
Abstract
Background
In our previous study, Citrobacter sp. XT1-2-2 was isolated from high cadmium-contaminated soils, and demonstrated an excellent ability to decrease the bioavailability of cadmium in the soil and inhibit cadmium uptake in rice. In addition, the strain XT1-2-2 could significantly promote rice growth and increase rice biomass. Therefore, the strain XT1-2-2 shows great potential for remediation of cadmium -contaminated soils. However, the genome sequence of this organism has not been reported so far.
Results
Here the basic characteristics and genetic diversity of the strain XT1-2-2 were described, together with the draft genome and comparative genomic results. The strain XT1-2-2 is 5040459 bp long with an average G + C content of 52.09%, and contains a total of 4801 genes. Putative genomic islands were predicted in the genome of Citrobacter sp. XT1-2-2. All genes of a complete set of sulfate reduction pathway and various putative heavy metal resistance genes in the genome were identified and analyzed.
Conclusions
These analytical results provide insights into the genomic basis of microbial immobilization of heavy metals.
Similar content being viewed by others


Background
The Citrobacter species belong to the domain Bacteria [1], the phylum Proteobacteria [2], the class Gammaproteobacteria [3], the order Enterobacteria [4], the Enterobacteriaceae family [5] and Citrobacter genus [6], and was introduced in 1932 by Werkman and Gillen [7]. The Citrobacter genus typically utilizes citric acid as the primary carbon source [8, 9]. Citrobacter species are commonly found in soil, water, sewage and food, sometimes exist as a normal flora in the gastrointestinal tract, also in human and animal feces, and sometimes as opportunistic pathogens isolated from clinical samples [10, 11].
Citrobacter sp. XT1-2-2 was isolated from high Cd-contaminated paddy soil. In our previous study, we found that the strain XT1-2-2 could tolerate a variety of heavy metals, and showed remarkable removal efficiency of Cd2+ in the solution compared with controls. Meanwhile, the strain could decrease the bioavailability of Cd in the soil and inhibit Cd uptake in rice plants. In addition, the strain could significantly promote rice growth and increase rice biomass [12]. These effects are mainly due to the strain's ability to reduce sulfate (SO42−) to sulfide ions (S2−), and then sulfide ions (S2−) can combine with cadmium ions (Cd2+) existing in the soil to produce cadmium sulfide (CdS) precipitation, thereby converting the highly active cadmium ions (Cd2+) into residual cadmium sulfide (CdS), and then reduces the absorption and transport of cadmium by rice [13, 14]. Therefore, these characteristics made the strain XT1-2-2 strong potential for application to remediate Cd-contaminated paddy soils. However, the genome sequence and basic properties of this organism have not been reported so far. Here we report the high quality draft genomic information of the strain XT1-2-2 and conduct comparative genomic analysis with the other relevant reference sequenced genomes.
Results
Organism classification and characteristics
The strain XT1-2-2 is Gram-negative, facultatively anaerobic, non-sporulating, motile and rod-shaped (Fig. 1). The colonies are circular, smooth and opaque with a regular slick edge on SRB agar plates [13]. The strain XT1-2-2 is a non-pathogenic and free-living bacterium. Growth occurs at 15–40℃ and at pH 5–10. Optimal growth occurs at 30℃ and at pH 6–8. The basic characteristics and classification of the strain XT1-2-2 are shown in Table S1. The results of previous studies showed that the strain XT1-2-2 exhibited high resistance to a variety of heavy metals, and the MIC of the strain XT1-2-2 for Cd2+ was as high as 400 mg/L [12].

Scanning electron micrograph depicting effect of Cd on cellular morphology of Citrobacter sp. XT1-2-2. a Citrobacter sp. XT1-2-2 in absence of Cd2+. b Citrobacter sp. XT1-2-2 in presence of 100mg/L Cd2+
SEM analysis
The scanning electron micrograph (SEM) analysis (Fig. 1) showed that cell shape was significantly influenced under high concentrations of Cd2+ of up to 100 mg/L. Compared with the control group (a), some cells in the treatment group (b) were twisted, lysed, or even broken. Oxidative damage and membrane permeability changes, caused by high Cd concentration, might be responsible for the cell morphology changes.
