
Abstract
Background
Plums are one of the most important economic crops of the Rosaceae family and are produced all over the world. China has many local varieties, but the genomic information is limited for genetic studies. Here, we first sequenced, assembled, and analyzed the plastomes of twelve plum cultivars and developed molecular markers to distinguish them.
Results
The twelve plastomes of plum cultivars have a circular structure of 157,863–157,952 bp containing a large single-copy region (LSC) of 86,109–86,287 bp, a small copy region (SSC) of 18,927–19,031 bp, and two inverted repeats (IR) of 26,353–26,387 bp each. The plastomes of plum cultivars encode 131 genes, including 86 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. We detected 50, 54, 54, 53, 53, 50, 54, 54, 54, 49, 50, 54 SSRs in the twelve analyzed varieties, respectively. For repeat sequences, we identified 553 tandem repeats, 204 direct repeats, and 270 palindromic repeats. We also analyzed the expansion/contraction of IR regions. The genes rpl22, rps19, rpl2, ycf1, ndhF, and the trnH span on or near the boundary of IR and single-copy regions. Phylogenetic analysis showed that the twelve cultivars were clustered with the P. salicina and P. domestica. We developed eight markers LZ01 to LZ08 based on whole plastomes and nuclear genes and validated them successfully with six repetitions.
Conclusions
The results obtained here could fill in the blanks of the plastomes of these twelve plum cultivars and provide a wider perspective based on the basis of the plastomes of Prunus to the molecular identification and phylogenetic construction accurately. The analysis from this study provides an important and valuable resource for studying the genetic basis for agronomic and adaptive differentiation of the Prunus species.
Similar content being viewed by others


Background
Plums are one of the most economically important crops of the Rosaceae family and are cultivated all over the world. Plums are one of the most important stone fruits consumed worldwide because of their high degree of acceptance by consumers [1]. Plums are rich in vitamin C, vitamin E, non-essential and essential amino acids, total phenols, flavonoids, and trace elements such as potassium, calcium, and magnesium. They have good antioxidant activity, and the active peptide extracted from it can effectively resist ABTS (2, 2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)) free radical and inhibit angiotensin-converting enzyme [2]. As a result, plums are widely recognized for their nutritional and economic value.
There are about 19 to 40 species of plums distributed across Asia, Europe, and North America [3, 4]. China is the original distribution center of Chinese plums, which are widely used. There are local varieties that have been cultivated for a long time in various regions of the country, such as the Wushan plum [5], the Fendai plum [6] and the Wanshuang plum [7] in Chongqing, the Sanhua plum [8] in Guangdong, Cuihong plum [9] and Jiangan plum [10] in Sichuan, the Fengtang plum [11] in Guizhou, Hongxin plum [12] in Zhejiang, among other regions. These fruits are mostly consumed while fresh because of their characteristic taste [13].
In recent years, the development of genomics and high-throughput sequencing technology has provided strong support for the study of plant plastomes. The interest in plant plastomes has increased since 1986, when the first whole plastomes were published for Nicotiana tabacum [14] and Marchantia polymorpha [15]. Compared with the nuclear genome, the plastome is characterized by small molecular weight, single copy, simple structure, highly conserved gene structure order and gene content, and low gene substitution rate [16, 17]. The plastome is uniparental, with gymnosperms inherited on the paternal line and angiosperms on the maternal line, therefore it will not be disturbed by genetic recombination; the evolutionary path of the plastome is relatively independent, and does not depend on other data to construct a phylogenetic tree [18]. So, the plastome is widely used in plant phylogeny and evolution, species identification, and taxonomy.
Lately, DNA markers were developed to authenticate Prunus genus. For example, Yamamoto T (2003) has analyzed peach germplasm resources using SSR markers and found that Japanese peaches are closely related to Chinese peaches [19]. Ortiz used the RAPD technique to detect hexaploid and diploid plum cultivars, and only three random primers were used to distinguish 31 plum varieties [20]. Twenty four Chinese plum varieties are proved from three types of production areas using 16 SSR primer pairs [21].
However, to date, genetic information is scarce, preventing in-depth molecular breeding. In this study, we selected twelve plum varieties that are most consumed in China, including Chinese plums: ‘Sanhua plum’, ‘Wanshuang plum’, ‘Wuyuecui’, ‘Oishiwase’, ‘Yinhong plum’, ‘Fengtang plum’, ‘Cuihong plum’, and ‘No.2 Guofeng’; European plums: ‘Richard Early’, ‘Bingtang plum’; Prunus cerasifera 'Hollywood' and Prunus simonii ‘Weiwang’. Our goal is to understand their taxonomic relationship and to develop high-resolution molecular markers for discrimination.
Results
General features of the twelve plastomes
Using Illumina NovaSeq 6000 sequencing platforms, we obtained 5.01 – 6.21 G clean data from each plum cultivar and the number of clean reads ranged from 16,709,174 to 20,713,829 (Table S1). The twelve plastomes of plum cultivars have a circular structure of 157,863–157,952 bp containing a large single-copy region (LSC) of 86,109–86,287 bp, a small copy region (SSC) of 18,927–19,031 bp, and two inverted repeats (IR) of 26,353–26,387 bp by each. In general, there were small differences in the length of plastomes of the plants in this study. The GC content analysis showed that the total GC content ranged from 36.72% to 36.76% in the twelve plastomes. The GC contents in IR regions (42.58%-42.62%) are significantly higher than those in LSC (34.51%-34.59%) and SSC regions (30.36%-30.54%) (Table 1). Since the cultivars belong to the same genus, there is little difference in GC content. The twelve plastomes were deposited to NCBI (Accession number: MW406457, MW406459, MW406460, MW406461, MW406463, MW406464, MW406465, MW406466, MW406468, MW406470, MW406471, MW406472).
