducks × 100 ♀ Mallard ducks were selected as parents. In the reciprocal cross, 4 ♂ Mallard ducks × 40 ♀ Pekin ducks were selected as parents, and nearly 2,000 F2 ducks were finally generated. All ducks had free access to feed and water and were managed in the same environment. In this study, 320 ducks randomly sampled.
Biochemical indicators’ measurements
Total 320 blood samples were collected from the wing vein of ducks and stored at 4 °C until centrifuged at 3000 rpm for 10 min to obtain serum. The levels of plasma parameters were measured using an automatic analyzer (Hitachi 7080, Japan) with a commercial kit (Maccura, China), including alanine aminotransferase (ALT), aspartate aminotransferase (AST), total protein (TP), albumin (ALB), total bilirubin (TBIL), alkaline phosphatase (ALP), glucose (GLU), urea nitrogen (BUN), uric acid (UA), phosphorus(P), total cholesterol (CHO), triglyceride (TG), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), lipoprotein(a) (LP(a)), creatine kinase (CK), lactate dehydrogenase (LDH), α- Hydroxybutyrate dehydrogenase (HBDH).
DNA isolation and sequencing
Genomic DNA was extracted from the blood using the standard phenol/chloroform extraction method. Nanodrop and agarose gel electrophoresis estimated the quality of DNA. Generate two paired-end libraries using standard procedures according to the manufacturer's protocol (Illumina, USA). The average insert size is 500 bp, and the read length is 150 bp. Ultimately, these libraries were sequenced on the Illumina® Hiseq X-Ten platform.
Variant detection and genotyping
The 2 × 150-bp paired-end reads were mapped to the Pekin duck reference genome (IASCAAS_Peking Duck_PBH1.5, GCF_003850225.1). After that, SNPs calling was performed using the GATK (version 3.5.0) HaplotypeCaller tool [41] with the following cut-off values: QUAL < 100.0, MQ < 40.0, QD < 2.0, SOR > 3.0, FS > 60.0, ReadPosRankSum < -8.0, and MQRankSum < -12.5. The output was further filtered using VCFtools (Version 0.1.15) [42], and the criteria were as follows: Only SNPs with minor allele frequencies above 0.05 and maximum allele frequencies below 0.99 were retained, and the maximum missing rate was set at < 0.1 and SNPs had to have only two alleles. After filtering, 320 ducks from an F2 segregating population mated by Pekin duck and Mallard were genotyped, and 8,234,067 SNPs were prepared for subsequent analysis.
GWAS
GWAS was performed on the phenotype Indicators with the mixed linear model program EMMAX [43]. Population structure and cryptic relationships were considered to minimize false positives and increase statistical power. The first three principal component values (PCA eigenvectors) are set as a fixed effect in the mixed model to correct population stratification [44]. The Random effect was the phylogenetic matrix estimated by all genome-wide SNPs. We defined the whole-genome significance cutoff as the Bonferroni threshold, 0.01/Total SNPs (− log 10 (P) = 8.916). The linear model is as follows:
where y is the vector of phenotypic values of serum biochemical indicators, Xα is the fixed effects; Zβ represents the effect of SNP, and β represents allele substitution effect; Wµ represents random animal effects with a variance–covariance structure based on the kinship matrix estimated using whole-genome SNP genotypes, and e is random residuals for perimysial thickness data.
Total RNA isolation and construction of RNA-seq libraries
Pekin duck and Mallard collected multiple tissues (breast muscle, skin fat, liver, belly fat, brain, heart, kidney, lung, spleen). In detail, 2 W, 4 W, 6 W, and 8 W, breast muscles of 3 Pekin ducks and 3 Mallards were collected, respectively. Other tissues were collected at 8 W. The total RNA was isolated with Trizol reagent (Takara), and then the integrity and concentration were estimated using a NanoDrop spectrophotometer (Thermo Fisher Scientific, USA), and verified using the agarose gel method. Only qualified samples were purified for RNA-seq library construction. The libraries meeting the quality criteria were sequenced using the Illumina Hiseq 4000 platform, which generated paired-end reads of 150 bp. RNA-seq paired-end reads were mapped to the Pekin duck reference genome (GCA_003850225.1) using TopHat version 2.0.11 software [45]. Subsequently, read counts per million (CPM) of the genes were obtained by running htseq-count [46]. CPM-mapped sequence reads for each gene were calculated by edgeR version 3.20.9 package, where CPM represents the gene expression level [47].
Candidate genes and functional annotation
To identify the positional candidate genes that are potentially associated with serum indicators, the genes located within the high-LD region (r2 > 0.4 and − log10(P) < 4) neighboring the significant locus also remained [48]. These regions were then referenced against the duck reference genome (IASCAAS_Peking Duck_PBH1.5, GCF_003850225.1) to find genes located in the vicinity of the significant SNPs. The candidate genes were performed GO enrichment analysis and KEGG enrichment analysis using the DAVID website (DAVID: Functional Annotation Tools (ncifcrf.gov)).