TYGS analysis and phylogenetic relation
The phylogenetic tree inferred from the intergenomic distance calculated from Genome BLAST Distance Phylogeny (GBDP) in the Type Strain Genome Server (TYGS) is shown in Fig. 2. Based on the 16S rDNA comparison, Citrobacter sp. XT1-2-2 is the closest relative to Citrobacter werkmanii BF-6 (CP019986.1) (Fig. 2). Similarly, the whole genome-based phylogeny also showed a cluster of the same species as the closest relatives of Citrobacter sp. XT1-2-2 (Fig. 2). All the Citrobacter species clustered together in a paraphyletic clade from the other type strains.

Genome BLAST Distance Phylogeny method (GBDP) for phylogenetic placement analysis using FastME 2.1.6.1 with 100 bootstrap values. A 16S rDNA gene sequence-based phylogeny of Citrobacter sp. XT1-2-2 with the closely related type strains and whole genomes with 93.5% average branch support. B Whole-genome sequence based phylogeny among the closely related type strains and whole genomes with 97.2% branch support. The numbers above branches represent the GBDP pseudo-bootstrap value, which is greater than 60%
Genome sequencing, annotation and features
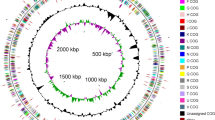
The strain XT1-2-2 was selected for sequencing particularly due to its multiple heavy metals resistance and heavy metal removal ability. Genome sequencing was performed by Shanghai Majorbio Bio-pharm Technology Co., Ltd (Shanghai, China). The project information is summarized in Table S2. The constructed standard shotgun library generated 165575 reads totaling 1164774594 bp and an average length of 7034.7 bp. The total size of the genome is 5,040,459 bp with 52.09% G + C content (Fig. 3). The genome properties and statistics are shown in Table S3. A total of 4801 genes, 4601 CDSs with protein, and 120 predicted RNA genes, including 84 tRNA, 25 rRNA and 11 ncRNA were predicted. In addition, 4383 (91.0%) genes are distributed into COG functional categories (Fig. 4).

A graphical circular map of Citrobacter sp. XT1-2-2. From outside to center, rings 1, 4 show protein-coding genes colored by COG categories on forward/reverse strand; rings 2, 3 denote genes on forward/reverse strand; ring 5 shows G + C % content plot; ring 6 shows GC skew; the innermost ring shows the marker of genome size

Number of genes associated with the 20 general COG functional categories. The gene number of the category B, W, Y, Z and X is zero
Identification of sulfate reduction pathway
According to the KEGG prediction analysis, the strain XT1-2-2 contains all genes of the complete set of sulfate reduction pathway (Fig. 5), including cysA, cysC, cysD, cysH, cysI, cysJ, cysN, cysP, cysU, cysW. which provides the genomic basis for the strain to reduce sulfate (SO42−) to sulfide (S2−) to form CdS precipitation, thereby reducing the uptake and transport of Cd2+ by rice. The basic information of sulfate reduction pathway genes including gene ID on chromosome, gene name, gene description has been analyzed, and heavy metal resistance genes have been already compared with the reference proteins in the swissprot database, and all the information has been shown in Table S4.

Putative sulfur metabolism pathway and assimilatory sulfate reduction genes in Citrobacter sp. XT1-2-2. a Putative sulfur metabolism pathway. b Assimilatory sulfate reduction gens in Citrobacter sp. XT1-2-2. c Putative sulfate transporter CysPUWA
Identification of heavy metal resistance genes
The results of previous studies showed that the strain XT1-2-2 could tolerate a variety of heavy metals (Cd2+, Pb2+, Zn2+, Mn2+ and Cr6+) and the removal rate of Cd2+ in solution is as high as 82.3 ± 2.1% within 240 min [12]. These results suggest that the strain XT1-2-2 has developed many evolutionary strategies to adapt the complex heavy metal pollution environment. According to the results of genome annotation, the strain XT1-2-2 contains multiple putative functional proteins, which are related to heavy metal resistance, including transporters, resistance proteins and metal reductases, and so on (Fig. 6). The basic information of heavy metal resistance genes including gene ID on chromosome, gene name, gene description has been analyzed, and heavy metal resistance genes have been already compared with the reference proteins in the swissprot database, and all the information has been shown in Table S5.