Genome annotation
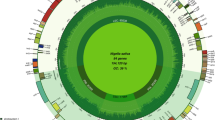
The plastomes of twelve plum cultivars all encoded 131 genes, among which, 110 are unique genes, including 78 protein-coding genes, 28 tRNA genes, and 4 rRNA genes (Table S2). An IR region contains 21 genes, four rRNA genes, nine tRNA genes, and eight protein-coding genes, respectively. The plastomes map is shown in Fig. 1 and Figures S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, S11. Among the 78 protein-coding genes annotated, the genes containing two introns were the ycf3 and clpP genes, and the genes containing one intron include the trnK-UUU, rps16, trnG-UCC, atpF, rpoC1, trnL-UAA, trnV-UAC, petB, petD, rpl16, ndhA, and two repeats of rpl2, ndhB, trnI-GAU, trnA-UGC (Table S3).

Genome map of P. salicina ‘Sanhua plum’ plastome. The map has four rings, from the center outward, with red and green arcs on the first circle connecting forward and reverse repeats, respectively; the second ring shows tandem repeats marked with dashes; the third ring is a MISA-identified microsatellite sequence; and the fourth ring shows the gene structure on the plastome. The colors of these genes are classified according to their function, as shown in the lower left corner
Repeats analysis
In plastomes of twelve plum cultivars, we identified three kinds of repeated sequences including tandem repeats, direct repeats, and palindromic repeats. The numbers of them are 553, 204 and 270, respectively. Among them, P. salicina 'Oishiwase' has the most repeats (95) including 52 tandem repeats, 19 direct repeats and 24 palindromic repeats. On the contrary, P. salicina 'Sanhua plum' has the least repeats (77) including 40 tandem repeats, 15 direct repeats and 22 palindromic repeats (Fig. 2A).

Comparison of the Repeats in the plastomes of 12 plum cultivars. A. Types and numbers of the interspersed repeats in the 12 plastomes; B. Types and numbers of SSRs detected in the 12 plastomes
Simple sequence repeats (SSRs), also known as microsatellite sequences, provide a large number of information about genetic variation. SSRs have high genetic polymorphism and are commonly used to develop molecular markers that play an important role in species identification. In this study, we detected 50, 54, 54, 53, 53, 50, 54, 54, 54, 49, 50, 54 SSRs in the twelve analyzed varieties, respectively (Fig. 2B, Table S4). Most SSRs are mononucleotide, particularly A/T motifs, which accounts for more than 80% of the total. Moreover, P. cerasifera ‘Hollywood’ has the least SSRs, but has the most dinucleotide. These SSRs have the potential in the identification of Prunus.
Contraction and expansion analysis of IR regions
The IR regions of the twelve plum cultivars plastomes are the most conserved regions, being 26,353 to 26,387 bp in length. However, the expansions and contractions of the IR boundary can cause the diversity of plastome length [22]. The LSC/IR and SSC/IR borders of the Prunus plastomes were compared (Fig. 3). We observed several genes span on or near the boundary of IR and single-copy regions. These are mainly the genes rpl22, rps19, rpl2, ycf1, ndhF and trnH. Among them, rps19 gene span the LSC/IRb boundary, but the P. domestica ‘Richard Early’, P. cerasifera ‘Hollywood’ and P. domestica ‘Bingtang plum’s’ rps19 gene in IRb (174 bp) is shorter than the other nine varieties (186 bp/187 bp). Similarly, in terms of two copies of ycf1 gene span the IRb/SSC and SSC/IRa, the above three varieties are also shorter than the other nine. It can be seen that the genomic structure has changed in Prunus.

Comparison of the borders among LSC, SSC, and IR regions of twelve analyzed plums. The genes around the borders are shown above or below the mainline. The JLB, JSB, JSA, and JLA represent junction sites of LSC/IRb, IRb/SSC, SSC/IRa, and IRa/LSC, respectively
Hypervariable Region Analysis
Hypervariable regions can be used to resolve phylogenies and to discriminate closely related plant species [23]. The pairwise comparison of intergenic spacer regions was conducted to identify divergence hotspot regions among the twelve plum cultivars using the Kimura 2-parameter (K2p) model. The average K2p distance ranged from 0.00 to 2.463. The IGS regions of rpl33-rps18, ndhC-trnV-UAC, rpl16-rps3, trnF-GAA-ndhJ, and petG-trnW-CCA showed the largest distances of 2.463, 1.915, 1.772, 1.64 and 1.615, respectively (Fig. 4).

Comparison of the variability of IGS regions among the plastomes of 12 plums. The X-axis indicates the IGS regions, and the Y-axis shows the range of K2p distances between different pairs of species. The diamond shows the average K2p distance of the IGS region, respectively
Phylogenetic analysis based on plastome data
To examine the phylogenetic position of the twelve plum cultivars, we constructed maximum likelihood (ML) trees based on complete plastome sequences (Fig. 5) and 71 common protein-coding genes shared among 32 species from Prunus (Figure S12A), including the twelve sequenced in this study (Table S5). Two trees had a similar topological structure. However, the varieties in this study have the same structure between the two results. They are distributed in three clades. The first clade is formed by ‘Hollywood’, Richard Early’ and ‘Bingtang plum’ with P. domestica. Besides, ‘Oshiwase’, Wuyuecui’ and ‘No.2 Guofeng’ were clustered with P. salicina. The other six are grouped into another clade. It indicated that these varieties are a most likely hybrid between P. domestica and P. salicina. Most nodes of the phylogenetic tree have high bootstrap support, indicating the reliability of the phylogenetic analysis.