Availability of data and materials
In this study, all sequences supporting the conclusions are deposited at the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) with the accession number PRJNA471401 and PRJNA450892. The genome assembly, sequence data, and SNP information were deposited in BIG Data Center (http://bigd.big.ac.cn/) with the accession numbers PRJCA000651, PRJCA000647, and GVM000015. The RNA-Seq datasets used in this study are available at BIG Data Center (http://bigd.big.ac.cn/) with the accession number PRJCA001307. For Other data supporting the results of this study, see the supplementary file.
Abbreviations
- LDH:
-
Lactate dehydrogenase
- HBDH:
-
α-Hydroxybutyrate dehydrogenase
- AST:
-
Aspartate aminotransferase
- ALT:
-
Alanine aminotransferase
- TP:
-
Total protein
- ALB:
-
Albumin
- HDL-C:
-
High-density lipoprotein cholesterol
- LDL-C:
-
Low density lipoprotein cholesterol
- CHO:
-
Cholesterol
- TG:
-
Triglyceride
- CK:
-
Creatine kinase
- GLU:
-
Glucose
- TBIL:
-
Total bilirubin
- BUN:
-
Urea nitrogen
- P:
-
Phosphorus
- GWAS:
-
Genome-wide association study
- UA:
-
Uric acid
- LD:
-
Linkage disequilibrium
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Gene and Genomes database
- PSE:
-
Pale, soft, and exudative meat
- QTL:
-
Quantitative trait loci
- LP(a):
-
Lipoproteins(a)
- QQ plot:
-
Quantile–Quantile plot
- SNP:
-
Single nucleotide polymorphism
- HCC:
-
Hepatocellular carcinoma
References
Goyal A, Terry MB, Siegel AB. Serum antioxidant nutrients, vitamin A, and mortality in U.S. Adults. Cancer Epidemiol Biomarkers Prev. 2013;22(12):2202–11.
Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, et al. The human serum metabolome. PLoS One. 2011;6(2):e16957.
Carlson GP. Electrolyte, and Acid-Base Balance: Clinical Biochemistry of Domestic Animals. 1997.
Crookshank HR, Elissalde MH, White RG, Clanton DC, Smalley HE. Effect of transportation and handling of calves upon blood serum composition. J Anim Sci. 1979;48(3):430–5.
Ma ZY, Gong YF, Zhuang HK, Zhou ZX, Huang SZ, Zou YP, Huang BW, Sun ZH, Zhang CZ, Tang YQ, et al. Pancreatic neuroendocrine tumors: a review of serum biomarkers, staging, and management. World J Gastroenterol. 2020;26(19):2305–22.
Martinez E, Martorell J, Riambau V. Review of serum biomarkers in carotid atherosclerosis. J Vasc Surg. 2020;71(1):329–41.
Reyer H, Oster M, Wittenburg D, Murani E, Ponsuksili S, Wimmers K. Genetic contribution to variation in blood calcium, phosphorus, and alkaline phosphatase activity in pigs. Front Genet. 2019;10:590.
Du T, Sun X, Yu X. Non-HDL cholesterol and LDL cholesterol in the dyslipidemic classification in patients with nonalcoholic fatty liver disease. Lipids Health Dis. 2017;16(1):229.
Qu D, Zhou X, Yang F, Tian S, Zhang X, Ma L, Han J. Development of class model based on blood biochemical parameters as a diagnostic tool of PSE meat. Meat Sci. 2017;128:24–9.
Dong JQ, Zhang H, Jiang XF, Wang SZ, Du ZQ, Wang ZP, Leng L, Cao ZP, Li YM, Luan P, et al. Comparison of serum biochemical parameters between two broiler chicken lines divergently selected for abdominal fat content. J Anim Sci. 2015;93(7):3278–86.
He Q, Ren P, Kong X, Wu Y, Wu G, Li P, Hao F, Tang H, Blachier F, Yin Y. Comparison of serum metabolite compositions between obese and lean growing pigs using an NMR-based metabonomic approach. J Nutr Biochem. 2012;23(2):133–9.
Huang Z, Zhong Z, Xiao T. Correlation Analysis on Egg-laying Performance and Blood Biochemical Parameters in Shanma Duck. Chinese Agricultural Science Bulletin. 2011.
Zhu F, Cui QQ, Yang YZ, Hao JP, Yang FX, Hou ZC. Genome-wide association study of the level of blood components in Pekin ducks. Genomics. 2020;112(1):379–87.
Bovo S, Mazzoni G, Bertolini F, Schiavo G, Galimberti G, Gallo M, Dall’Olio S, Fontanesi L. Genome-wide association studies for 30 haematological and blood clinical-biochemical traits in Large White pigs reveal genomic regions affecting intermediate phenotypes. Sci Rep. 2019;9(1):7003.