Heavy metal resistance genes distributed in Citrobacter sp. XT1-2-2. a Zinc or cadmium transporter, b Chromate transporter, c Zinc/ cadmium /mercury /lead-transporting ATPase, d Zinc ABC transporter permease, e Arsenical resistance protein, f copper resistance system, g Mercury transport system, h Cobalt ECF transporter complex
Identification of the heavy metal resisitance genomic island
The genomic islands in Citrobacter sp. XT1-2-2 were predicted by genome annotation combined with Island Viewer 4 software (Fig. 7), and all the genes (Table S6) on the genomic islands were further analyzed. Among all heavy metal resistance genes present on the genome, the membrane transporter chrA and mercury transport system (merR, merT, merP, merC, merA, merD, and merE) were present on the same gene island. The region of the gene island ranging from nucleotide positions 3469026 to 3490287 were annotated as heavy metal resistance genomic island by SIGIHMM and IslandPath-DIMOB analysis.

Identification of heavy metal resistance genomic island in the genome of Citrobacter sp. XT1-2-2. a The genomic islands were predicted by Island Viewer 4. Putative genomic islands predicted by the SIGI-HMM method (blue squares) or IslandPath-DIMOB method (yellow squares) or IslandPich (green squares). The integrated results are indicated by red squares. The inner ring indicated the G + C contents. b A vertical view of the genes and their description. c The gene arrangement of heavy metal resistance genes
Features of the core and pan-genomes
In order to assess genetic diversity, we constructed Citrobacter genus core and pan genomes and compared the gene content of Citrobacter sp. XT 1-2-2 with other relevant reference strains (Fig. 8). The basic information of the strains used for pan genome analysis has been indicated in Table S7, including the strain name, G + C content, number of proteins, genome size and the accession numbers of the Citrobacter species. From the alignment results, 13,614 gene families were found in 16 genomes, of which 2,449 genes constitute the core genome. The functional categories of the core gene families were further determined via the Cluster of Orthologous Group (COG) assignments among all the related species. The results showed that the core gene family presented an uneven distribution among functional categories (Fig. 4). We further analyzed the core, accessory and specific genes (Tables S8, S9 and S10), carefully checked the classification of the heavy metal resistance in the gene category, and the results are shown in Table S11.

The Citrobacter core and pan-genome plotted were constructed for 16 genome sequences of Citrobacter related species
Comparative genomics analysis
The amino acid sequences of the involved twenty species were aligned via the OrthoMCL, and a certain threshold (E-Value: 1e-5, Percent Identity Cutoff: 0, Markov Inflation Index: 1.5) was selected for similarity clustering to obtain homologous genes. With the help of Venn diagram, the common and unique homologous genes between species are displayed intuitively. The strain XT1-2-2 shares 2285 proteins with the other genomes and has 342 specific proteins. The 2285 core genes include the genes in the whole sulfate reduction pathway and some of the heavy metal resistance genes (Fig. 9).

The Venn diagram depicting the core and unique genes between Citrobacter sp. XT1-2-2 and other 19 relevant reference species
Discussion
In this study, the complete genome of Citrobacter sp. XT1-2-2 was sequenced and comparative genomics analysis was also conducted with the other relevant reference sequenced genomes. In our previous study, the strain XT1-2-2 was isolated from high Cd-contaminated soils, and demonstrated an excellent ability to decrease the bioavailability of Cd in the soil and inhibit Cd uptake in rice. In addition, the strain XT1-2-2 could significantly promote rice growth and increase rice biomass. However, the genome sequence of this organism has not been reported so far.