Phylogenetic relationships of species from Prunus (Rosaceae) inferred using Maximum likelihood (ML) method. The phylogenetic tree was constructed using the complete plastome sequences among the 32 plastomes. The number at the bottom of the scale, 0.001, means that the length of the branch represents the replacement frequency of bases at each site of the genome at 0.001. Bootstrap values were calculated from 1000 replicates
Identification and validation of plastome-based markers
To distinguish the twelve cultivars, we selected six hypervariable regions manually based on plastome to develop six molecular markers named LZ01 to LZ06. The primers used for validation are shown in Table S6. All of PCR amplification results have single and bright band. The DNA fragments were extracted from each band and then sent for Sanger sequencing. The sequencing results were identical to the previous sequences.
Marker LZ01 can divide the twelve varieties into five groups, named Group1 to Group5 (Group1: ‘Sanhua plum’ (SH), ‘Wanshuang plum’ (WS), ‘Weiwang’ (WW), ‘Yinhong plum’ (YH), ‘Fengtang plum’ (FT) and ‘Cuihong plum’ (CH); Group2: ‘Oishiwase’ (OW); Group3: ‘Wuyuecui’(WY) and ‘No.2 Guofeng’ (GF); Group4: ‘Hollywood’ (HW); Group5: ‘Richard Early’ (RE) and ‘Bingtang plum’ (BT)). And two varieties: ‘Oishiwase’ (OW) and ‘Hollywood’ (HW) can be distinguished with three Indels (Fig. 6A). For the Group1, Marker LZ02, LZ03,LZ04, LZ05 and LZ06 can distinguish ‘Fengtang plum’ (FT) (Fig. 6B), ‘Cuihong plum’ (CH) (Fig. 6C), ‘Weiwang’ (WW) (Fig. 6D), ‘Sanhua plum’ (SH) (Fig. 6E) and ‘Yinhong plum’ (YH) (Fig. 6F), respectively. Unfortunately, for the Group3 and Group5, their plastome sequences are the same as another. As a result, we further developed markers for these cultivars based on nuclear genome.

The alignment of the sequencing of the PCR products amplified using the primer LZ01 to LZ08. A-H represents the alignment using LZ01, LZ02, LZ03, LZ04, LZ05, LZ06, LZ07 and LZ08, respectively. The SNP and Indel regions are highlighted with red squares. The nucleotides identical across all plastomes are shaded in black, whereas those conserved in 60% of the sequences are shaded in gray. SH: P. salicina 'Sanhua plum'; WS: P. salicina 'Wanshuang plum'; WY: P. salicina 'Wuyuecui'; OW: P. salicina 'Oishiwase'; WW: P. simonii 'Weiwang'; RE: P. domestica 'Richard Early'; YH: P. salicina 'Yinhong plum'; FT: P. salicina 'Fengtang plum'; CH: P. salicina 'Cuihong plum'; HW: P. cerasifera 'Hollywood'; BT: P. domestica 'Bingtang plum'; GF: P. salicina 'No.2 Guofeng'. Arabic numerals represent different individuals
Identification and validation of nuclear genome-based markers
To identify the remaining four cultivars, we extracted nuclear genes from sequence data among the Angiosperms-mega 353 gene set [24]. Among these genes, 342, 295, 331, 339 genes had extracted for ‘Wuyuecui’, ‘Richard Early’, ‘Bingtang plum’ and ‘No.2 Guofeng’, respectively. Among these coding sequences, 254 genes were shared among the four cultivars. These common genes were used to construct a phylogenetic tree using the same method as that for the complete plastome sequences. The relationships in both the nuclear and plastome trees were consistent (Figure S12B). We selected two hypervariable regions from two genes (AT2G45770 and AT4G02790) to develop two molecular markers named LZ07 and LZ08. The same method for PCR amplification and Sanger sequencing as the above. All of the PCR amplification results have single and bright band. The sequencing results were identical to the previous sequences (Fig. 6G, H).
To verify the reliability of the markers, we also collected plant materials for three individuals from another region (Table 2). All individuals used the same method for DNA extraction, PCR amplification and Sanger sequencing as the above. These markers can discriminate all six individuals from two regions (Figures S13, S14, S15, S16, S17, S18, S19, S20, S21, S22, S23, S24, S25, S26, S27, S28). The identification scheme is shown in the Fig. 7.

The identification scheme for this study. The boxes represent varieties names. The arrows represent the primers names
Discussion
We first reported the plastome sequences of twelve plum cultivars. Our assembly results showed that the length of twelve plastomes ranged from 157,863 bp to 157,952 bp. This result is similar to most Rosaceae plant plastomes [25, 26]. In our study, the longest and shortest plastome sequences were 158,955 bp (Prunus padus) and 157,395 bp (Prunus domeatica), respectively. This suggests that the plastomes of Prunus may be evolutionarily different. The plastome of angiosperms evolves faster, with inversions and gene loss occurring during evolution [27]. Among our study, the longest genome sequence was found in P. cerasifera ‘Hollywood’ (157,952 bp) and the shortest was that of P. salicina ‘Sanhua plum’ (157,863 bp). They have a similar structure to most angiosperms, and we didn’t find gene gain/loss, thus, the plastomes were still relatively conserved. In terms of gene composition, these species encoded 131 genes, including 86 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. The plastomes among Prunus varieties were similar in intron and GC contents, but the GC contents in LSC and SSC regions were significantly lower than that in the IR region. These results are similar to those reported previously [28,29,30,31].