Shi K, Niu F, Zhang Q, Ning C, Yue S, Hu C, Xu Z, Wang S, Li R, Hou Q, et al. Identification of whole-genome significant single nucleotide polymorphisms in candidate genes associated with serum biochemical traits in Chinese Holstein Cattle. Front Genet. 2020;11:163.
Hu ZL, Park CA, Reecy JM. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2019;47(D1):D701-d710.
Bishop SC. A consideration of resistance and tolerance for ruminant nematode infections. Front Genet. 2012;3:168.
Zhang H, Shen L, Li Y, Xu Z, Zhang X, Yu J, Cao Z, Luan P. Genome-wide association study for plasma very low-density lipoprotein concentration in chicken. J Anim Breed Genet. 2019;136(5):351–61.
Lin JP, Zheng G, Joo J, Cupples LA. Genome-wide linkage and association scans for quantitative trait Loci of serum lactate dehydrogenase-the framingham heart study. Hum Genomics Proteomics. 2010;2010:905237.
Vuppalanchi R, Chalasani N. Laboratory Tests in Liver Disease. In: Practical Hepatic Pathology: a Diagnostic Approach (Second Edition). 2018. p. 43–53.
Wang F, Hou W, Chitsike L, Xu Y, Bettler C, Perera A, Bank T, Cotler SJ, Dhanarajan A, Denning MF, et al. ABL1, Overexpressed in Hepatocellular Carcinomas, Regulates Expression of NOTCH1 and Promotes Development of Liver Tumors in Mice. Gastroenterology. 2020;159(1):289-305.e216.
Liu R, Kong W, Jiang Z, Zheng S, Yuan X, Ye L. ABL1 Is a Prognostic Marker and Associated with Immune Infiltration in Hepatocellular Carcinoma. J Oncol. 2021;2021:1379706.
Huang Y, Tan N, Jia D, Jing Y, Wang Q, Li Z, Zhang J, Liu L, Li J, Chen Z, et al. Speckle-type POZ protein is negatively associated with malignancies and inhibits cell proliferation and migration in liver cancer. Tumour Biol. 2015;36(12):9753–61.
Song Y, Xu Y, Pan C, Yan L, Wang ZW, Zhu X. The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol Cancer. 2020;19(1):2.
Bai Z, Xia X, Lu J. MicroRNA-639 is Down-Regulated in Hepatocellular Carcinoma Tumor Tissue and Inhibits Proliferation and Migration of Human Hepatocellular Carcinoma Cells Through the KAT7/Wnt/β-Catenin Pathway. Med Sci Monit. 2020;26:e919241.
Liang S, Wang MS, Zhang B, Feng Y, Tang J, Xie M, Huang W, Zhang Q, Zhang D, Hou S. NOD1 Is Associated With the Susceptibility of Pekin Duck Flock to Duck Hepatitis A Virus Genotype 3. Front Immunol. 2021;12:766740.
Bhattarai T, Bhattacharya K, Chaudhuri P, Sengupta P. Correlation of common biochemical markers for bone turnover, serum calcium, and alkaline phosphatase in post-menopausal women. Malays J Med Sci. 2014;21(1):58–61.
Schoppet M, Shanahan CM. Role for alkaline phosphatase as an inducer of vascular calcification in renal failure? Kidney Int. 2008;73(9):989–91.
Gibbons RJ, Brueton L, Buckle VJ, Burn J, Clayton-Smith J, Davison BC, Gardner RJ, Homfray T, Kearney L, Kingston HM, et al. Clinical and hematologic aspects of the X-linked alpha-thalassemia/mental retardation syndrome (ATR-X). Am J Med Genet. 1995;55(3):288–99.
Gibbons RJ, Higgs DR. Molecular-clinical spectrum of the ATR-X syndrome. Am J Med Genet. 2000;97(3):204–12.
Liu T, Li B, Zheng XF, Jiang SD, Zhou ZZ, Xu WN, Zheng HL, Wang CD, Zhang XL, Jiang LS. Chordin-Like 1 Improves Osteogenesis of Bone Marrow Mesenchymal Stem Cells Through Enhancing BMP4-SMAD Pathway. Front Endocrinol. 2019;10:360.
Moysés-Oliveira M, Giannuzzi G, Fish RJ, Rosenfeld JA, Petit F, Soares MF, Kulikowski LD, Di-Battista A, Zamariolli M, Xia F, et al. Inactivation of AMMECR1 is associated with growth, bone, and heart alterations. Hum Mutat. 2018;39(2):281–91.
Fathallah-Shaykh SA, Cramer MT. Uric acid and the kidney. Pediatr Nephrol. 2014;29(6):999–1008.