The antigenic system of the Bethesna-Ballerup group bacteria was established by West and Edwards in 1954 [15]. This group of bacteria is now called Citrobacter freundii [16]. So far, Citrobacter genus contains eleven species: Citrobacter freundii, Citrobacter koseri, Citrobacter amalonaticus, Citrobacter farmeri, Citrobacter youngae, Citrobacter braakii, Citrobacter werkmanii, Citrobacter sedlakii, Citrobacter rodentium, Citrobacter genomospecies 10, Citrobacter genomospecies 11 [17, 18]. According to the results of TYGS analysis and phylogenetic relation, Citrobacter sp. XT1-2-2 is the closest relative to Citrobacter werkmanii BF-6 (CP019986.1) (Fig. 2). According to the physicochemical properties of these strains, some Citrobacter species immobilized biofilms were used to bioremediate heavy metal contaminated soils through an acid-type phosphatase enzymatic activity or their ability to accumulate heavy metals [19,20,21]. In this study, genome analysis of the strain XT1-2-2 revealed all genes of a complete set of sulfate reduction pathway according to the KEGG analysis (Fig. 5). The occurrence of metabolic pathways involves the following steps: (1) Sulfate (SO42−) from outside is taken up into cells by putative sulfate transporter CysPUWA; (2) Sulfate (SO42−) entering the cell is first acetylated to adenylylsulphate (APS) by sulfate adenylyltransferases CysN and CysD; (3) The resulting APS is then phosphorylated to phosphoadenylyl-sulphate (PAPS) by the APS kinase CysC; (4) The resulting PAPS is further reduced to sulfite (SO32−) by PAPS reductase CysH; (5) The resulting sulfite (SO32−) is finally reduced to sulfide (S2−) by sulfite reductase CysIJ [14]. The reason why the strain XT1-2-2 has a significant effect of removing cadmium is mainly because the strain generates sulfide (S2−) via the sulfur metabolism pathway, which can combine with Cd2+ in the soil to form the precipitated CdS, thereby reducing the uptake and transport of cadmium in the soil by rice plant.
Meanwhile, the strain XT1-2-2 also revealed various genes responsible for multiple heavy metal resistance (Fig. 6), which provided the genomic basis for the strain to adapt to the external complex harmful environment. CzcD is involved in resistance to the heavy metals Cd2+, Zn2+ and Co2+ [22]. The membrane transporter ChrA is responsible for the efflux of intracellular Cr(VI) from the cell [23]. Heavy metal-transporting ATPase (ZntA) is responsible for the efflux of Pb2+, Zn2+ and Cd2+ [24]. The metal ABC transport system (ZnuABC) are involved in Zn2+ uptake [25]. ArsB, ArsC, and ArsH proteins are involved in the functions of arsenical pump membrane protein, arsenate reductase and arsenical resistance protein, respectively [26]. Cus copper resistance system consists of CusCBA efflux pump, CusF periplasmic protein and CusS regulatory protein [27]. Mercury transport system (mer operon) encodes a group of proteins consisting of MerR mercury regulatory proteins, MerT, MerC, MerP mercury transport proteins and MerA, MerD, MerE mercury resistance proteins [28]. The Co2+ ECF transporter complex is involved in Co2+ resistance and transmembrane transport [29].
The analysis of the core and pan genomes showed an uneven distribution among functional categories (Fig. 4). There were several notable differences in the numbers of genes, such as amino acid transport and metabolism (category E), transport and metabolism of carbohydrates (category G), translation (category K) and inorganic ion transport and metabolism (category P). In particular, this difference in the number of genes belonging to the same COG category was mainly reflected in transport and metabolism [5]. For KEGG annotations [30,31,32], two gene functional categories were enriched in core gene families including metabolism and environmental information processing (Fig. 10). It is noteworthy that the uneven distribution of genes in the COG and KEGG categories was related to transport, metabolism and signal transduction system [18]. The signal transduction systems are responsible for sensing environmental cues and adjusting cellular behavior. Microbial metabolism and transport involve complex metabolic pathway, gene regulation network, and environmental cues. These gene functional categories were enriched among the core gene families in response to complex environmental stimuli. Due to the complex and changeable external environment, strains need to respond quickly to adapt to the environmental changes. So we hypothesized that these gene categories related to transport, metabolism and signal transduction system might provide a competitive advantage to Citrobacter sp. XT1-2-2 adapt to the environmental changes.

Determining the taxonomic position is crucial for classifcation, characterization and identifcation of bacteria. The genome of Citrobacter sp. XT1-2-2 was submitted to Type Strain Genome Server (TYGS) for whole genome based taxonomic analysis. TYGS compares the query genome with all type strain genomes available in the TYGS database [33] where the intergenomic or intragenomic relations can be inferred through the auto-generated phylogeny and digital DNA-DNA hybridization (dDDH) values. The pairwise comparison between Citrobacter sp. XT1-2-2 and the closest type strains using dDDH is shown in Table S12. The table contains dDDH values and confidence intervals for species and subspecies close to Citrobacter sp. XT1-2-2 using three different Genome-to-Genome Distance calculator (GGDC) formulas [34].