In this study, we examined the SSRs and repetitive sequences of twelve plastomes. 593 of the 629 SSRs were mononucleotide repeats, accounting for the majority of all SSRs (94.28%). These mononucleotide repeats were mainly A/T repeats, which had a significant effect on the overall G/C content of the genome [32, 33]. They are often used as molecular markers due to the length of polymorphism in different species. Plastomes are rich in SSR loci and have been recommended for species identification [34, 35]. Besides, we also detected three kinds of repeated sequences in twelve plastomes. Among them, P. salicina 'Oishiwase' had the most repeats. Genomic recombination and sequence variation were mainly caused by slip-strand mismatches and inappropriate recombination of repetitive sequences [35, 36]. These repeats are the genetic markers that are the basis of population and phylogenetic studies and are widely used because of their high polymorphism rates [37,38,39,40].
Typically, the IR region is the most conserved region of the chloroplast genome [41]. The expansion and contraction of IR, LSC and SSC regions are common during the evolutionary process and are the main reasons for the differences in plastomes length [42, 43]. For example, Cicer arietinum and Pisum sativum were found to lack a copy of the IR region [44, 45], Cephalotaxus oliveri was no IR region [36], and gene loss events were identified in the plastome of Astragalus membranaceus [27]. Pelargonium hortorum and Pinus thunbergii plastome showed expansion/ contraction events in the IR region leading to the length of the plastome being unusual [46, 47]. A comparative map of chloroplast genome boundary regions was obtained by analyzing the boundary genes of the IR, LSC and SSC regions of the plastome [48, 49]. In our study, all species have two copies of the IR regions. Gene ycf1 is located in the SSC and IRa regions, the length in IRa is between 391 bp to 1,051 bp. These overlapping segments resulted in a pseudogene fragment of ycf1 at the IRb/SSC boundary except P. padus and P. mume. Especially, pseudogene ycf1 of P. persica is all in the IRb region. However, except for the P. avium and rps19 genes that are located in the boundary of LSC/IRb, the length in the IR region is between 39 bp (P. padus) and 197 bp (P. mume). Thus, the length of the IR region of P. padus is shorter than others.
The phylogenetic relationships of Rosaceae have long been problematic because of frequent hybridization, asexual reproduction, presumed rapid radiation, and historical diversification [50]. In this study, we obtained identical phylogenetic relationships for the twelve cultivars using complete plastomes and common protein-coding genes. Three cultivars: ‘Hollywood’, ‘Bingtang plum’ and ‘Richard Early’ are close to the European plum (P. domestica). The others are close to the Chinese plum (P. salicina). This also can confirm their breeding background [7, 11, 51,52,53,54,55].
Currently, there are many studies on molecular markers for the Prunus. But there are few studies on the identification of Prunus based on the plastome, which is extremely conserved and has many variant loci and is ideal data for molecular marker development. In this study, we identified 12 plum cultivars based on the plastomes and used nuclear genes to identify some of the plants that could not be distinguished from the chloroplast genome. In this experiment, the plastomes of the twelve plum cultivars differed very little, and the highly variable regions screened by the K2p model could not achieve the purpose of distinguishing the individual resources by sequence comparison; therefore, we manually screened the regions with large variation and validated eight molecular markers that could identify them.
Over all, we first developed markers to identify the twelve plum cultivars. DNA markers can comprehensively compare genetic material between populations and individuals, and improve the accuracy and reliability of plant classification. The genetic distance is related to the sequence divergence [56]. In this study, the plastomes of twelve plum cultivars differ slightly. According to the hypervariable region using the K2p module, the most variable regions can’t distinguish each variety. Thus, we selected the different segments manually and verified eight markers that could separate each of them.
Conclusions
The complete plastomes of twelve plum cultivars are reported for the first time in this study. These twelve cultivars are closely related to P. salicina and P. domestica. In addition, we successfully developed a scheme using eight molecular makers in plastome and nuclear genome. Our results provide a wider perspective on the basis of the plastomes of Prunus to the molecular identification and phylogenetic construction.
Methods
Plant material, DNA extraction and Sequencing
The fresh leaves of twelve plants were collected from Chongqing, Guangdong, Sichuan, Liaoning, Guizhou. All the samples were saved deposited at the Herbarium of Southwest University, Chongqing, China. The detailed information for the plant samples is shown in Table 1. The total genomic DNA was extracted by using the CTAB method [57]. The DNA library was constructed using the Agilent 2100 and sequenced using the Illumina NovaSeq 6000 sequencing platform. Sequencing produced a total of 5.04 – 6.26 G raw data per sample. Clean data were obtained by removing low-quality sequences: sequences with a quality value of Q < = 5 accounted for more than 50% of the total base, sequences with more than 10% bases being “N”, and sequences having an adapter.
Genome assembly and annotation
The chloroplast genome was assembled from the clean data by GetOrganelle (v. 1.6.4) [58]. The correctness of the assembly was confirmed by using Bowtie2 (v2.0.1) [59] to manually edit and map all raw reads to the assembled genome sequence under the default settings. The annotation of the plastome was conducted initially using CpGAVAS2 [60]. Geseq was then used to confirm the annotation results [61]. Furthermore, the annotations with problems were manually edited by using Apollo [62]. The genome sequence and annotations have been deposited in the GenBank with accession numbers MW406457, MW406459, MW406460, MW406461, MW406463, MW406464, MW406465, MW406466, MW406468, MW406470, MW406471, MW406472.
Repeats and SSR analysis
The GC content was conducted by using the cusp program provided by EMBOSS (v6.3.1) [63]. The simple sequence repeats (SSRs) were identified using the Online website MISA (https://webblast.ipk-gatersleben.de/misa/), including mono-, di-, tri-, tetra-, penta-, and hexanucleotides with the minimum numbers were 10, 5, 4, 3, 3, and 3, respectively [64]. Additionally, REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) was used to calculate palindromic repeats, forward repeats, reverse repeats, and complement repeats with the settings: Hamming Distance was three, and Minimal Repeat Size was 30 bp [65].