Feig DI, Kang DH, Nakagawa T, Mazzali M, Johnson RJ. Uric acid and hypertension. Curr Hypertens Rep. 2006;8(2):111–5.
Stoetzel C, Bär S, De Craene JO, Scheidecker S, Etard C, Chicher J, Reck JR, Perrault I, Geoffroy V, Chennen K, et al. A mutation in VPS15 (PIK3R4) causes a ciliopathy and affects IFT20 release from the cis-Golgi. Nat Commun. 2016;7:13586.
Keene KL, Hyacinth HI, Bis JC, Kittner SJ, Mitchell BD, Cheng YC, Pare G, Chong M, O’Donnell M, Meschia JF, et al. Genome-Wide Association Study Meta-Analysis of Stroke in 22 000 Individuals of African Descent Identifies Novel Associations With Stroke. Stroke. 2020;51(8):2454–63.
Meenakshi B, Chandran CR, Aravindhan TR, Devaraj N, Rama KV, Valarmathi S. A Comparative Study on the Levels of Alkaline Phosphatase and Trace Elements in Gingival Crevicular Fluid and Serum of Periodontitis and Gingivitis Patients with Healthy Population. Iosr Journal of Dental & Medical Sciences. 2017;16(1):14–7.
Pi S, Mao X, Long H, Wang H. A de novo inframe deletion variant in CAPZA2 tentacle domain with global developmental delay and secondary microcephaly. Clin Genet. 2022;102(4):355–6.
Kumagai S, Koyama S, Nishikawa H. Antitumour immunity regulated by aberrant ERBB family signalling. Nat Rev Cancer. 2021;21(3):181–97.
Zhou Z, Li M, Cheng H, Fan W, Yuan Z, Gao Q, Xu Y, Guo Z, Zhang Y, Hu J, et al. An intercross population study reveals genes associated with body size and plumage color in ducks. Nat Commun. 2018;9(1):2648.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer N, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42(4):348–54.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–9.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11.
Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Zhang Y, Wang Y, Li Y, Wu J, Wang X, Bian C, Tian Y, Sun G, Han R, Liu X, et al. Genome-wide association study reveals the genetic determinism of growth traits in a Gushi-Anka F(2) chicken population. Heredity (Edinb). 2021;126(2):293–307.
Perciedusert N, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020;18(7):e3000411.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31972523), the Young Top-notch Talent Project of the National Ten Thousand Talent Program, the China Agriculture Research System of MOF and MARA (CARS-42), and the CAAS Innovation Team Project (ASTIP-IAS-9, CAAS-ZDRW202104). The funders had no role in collecting, analyzing, and studying data or writing the manuscript.
Author information
Authors and Affiliations
Contributions
ZZ conceived and designed the experiments. HT prepared the samples, collected the phenotypes, prepared the Figures, and wrote the manuscript. HZ and DL performed the data analysis and prepared the Tables. ZW, DY, WF, and ZG prepared the samples, collected the phenotypes and revised all Figures and Tables. WH and SH revised the manuscript. All authors reviewed the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animal experiments in this study are according to the Regulations of the Chinese National Research Council and approved by the Science Research Department of the Institute of Animal Sciences, Chinese Academy of Agricultural Sciences (CAAS) (Beijing, China). All experimental procedures and methods were carried out following approved guidelines to ensure animal welfare. The study was carried out in compliance with the ARRIVE guidelines [49].
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Quantile-quantile (QQ) plot on Serum AST, ALP, LDH, HBDH, UA and TG.
Additional file 2:
Figure S2. Manhattan and quantile-quantile (QQ) plot on Serum ALT, TP, ALB, TBIL, GLU, and BUN.
Additional file 3:
Figure S3. Manhattan and quantile-quantile (QQ) plot on Serum P, CHO, HDL-C, LDL-C, LP(a) and CK.
Additional file 4:
Table S1. Determination results of 18 blood biochemical indicators.
Additional file 5:
Table S2. SNPs with a pairwise r2 > 0.6 with the leader SNP at chr2: 44113911 bp.
Additional file 6:
Table S3. SNPs with a pairwise r2 > 0.6 with the leader SNP at chr20: 2228453 bp.
Additional file 7:
Table S4. Candidate genes for enrichment analysis. The putative candidate genes, including the genes annotated on potential candidate SNPs (top 10) and genes located in the genomic region (r2 > 0.4 and -log10(P) < 4). A total of 85 unique potential candidate genes were identified for KEGG enrichment analysis.
Additional file 8:
Table S5. The result of GO enrichment analysis.
Additional file 9:
Table S6. The result of the KEGG enrichment analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tang, H., Zhang, H., Liu, D. et al. Genome-wide association study reveals the genetic determinism of serum biochemical indicators in ducks. BMC Genomics 23, 856 (2022). https://doi.org/10.1186/s12864-022-09080-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-09080-9