Whole-genome sequencing technology increased the identification of genomic islands in bacterial genomes. The genomic islands are considered to be the major elements for disseminating resistance genes among bacteria, though the mechanism of transfer was rarely determined [35]. An increasing number of evidences indicated that some genomic islands can transfer between bacteria by conjugation autonomously or with the help of other mobile genetic elements (e.g. conjugative plasmid) [36]. In this study, we discovered a heavy metal resistance genomic island in the chromosome of Citrobacter sp. XT1-2-2. The G + C content of chrA is 58.87%, and G + C contents of merR, merT, merP, merC, merA, merD and merE are 61.25%,61.25%, 62.68%, 65.37%, 65.68%, 69.70% and 69.20%, respectively, and they differ from that of the XT1-2-2 overall genome (52.09%), suggesting that these resistance genes may be horizontally transferred genes obtained from other bacteria under the stress of external environmental stress.
Conclusions
Results of comparative genomic analysis from Citrobacter sp. XT1-2-2 revealed correlations between genotype and phenotype. Genome analysis revealed all genes of a complete set of sulfate reduction pathway according to the KEGG analysis, which provides the genomic basis for the strain to reduce sulfate (SO42−) to sulfide (S2−) to form CdS precipitation, thereby reducing the uptake and transport of Cd2+ by rice plants. Meanwhile, the strain also revealed various genes responsible for multiple heavy metal resistance, which provided the genomic basis for the strain to adapt to the external complex harmful environment. These analytical results provide insights into the genomic basis of microbial immobilization of heavy metals.
Materials and methods
Bacterial strain and DNA extraction
The strain XT1-2-2 was initially isolated from high Cd- contaminated paddy soils (~ 220 mg/kg) in Liuyang city, Hunan Province, China (28°01’N, 113°34’E). Based on previous morphological and molecular characterization, the strain XT1-2-2 was identified as the genus Citrobacter. The genomic DNA of the strain XT1-2-2 was extracted by QIAamp DNA Mini Kit (Qiagen, CA, USA) according to the manufacturer’s protocol.
Bacterial morphological characterization
The selected bacterial strain (Citrobacter sp. XT1-2-2) was cultivated in the liquid medium in the absence or presence of 100 mg/L Cd2+, and then bacteria were prepared for scanning electron microscopy (SEM), by centrifugation at 12000 rpm for 10 min to pellet bacterial cells. The pellet was resuspended in 4% p-formaldehyde (PFA) to fix the cells for 1 h. Then bacterial cells were resuspended in 200 µl of hexamethyldisilazane (HMDS), and 2 µl suspension was mounted onto a silicon wafer and dried overnight. The samples were investigated using an Quanta 400 FEG (Thermo Scientific, USA) in high-vacuum conditions at 5-kV accelerating voltage.
Genome sequencing and assembly
Genome sequencing was performed by Shanghai Majorbio Bio-pharm Technology Co., Ltd (Shanghai, China). The genome sequence of the strain XT1-2-2 was obtained via the Illumina Hiseq×10 and Pacbio platforms, with a depth of ~ 100-fold coverage in both platforms. The previously extracted genomic DNA was randomly fragmented through Covaris or Bioruptor method. Fragmented DNA was purified by the QIAquick Nucleotide Removal Kit (Qiagen, Crawley, United Kingdom). Sequencing adaptors were ligated to A-tailed 3’ends according to the manufacturer’s instructions. A library for Illumina Paired-End sequencing was prepared. The sequencing library was sequenced via the combined sequencing method of Illumina Hiseq ×10 + PacBio, and each sample provides at least 100× PacBio sequencing data and 100× Illumina sequencing data of the genome to ensure a more complete and accurate assembly. The constructed standard shotgun library generated 165575 reads totaling 164774594 bp and an average length of 7034.7 bp. The resulting reads were de novo assembled with the help of SOAPdenovo v1.05 [37]. The genome was annotated using the NCBI Pro-karyotic Genome Annotation Pipeline (PGAP), and genes were identified by the gene caller GeneMarkS (Version 4.3). The genomic islands were predicted by Island Viewer 4 [38].
Identification of gene orthologous groups
OrthoMCL (version 2.0.9) was exploited to determine orthologous families in the pan-genome with default parameter (E-Value: 1e-5, Percent Identity Cutoff: 0, Markov Inflation Index: 1.5). The single-copy core gene and pan gene were extracted with the help of the OrthoMCL (http://www.orthomcl.org/common/downloads/software/v2.0/). Their nucleotide sequences were extracted on the basis of protein ID.