Genome comparison
The multiple sequences were aligned using ClustalW2 [66]. The intergenic regions were extracted with extractseq from EMOSS [63]. The distances of intergenic spacers were conducted using the program distmat from EMBOSS [63]. IRscope (https://irscope.shinyapps.io/irapp/) was used for visualizing the IR boundaries in these plastomes [67].
Phylogenetic analysis
Except for the twelve sequences in this study, the plastome sequences of 19 species belonging to the genus Prunus were downloaded from GenBank (NCBI, https://www.ncbi.nlm.nih.gov/). Malus baccata (Rosaceae) was used as an outgroup. The details are shown in Table S1. The complete plastome sequences were aligned by using MAFFT (https://mafft.cbrc.jp/alignment/server/) [68]. These aligned sequences were used to construct the phylogenetic trees by using the Maximum Likelihood (ML) method implemented in RaxML (v8.2.4) [69]. The parameters were “raxmlHPC-PTHREADS-SSE3 -f a -N 1000 -m GTRGAMMA—× 551,314,260 -p 551,314,260”. The bootstrap analysis was performed with 1,000 replications. As for the common genes, we extracted 71 protein-coding genes from 32 species. The method to construct the tree is the same with the above.
Identification of nuclear markers for phylogenetic analysis
To distinguish the four varieties whose plastome sequences are pairwise consistent, we used the pipeline HybPiper (v1.2) (https://github.com/mossmatters/HybPiper) to identify nuclear markers with the default settings to process our cleaned data [70]. The HybPiper package contains an internal reference set of 353 genes [24]. This Angiosperms-mega 353 gene set can capture loci in our sequence reads. The identified contigs matching probe can be extract using the command line “./reads_first.py -b mega353.fasta -r sample_R1.fastq sample_R2.fastq –prefix sample_result –bwa”. And we selected the common genes among the four varieties to construct the phylogenetic tree using RaxML with 1000 bootstrap replicates. Oryza sativa is the outgroup.
Identification and validation of molecular markers for discrimination
We selected different segments manually to develop molecular markers. Primers were designed using the IDT website (https://sg.idtdna.com/pages/tools/primerquest?returnurl=%2FPrimerquest%2FHome%2FIndex). We collected three individuals from each variety. DNA samples were extracted and then subjected to PCR amplification on a Pro Flex PCR system (Applied Biosystems, Waltham, MA, USA). PCR amplifications were performed in a final volume of 25μL with 2 μL template DNA, 1 μl of forwarding primer, 1 μl of reverse primer, 12.5 μL 2 × Taq PCR Master Mix and 8.5 μL ddH20. PCR experiments were conducted under the following conditions: pre-denaturation at 94 ˚C for 5 min, 30 cycles of amplification at 94 ˚C for 30 s, 58 ˚C for 30 s, and 72 ˚C for 60 s, followed by a final extension at 72 ˚C for 5 min. The PCR products were evaluated with 1% agarose gelelectrophoresis. Only single bands were subjected to Sanger sequencing.
Availability of data and materials
The annotated chloroplast genome sequences of twelve plum cultivars were deposited in GeneBank (https://www.ncbi.nlm.nih.gov/) with Accession number: MW406457 (P. salicina 'Oishiwase'), MW406459 (P. salicina ‘Sanhua plum’), MW406460 (P. salicina ‘Wanshuang plum’), MW406461 (P. salicina ‘Wuyuecui’), MW406463 (P. simonii 'Weiwang'), MW406464 (P. domestica 'Richard Early'), MW406465 (P. salicina 'Yinhong plum'), MW406466 (P. salicina 'Fengtang plum'), MW406468 (P. salicina 'Cuihong plum'), MW406470 (P. cerasifera 'Hollywood'), MW406471 (P. domestica 'Bingtang plum') and MW406472 (P. salicina 'No.2 Guofeng'). Raw sequence data for this study also can be found in GenBank. The associated BioProject, SRA, Bio-Sample numbers are PRJNA795302, SRR17478141, SAMN24706757 (P. salicina 'Oishiwase'); PRJNA795262, SRR17477716, SAMN24699028 (P. salicina ‘Sanhua plum’); PRJNA795282, SRR17477883, SAMN24704169 (P. salicina ‘Wanshuang plum’); PRJNA795299, SRR17478140, SAMN24705967(P. salicina ‘Wuyuecui’); PRJNA719267, SRR14133449, SAMN18593728 (P. simonii 'Weiwang'); PRJNA795321, SRR17478255, SAMN24707166 (P. domestica 'Richard Early'); PRJNA795322, SRR17478256, SAMN24707171 (P. salicina 'Yinhong plum'); PRJNA795338, SRR17480298, SAMN24708705 (P. salicina 'Fengtang plum'); PRJNA795340, SRR17480297, SAMN24709469 (P. salicina 'Cuihong plum'); PRJNA795572, SRR17498860, SAMN24731932 (P. cerasifera 'Hollywood'); PRJNA795571, SRR17498195, SAMN24731931 (P. domestica 'Bingtang plum'); PRJNA795581, SRR17498228, SAMN24734651 (P. salicina 'No.2 Guofeng'). All the samples are saved at the Herbarium of Southwest University, Chongqing, China. All other data and material generated in this manuscript are available from the corresponding author upon reasonable request.
Abbreviations
- SSRs:
-
Simple sequence repeats
- IR:
-
Inverted repeat
- LSC:
-
Long Single Copy
- SSC:
-
Short Single Copy
- IGS:
-
Intergenic spacer
- tRNA:
-
Transfer RNA
- rRNA:
-
Ribosomal RNA
References
Huertas M, Díaz-Mula P, Javier Z, Fabián G, Salvador C. Changes in physicochemical and nutritive parameters and bioactive compounds during development and on-tree ripening of eight plum cultivars: a comparative study. J Sci Food Agric. 2008;14:2499–507.