TYGS analysis
The whole genome sequence of Citrobacter sp. XT1-2-2 was uploaded to the Type Strain Genome Server (TYGS) for in silico based taxonomic analysis [33]. The pairwise comparison of the user strain with the type strains were performed using GBDP and accurate intergenomic distances inferred under the “trimming” algorithm and distance formula d5. Digital DDH values and confidence intervals were calculated following the recommended settings of GGDC 2.1 [33]. The intergenomic distances were used to create a balanced minimum evolution tree using FASTME 2.1.4 with 100 pseudobootstrap replicates for branch support [33].
Core and pan-genome analysis
The comparative study on the core and pan-genome analysis was manipulated by the 16 genome sequences of Citrobacter related species according to the previously reported methods [5, 39]. Briefly, the gene set in Citrobacter sedlakii NBRC 105722 was selected and regarded as the Reference and the gene sets in the other 15 Citrobacter sp. genomes were considered as the Query. The Query genes in each genome were aligned against the Reference genes in reference strain using BLAST v2.2.26 (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and the blast results were filtered by their length and identity. The regression analysis for the core gene cluster curve was performed using a weighted least square regression by fitting the power law n = κexp (m×N) + ϴ to means [40]. N is the number of genomes, n is the number of core gene clusters, ϴ is a constant value representing the predicted minimum number of core genes, and κ and m are parameters.
Gene functional category
The functional category of the core gene families was analyzed and classified by different database (COG/GO/KEGG). The numbers of corresponding proteins were computed for each term of COG/GO/KEGG.
The main biological functions of different proteins were determined by functional enrichment analysis, and then the resulting results were visualized by GraphPad Prism 7.0.
Availability of data and materials
The genome sequence of Citrobacter sp. XT1-2-2 has been deposited in GenBank under the BioSample number SAMN28157541. https://www.ncbi.nlm.nih.gov/biosample/SAMN28157541/.
Abbreviations
- NCBI:
-
National Center for Biotechnology Information
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- COG:
-
Cluster of Orthologous Groups of Proteins
- GBDP:
-
Genome BLAST Distance Phylogeny
- dDDH:
-
digital DNA:DNA Hybridization
- TYGS:
-
Type Strain Genome Server
- PAPS:
-
Phosphoadenylyl-sulphate
- HMDS:
-
Hexamethyldisilazane
- APS:
-
Adenylylsulphate
- GO:
-
Gene Ontology
- Cd:
-
Cadmium
References
Woese CR, Kandler O, Weelis ML. Towards a natural system of organisms: proposal for the domains archaea, bacteria and eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–9.
Stackebrandt E, Murray RGE, Trüper HG. Proteobacteria classis nov., a name for the phylogenetic taxon that includes the “purple bacteria and their relatives”. Int J Syst Evol Microbiol. 1988;38:321–5.
Validation of publication of new names. and new combinations previously effectively published outside the IJSEM. Int J Syst Evol Microbiol. 2005;55:1395–97.
Ramos-Vivas J, Chapartegui-González I, Fernández-Martínez M, González-Rico C, Barrett J, Fortún J, Escudero R, Marco F, Linares L, Nieto J, Aranzamendi M, Muñoz P, Valerio M, Aguado JM, Chaves F, Gracia-Ahufinger I, Paez-Vega A, Martínez-Martínez L, Fariñas MC. Adherence to human colon cells by multidrug resistant Enterobacterales strains isolated from solid organ transplant recipients with a focus on Citrobacter freundii. Front Cell Infect Microbiol. 2020;10:447.
Zhou G, Peng H, Wang YS, Huang XM, Xie XB, Shi QS. Complete genome sequence of Citrobacter werkmanii strain BF-6 isolated from industrial putrefaction. BMC Genomics. 2017;18:765.
Wang H, Hou H, Huang J. Citrobacter arsenatis sp. nov., an arsenate-reducing bacterium isolated from freshwater sediment. Antonie Van Leeuwenhoek. 2021;114:1285–92.
Werkman CH, Gillen GF. Bacteria producing trimethylene glycol. J Bacteriol. 1932;23:167–82.
Lipsky BA, Hook EW, Smith AA, Plorde JJ. Citrobacter infections in humans: experience at the Seattle veterans administration medical center and a review of the literature. Rev Infect Dis. 1980;2:746–60.
Janda JM, Abbott SL, Cheung WK, Hanson DF. Biochemical identification of Citrobacteria in the clinical laboratory. J Clin Microbiol. 1994;32:1850–4.
Arens S, Verbist L. Differentiation and susceptibility of Citrobacter isolates from patients in a university hospital. Clin Microbiol Infect. 1997;3:53–7.
Liu LH, Wang NY, Wu AY, Lin CC, Lee CM, Liu CP. Citrobacter freundii bacteremia: risk factors of mortality and prevalence of resistance genes. J Microbiol Immunol Infect. 2018;51:565–72.
Shan S, Guo Z, Lei P, Li Y, Wang Y, Zhang M, Cheng W, Wu S, Wu M, Du D. Increased biomass and reduced tissue cadmium accumulation in rice via indigenous Citrobacter sp. XT1-2-2 and its mechanisms. Sci Total Environ. 2020;708:135224.
Shan S, Guo Z, Lei P, Wang Y, Li Y, Cheng W, Zhang M, Wu S, Yi H. Simultaneous mitigation of tissue cadmium and lead accumulation in rice via sulfate-reducing bacterium. Ecotoxicol Environ Saf. 2019;169:292–300.
Xia X, Li J, Liao S, Zhou G, Wang H, Li L, Xu B, Wang G. Draft genomic sequence of a chromate- and sulfate-reducing Alishewanella strain with the ability to bioremediate Cr and Cd contamination. Stand Genomic Sci. 2016;11:48.
West MG, Edwards PR. The Bethesda-Ballerup group of paracolon bacteria. Public Health Monogr. 1954;22:1–35.
Miki K, Tamura K, Sakazaki R, Kosako Y. Re-speciation of the original reference strains of serovars in the Citrobacter freundii (Bethesda-Ballerup group) antigenic scheme of West and edwards. Microbiol Immunol. 1996;40:915–21.
Brenner DJ, O'Hara CM, Grimont PA, Janda JM, Falsen E, Aldova E, Ageron E, Schindler J, Abbott SL, Steigerwalt AG. Biochemical identification of Citrobacter species defined by DNA hybridization and description of Citrobacter gillenii sp. nov. (formerly Citrobacter genomo species 10) and Citrobacter murliniae sp. nov. (formerly Citrobacter genomospecies 11). J Clin Microbiol. 1999;37:2619–24.
Yuan C, Yin Z, Wang J, Qian C, Wei Y, Zhang S, Jiang L, Liu B. Comparative genomic analysis of Citrobacter and key genes essential for the pathogenicity of Citrobacter koseri. Front Microbiol. 2019;10:2774.
Macaskie LE, Empson RM, Lin F, Tolley MR. Enzymatically-mediated uranium accumulation and uranium recovery using a Citrobacter sp. immobilised as a biofilm within a plug-flow reactor. J Chem Technol Biotechnol. 1995;63:1–16.
Finlay JA, Allan VJM, Conner A, Callow ME, Basnakova G, Macaskie LE. Phosphate release and heavy metal accumulation by biofilm-immobilized and chemically-coupled cells of a Citrobacter sp. pre-grown in continuous culture. Biotechnol Bioeng. 1999;63:87–97.
Zinicovscaia I, Safonov A, Boldyrev K, Gundorina S, Yushin N, Petuhov O, Popova N. Selective metal removal from chromium-containing synthetic effluents using Shewanella xiamenensis biofilm supported on zeolite. Environ Sci Pollut Res Int. 2020;27:10495–505.
Anton A, Grosse C, Reissmann J, Pribyl T, Nies DH. CzcD is a heavy metal ion transporter involved in regulation of heavy metal resistance in Ralstonia sp. strain CH34. J Bacteriol. 1999;181:6876–81.
Gu R, Gao J, Dong L, Liu Y, Li X, Bai Q, Jia Y, Xiao H. Chromium metabolism characteristics of coexpression of ChrA and ChrT gene. Ecotoxicol Environ Saf. 2020;204:111060.
Chien CC, Huang CH, Lin YW. Characterization of a heavy metal translocating P-type ATPase gene from an environmental heavy metal resistance Enterobacter sp. isolate. Appl Biochem Biotechnol. 2013;169:1837–46.