González-García E, Marina ML, García M. Plum (Prunus Domestica L.) by-product as a new and cheap source of bioactive peptides: Extraction method and peptides characterization. J Funct Foods. 2014;11:428–37.
Liu C, Feng C, Peng W, Hao J, He Y: Chromosome-level draft genome of a diploid plum ( Prunus salicina ). GigaScience 2020, 9(12).
Wen LJ. A phylogenetic analysis of Prunus and the Amygdaloideae (Rosaceae) using ITS sequences of nuclear ribosomal DNA. Am J Bot. 2001;88(1):150–60.
Huang M. Key points of high quality and high yield cultivation techniques of Wushan Plum South China. Agriculture. 2009;3(04):3–5.
He C, Pu C, Zhang N, Wang X. Breeding and cultivation techniques of a new plum variety “Fendai Plum.” South China Fruit. 2019;48(2):144–6.
Zhou W, He C, Zhang N, Pu C, Wang X. Breeding and cultivation techniques of a new plum variety “Wanshuang Plum.” China Fruit News. 2019;36(03):55–7.
Li Y. “Sanhua plum” in Guangdong. Shanxi Fruits. 1989;03:44–5.
Xu Y, Zheng X, Li W. The excellent variety of local Plum crisp hongli and its cultivation techniques. China Fruit News. 2002;05:39–40.
Tao Y, Cheng Y. Sichuan famous fruit – Jiang ’an plum. Sichuan Fruit Tree Technology. 1986;01:37–8.
Zhang Y, Li Y. XIao Y, Cheng L, Wu J: New medium ripe plum variety “Fengtang Plum.” Northern Fruits. 2018;05:56.
Lou S. Fruit growth and meteorological conditions of Plum. Zhejiang Meteorological Technology. 1989;02:35–6.
Xiao W, Qiuping Z, Weisheng L. Research Progress on Plum Germplasm Resources in China. Acta Horticultural Sinica. 2020;47(06):1203–12.
Shinozaki O. Tanaka, Wakasugi, Sugiura: The complete nucleotide sequence of the tobacco chloroplast genome. Plant Mol Biol Report. 1986;4(3):110–47.
Ohyama K, Fukuzawa H, Kohchi T, Shirai H, Sano T, Sano S, Umesono K, Shiki Y, Takeuchi M, Chang Z. Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature. 1986;322(6079):572–4.
Korpelainen H. The evolutionary processes of mitochondrial and chloroplast genomes differ from those of nuclear genomes. The Science of Nature. 2004;91(11):505–18.
Wolfe KH, Li WH, Sharp PM. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci U S A. 1987;84(24):9054–8.
Badenes ML, Parfitt DE. Phylogenetic relationships of cultivated Prunus species from an analysis of chloroplast DNA variation. Theor Appl Genet. 1995;90(7):1035–41.
Yamamoto T, Mochida K, Imai T, Haji T, Yaegaki H, Yamaguchi M, Matsuta N, Ogiwara I, Hayashi T. Parentage Analysis in Japanese Peaches using SSR Markers. Japanese Journal of Breeding. 2003;53(1):35–40.
Sm A, Yg B, Jmo A. The use of RAPD markers for identification of cultivated grapevine ( Vitis vinifera L.). Sci Hortic. 1995;62(4):237–43.
Zuo L, Han Z, Liang H, Yang M. Analysis of Genetic Diversity of Prunus salicina from Different Producing Areas by SSR Markers. Acta Horticulturae Sinica. 2015;42(1):111–8.
Goulding SE, Wolfe KH, Olmstead RG, Morden CW. Ebb and flow of the chloroplast inverted repeat. Mol Gen Genet MGG. 1996;252(1):195–206.
Dong W, Liu J, Yu J, Wang L, Zhou S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. Plos One. 2012;7(4):e35071.
McLay TGB, Birch JL, Gunn BF, Ning W, Tate JA, Nauheimer L, Joyce EM, Simpson L, Schmidt-Lebuhn AN, Baker WJ et al: New targets acquired: Improving locus recovery from the Angiosperms353 probe set. Appl Plant Sci 2021, 9(7).
Duan C, Zhang K, Duan Y. Comparison of complete chloroplast genome sequences of Amygdalus pedunculata Pall. Chin J Biotechnol. 2020;36(12):2850–9.
Li C, Zheng Y, Huang P. Molecular markers from the chloroplast genome of rose provide a complementary tool for variety discrimination and profiling. Sci Rep. 2020;10(1):12188.
Lei W, Ni D, Wang Y, Shao J, Chang L. Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci Rep. 2016;6:21669.
Katayama H, Uematsu C. Structural analysis of chloroplast DNA in Prunus (Rosaceae): evolution, genetic diversity and unequal mutations. Theor Appl Genet. 2005;111(7):1430–9.
Huo Y, Yan M, Zhao X, Zhu Z, Yuan Z. The complete chloroplast genome sequence of Prunus Cerasifera Ehrh. ’Pissardii’ (Rosaceae). Mitochondrial DNA B Resour. 2019;4(2):3744–5.
Geng W, Ouyang L, Xu M, Jing J. The complete chloroplast genome of Prunus domestica L. (Rosaceae) and its phylogenetic implication. Mitochondrial DNA B Resour. 2020;5(3):2783–4.
Xue S, Shi T, Luo W, Ni X, Iqbal S, Ni Z, Huang X, Yao D, Shen Z, Gao Z: Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic Res 2019, 6:89.
Gichira AW, Avoga S, Li Z, Hu G, Wang Q, Chen J. Comparative genomics of 11 complete chloroplast genomes of Senecioneae (Asteraceae) species: DNA barcodes and phylogenetics. Botanical Studies. 2019;60:17.
Li W, Zhang C, Guo X, Liu Q, Wang K. Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis. PLoS ONE. 2019;14(5): e0216645.
Jiang M, Chen H, He S, Wang L, Chen A, Chang L. Sequencing, Characterization, and Comparative Analyses of the Plastome of Caragana rosea var. rosea. Int J Mol Sci. 2018;19(5):1419.
Su Y, Liu Y, Li Z, Fang Z, Yang L, Zhuang M, Zhang Y. QTL Analysis of head splitting resistance in cabbage (Brassica oleracea L. var. capitata) Using SSR and InDel makers based on whole-genome re-sequencing. Plos One. 2015;10(9):e0138073.
Yi X, Lei G, Wang B, Su YJ, Wang T: The Complete Chloroplast Genome Sequence of Cephalotaxus oliveri (Cephalotaxaceae): Evolutionary Comparison of Cephalotaxus Chloroplast DNAs and Insights into the Loss of Inverted Repeat Copies in Gymnosperms. Genome Biology and Evolution,5,4(2013–3–28) 2013(4):688–698.
Zhang Y, Li L, Yan TL, Liu Q. Complete chloroplast genome sequences of Praxelis (Eupatorium catarium Veldkamp), an important invasive species. Gene. 2014;549(1):58–69.
Nie X, Lv S, Zhang Y, Du X, Wang L, Biradar SS, Tan X, Wan F, Song W, Sergios-Orestis K. Complete Chloroplast Genome Sequence of a Major Invasive Species, Crofton Weed (Ageratina adenophora). PLoS ONE. 2012;7(5): e36869.
Pauwels M, Vekemans X, Godé C, Frérot H, Castric V, Saumitou-Laprade P: Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, <em>Arabidopsis halleri</em> (Brassicaceae). 2012, 193(4):916–928.
Liu L, Wang Y, He P, Li P, Lee J, Soltis DE, Fu C. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genomics. 2018;19(1):235.
Li R, Ma PF, Wen J, Yi TS. Complete Sequencing of Five Araliaceae Chloroplast Genomes and the Phylogenetic Implications. PLoS ONE. 2013;8(10): e78568.
Zhang Y, Du L, Ao L, Chen J, Li W, Hu W, Zhang W, Kyunghee K, Sang-Choon L, Yang TJ. The complete chloroplast genome sequences of Five Epimedium Species: lights into Phylogenetic and Taxonomic Analyses. Front Plant Sci. 2016;7:306.
Ki-Joong K, Hae-Lim L. Complete Chloroplast Genome Sequences from Korean Ginseng (Panax schinseng Nees) and Comparative Analysis of Sequence Evolution among 17 Vascular Plants. DNA Res. 2005;4:247–61.
Jansen RK, Wojciechowski MF, Sanniyasi E, Lee SB, Daniell H. Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol Phylogenet Evol. 2008;48(3):1204–17.
Magee AM, Aspinall S, Rice DW, Cusack BP, Semon M, Perry AS, Stefanovic S, Milbourne D, Barth S, Palmer JD. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010;20(12):1700–10.
Chumley TW, Palmer JD, Mower JP, Matthew FH, Calie PJ, Boore JL, Jansen RK. The complete Chloroplast Genome Sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol. 2006;11:2175–90.
Wakasugi T, Tsudzuki J, Ito S, Nakashima K, Tsudzuki T, Sugiura M. Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc Natl Acad Sci U S A. 1994;91(21):9794–8.
Hui C, Li J, Hong Z, Cai B, Lin M. The complete chloroplast genome sequence of strawberry (Fragaria ×ananassa Duch.) and comparison with related species of Rosaceae. Peerj. 2017;5(10):e3919.
Ni L, Zhao Z, Gaawe D, Mi M, Chen S. The Complete Chloroplast Genome of Ye-Xing-Ba (Scrophularia dentata; Scrophulariaceae), an Alpine Tibetan Herb. PLoS ONE. 2016;11(7): e0158488.
Hummer KE, Janick J: Rosaceae: Taxonomy, Economic Importance, Genomics: Rosaceae: Taxonomy, Economic Importance, Genomics; 2009.
Zhang Q, Liu W, He Y, Luo X. Development of “Sanhua Plum” in Xinyi city. Guangdong Province Northern Fruits. 2019;05:52–3.
Zhang J, He F. Introduction, cultivation and research of Plum in China. Hebei Fruits. 2003;03:1–3.
Du H, Li F, Fu D, Yang S, Du L, Fu J, Li F. A new interspecific hybrid of apricot and plum at middle and late maturity “Weiwang.” Acta Horticulturae Sinica. 2005;01:174.
Yu X, Liu W, Liu N, Sun M, Zhang Y, Xu M. Report on the introduction and trial planting of “Richard Early.” Forest By-Product and Speciality in China. 2007;04:51–2.
Ou Y, Cheng Y, Dai Z. Cultivation technology of crisp red plum with high quality and high yield. Southwest Horticulture. 2005;06:56–7.
Wang F, Fang C, Lin S. AFLP technique and its application in fruit genetics and breeding. China Fruits. 2006;03:50–3.
Arseneau J-R, Steeves R, Laflamme M: Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Molecular Ecology Resources 2017.
Jin JJ, Yu WB, Yang JB, Song Y, Li DZ. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020;21(1):241.
Langmead B. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.
Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nuclc Acids Research. 2019;47(W1):W65–73.
Michael, Tillich, Pascal, Lehwark, Tommaso, Pellizzer, Elena, Ulbricht-Jones, Axel: GeSeq?- versatile and accurate annotation of organelle genomes. Nucleic Acids Research 2017.