Santos RERS, da Silva Júnior WP, Harrison S, Skaar EP, Chazin WJ, da Silva Neto JF. The zinc transporter ZnuABC is critical for the virulence of chromobacterium violaceum and contributes to diverse zinc-dependent physiological processes. Infect Immun. 2021;89:e0031121.
Chauhan NS, Nain S, Sharma R. Identification of arsenic resistance genes from marine sediment metagenome. Indian J Microbiol. 2017;57:299–306.
Franke S, Grass G, Rensing C, Nies DH. Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J Bacteriol. 2003;185:3804–12.
Barkay T, Miller SM, Summers AO. Bacterial mercury resistance from atoms to ecosystems. FEMS Microbiol Rev. 2003;27:355–84.
Siche S, Neubauer O, Hebbeln P, Eitinger T. A bipartite S unit of an ECF-type cobalt transporter. Res Microbiol. 2010;161:824–9.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49(D1):D545–51.
Meier-Kolthoff JP, Göker M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun. 2019;10(1):2182.
Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60.
Guglielmini J, Quintais L, Garcillán-Barcia MP, de la Cruz F, Rocha EP. The repertoire of ICE in prokaryotes underscores the unity, diversity, and ubiquity of conjugation. PLoS Genet. 2011;7:e1002222.
Partridge SR, Kwong SM, Firth N, Jensen SO. Mobile genetic elements associated with antimicrobial resistance. Clin Microbiol Rev. 2018;31:e00088-17.
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9.
Bertelli C, Laird MR, Williams KP, Simon Fraser University Research Computing Group, Lau BY, Hoad G, Winsor GL, Brinkman FSL. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017;45(W1):W30–5.
Xiao B, Sun YF, Lian B, Chen TM. Complete genome sequence and comparative genome analysis of the Paenibacillus mucilaginosus K02. Microb Pathog. 2016;93:194–203.
Bottacini F, Medini D, Pavesi A, Turroni F, Foroni E, Riley D, Giubellini V, Tettelin H, van Sinderen D, Ventura M. Comparative genomics of the genus Bifidobacterium. Microbiol (Reading). 2010;156(Pt11):3243–54.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Project No.32171633), Natural Science Foundation of Hunan Province of China (Project No. 2021JJ40307) and Key Technology Research Project of Hunan Province of China (Project No. 2021NK1040).
Author information
Authors and Affiliations
Contributions
SS, YL,WC and MZ wrote manuscript. XW, YW and SW collected and analyzed data. DD and ZG interpreted the data and reviewed manuscript. ZL and ZF edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Table S1. Classification and general features of Citrobacter sp. XT1-2-2.
Additional file 2:
Supplementary Table S2. Project information of Citrobacter sp. XT1-2-2.
Additional file 3:
Supplementary Table S3. The genome properties and statistics of Citrobacter sp. XT1-2-2
Additional file 4:
Supplementary Table S4. Basic information of sulfate reduction pathwaygenes on Chromosomal and BLASTP analysis in swissprot database.
Additional file 5:
Supplementary Table S5. Basic information of heavy metal resistancegenes on Chromosomal and BLASTP analysis in swissprot database.
Additional file 6:
Supplementary Table S6. all the genes on the genomic islands of Citrobacter sp. XT1-2-2.
Additional file 7:
Supplementary Table S7. Basic information of the Citrobacter species used for the pan genome analysis.
Additional file 8:
Supplementary Table S8. The core genes in gene category of Citrobacter sp. XT1-2-2.
Additional file 9:
Supplementary Table S9. The specific genes in gene category of Citrobacter sp. XT1-2-2.
Additional file 10:
Supplementary Table S10. The accessory genes in gene category of Citrobacter sp. XT1-2-2.
Additional file 11:
Supplementary Table S11. Classification of the heavy metal resistancegenes in the gene category.
Additional file 12:
Supplementary Table S12. Pairwise digital DNA-DNA hybridization valuesbetween query genome and the selected type strains and whole genomes by Typestrain genome server.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shan, S., Cheng, W., Li, Y. et al. Comparative genomic analysis of Citrobacter sp. XT1-2-2 reveals insights into the molecular mechanism of microbial immobilization of heavy metals. BMC Genomics 23, 838 (2022). https://doi.org/10.1186/s12864-022-09069-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-09069-4