Misra S, Harris N: Using Apollo to browse and edit genome annotations. Curr Protoc Bioinformatics 2006.
Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–7.
Sebastian B, Thomas T, Thomas M, Uwe S, Martin M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;16:2583.
Stefan K, Choudhuri JV, Enno O, Chris S, Jens S, Robert G. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;22:4633–42.
Thompson JD, Gibson TJ, Higgins DG: Multiple Sequence Alignment Using ClustalW and ClustalX. Current Protocols in Bioinformatics 2003, Chapter 2, Unit 2 3,.
Amiryousefi, Ali, Hyvonen, Jaakko, Poczai, Peter: IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018.
John R, Songling L, Mar AK, Standley DM, Kazutaka K. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 2019;W1:W5–10.
Alexandros S. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;9:1312–3.
Johnson MG, Gardner EM, Liu Y, Medina R, Goffinet B, Shaw AJ, Zerega NJ, Wickett NJ. HybPiper: Extracting coding sequence and introns for phylogenetics from high-throughput sequencing reads using target enrichment. Appl Plant Sci. 2016;4(7):apps.1600016.
Acknowledgements
The authors appreciate the taxonomists of the Chongqing Academy of Agricultural Sciences who helped collect samples.
Funding
The National Natural Science Foundation of China (31772260). Chongqing Study Abroad Innovation Project (cx2019052). The Youth Innovation Team Project of the Chongqing Academy of Agricultural Sciences (NKY-2019QC08).
Author information
Authors and Affiliations
Contributions
CL and JY contributed to the conception and planning of the research. YCX and JTL performed the experiments and analyzed the data. BF, JY and YWW collected samples. JLL and YCX assembled the plastomes. YCX wrote the paper. CL and JY critically reviewed the manuscript. All authors have read and agreed on the final contents of the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The twelve collected plum cultivars are widely distributed in China. All samples in this study were collected with permission from the garden authorities. All samples were identified by Professor Jie Yu of Southwest University. The voucher specimens were deposited in the herbarium of the College of Horticulture and Landscape Architecture in Southwest University. The voucher numbers were swu202003 (P. salicina 'Sanhua plum'), swu202005 (P. salicina 'Wanshuang plum'), swu202006 (P. salicina 'Wuyuecui'), swu202015 (P. salicina 'Oishiwase'), swu202025 (P. simonii 'Weiwang'), swu202034(P. domestica 'Richard Early'), swu202052 (P. salicina 'Yinhong plum'), swu202053 (P. salicina 'Fengtang plum'), swu202055 (P. salicina 'Cuihong plum'), swu202064 (P. cerasifera 'Hollywood'), swu202066 (P. domestica 'Bingtang plum'), swu202068 (P. salicina 'No.2 Guofeng').
This study complies with relevant institutional, national, and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
TableS1. Summary of sequencing data quality. Table S2. Gene composition in the plastomes of twelve plum cultivars. Table S3. Length of introns and exons inthe plastomes of twelve plumcultivars. Table S4. Statistics on simple sequencerepeats (SSRs) in the twelve plastomes. TableS5. The list of accession numbers of the plastome sequences used in thephylogenetic analyses of the Prunus. FigureS1. Genome map of P. salicina ‘Wanshuang plum’ plastome. Figure S2. Genome map of P. salicina ‘Wuyuecui’ plastome. Figure S3. Genome map of P. salicina ‘Oishiwase’ plastome. Figure S4. Genome map of P. simonii 'Weiwang' plastome. Figure S5. Genome map of P. domestica 'Richard Early' plastome. Figure S6. Genome map of P. salicina 'Yinhong plum' plastome. Figure S7. Genome map of P. salicina ' Fengtang plum' plastome. Figure S8. Genome map of P. salicina ' Cuihong plum' plastome. Figure S9. Genome map of P. cerasifera 'Hollywood' plastome. Figure S10. Genome map of P. domestica 'Bingtang plum' plastome. Figure S11. Genome map of P. salicina 'No.2 Guofeng' plastome. Figure S12. Phylogenetic relationshipsof species from Prunus (Rosaceae)inferred using Maximum likelihood (ML) method. Figure S13. The gel electrophoresis results of the amplificationof DNA barcodes using designed primer LZ01. Figure S14. The gel electrophoresis results of the amplification ofDNA barcodes using designed primer LZ02. FigureS15. The gel electrophoresis results of the amplification of DNA barcodesusing designed primer LZ03. Figure S16.The gel electrophoresis results of the amplification of DNA barcodes usingdesigned primer LZ04. Figure S17.The gel electrophoresis results of the amplification of DNA barcodes usingdesigned primer LZ05. Figure S18.The gel electrophoresis results of the amplification of DNA barcodes usingdesigned primer LZ06. Figure S19.The gel electrophoresis results of the amplification of DNA barcodes usingdesigned primer LZ07. Figure S20.The gel electrophoresis results of the amplification of DNA barcodes usingdesigned primer LZ08. Figure S21.The alignment of amplicons produced by designed LZ01 primer. Figure S22. The alignment of ampliconsproduced by designed LZ02 primer. FigureS23. The alignment of amplicons produced by designed LZ03 primer. Figure S24. The alignment of ampliconsproduced by designed LZ04 primer. FigureS25. The alignment of amplicons produced by designed LZ05 primer. Figure S26. The alignment of ampliconsproduced by designed LZ06 primer. FigureS27. The alignment of amplicons produced by designed LZ07 primer. Figure S28. The alignment of ampliconsproduced by designed LZ08 primer.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, Y., Fang, B., Li, J. et al. Phylogenomic analysis and development of molecular markers for the determination of twelve plum cultivars (Prunus, Rosaceae). BMC Genomics 23, 745 (2022). https://doi.org/10.1186/s12864-022-08965-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08965-z