
Abstract
Background
With the expansion of animal production, parasitic helminths are gaining increasing economic importance. However, application of several established deworming agents can harm treated hosts and environment due to their low specificity. Furthermore, the number of parasite strains showing resistance is growing, while hardly any new anthelminthics are being developed. Here, we present a bioinformatics workflow designed to reduce the time and cost in the development of new strategies against parasites. The workflow includes quantitative transcriptomics and proteomics, 3D structure modeling, binding site prediction, and virtual ligand screening. Its use is demonstrated for Acanthocephala (thorny-headed worms) which are an emerging pest in fish aquaculture. We included three acanthocephalans (Pomphorhynchus laevis, Neoechinorhynchus agilis, Neoechinorhynchus buttnerae) from four fish species (common barbel, European eel, thinlip mullet, tambaqui).
Results
The workflow led to eleven highly specific candidate targets in acanthocephalans. The candidate targets showed constant and elevated transcript abundances across definitive and accidental hosts, suggestive of constitutive expression and functional importance. Hence, the impairment of the corresponding proteins should enable specific and effective killing of acanthocephalans. Candidate targets were also highly abundant in the acanthocephalan body wall, through which these gutless parasites take up nutrients. Thus, the candidate targets are likely to be accessible to compounds that are orally administered to fish. Virtual ligand screening led to ten compounds, of which five appeared to be especially promising according to ADMET, GHS, and RO5 criteria: tadalafil, pranazepide, piketoprofen, heliomycin, and the nematicide derquantel.
Conclusions
The combination of genomics, transcriptomics, and proteomics led to a broadly applicable procedure for the cost- and time-saving identification of candidate target proteins in parasites. The ligands predicted to bind can now be further evaluated for their suitability in the control of acanthocephalans. The workflow has been deposited at the Galaxy workflow server under the URL tinyurl.com/yx72rda7.
Similar content being viewed by others


Background
The global market for antiparasitics, or parasiticides, currently amounts to about seven billion euros per year [1]. Most of this is spent on meat production but the share of expenditure in the aquaculture of fish is very likely to significantly grow in near future. With about 50 million tons per year, the production of animal protein in aquaculture is now already of great importance for many countries. However, many of these follow the agenda to increase the aquaculture branch, for securing high-quality food supply to their often growing populations [2]. As a result, revenues in fish aquaculture, currently estimated at around 140 billion dollars per year, will also rise [3], as will the losses due to diseases of yearly around 6 billion US dollars [4]. Accordingly, the control of parasites of taxa such as Nematoda (roundworms), Platyhelminthes (flatworms), and Acanthocephala (thorny-headed worms) in fish aquaculture is a major issue.
Extracts from garlic [5], thyme [6] and different species of the Fabaceae genus Copaifera [7] might have potential in the control of Acanthocephala (reviewed in [8]). However, to date no reliable agent has passed clinical evaluation. In addition to phytoextracts, a whole arsenal of chemical anthelmintics is available [9, 10]. Although effective, a widespread disadvantage is a low specificity as reflected in the use against a broad range of taxa (survey e.g. in [11]). Well-known examples are niclosamide and benzimidazole derivates, which are used against phylogenetically distant parasites [9, 12, 13]. But snails, unicellular species, invertebrate metazoans, and algae can be negatively affected as well [12, 14, 15]. Their pro-apoptotic activity in the broadest sense even confers potential on diverse anthelmintics as cytostatic agents in cancer therapy [16,17,18,19,20,21,22,23,24]. Consequently, when dissipated into surrounding waters, deworming agents and their mostly under-investigated metabolites might cause harm to a broad range of species (e.g., [25]). Also, detrimental long-term effects appear possible since anthelmintic metabolites can accumulate and persist in the environment [26, 27]. Additional concerns arise from the growing number of resistant parasite strains (e.g., [28,29,30]) while almost no new anthelmintics are being developed [31, 32]. Thus, there is a need for the development of novel strategies in parasite control.
A time- and cost-saving approach lies in the mechanism-based screening of compound libraries for ligands to parasitic target molecules. An important point here is that advances in genomics, transcriptomics, and proteomics target now enable the determination of functionally highly important and specific target molecules in parasites. Favorable for ligand-screening is further the recent leap in protein structure prediction. Indeed, with the recent progress in 3D structure modelling (AlphaFold2) it is now possible to predict 3D models with much greater precision de novo than before [31]. Nevertheless, traditional methods were prevailing in the development of drugs against parasites until recently [32,33,34], with comparably few exceptions so far. For example, several studies used omics for identifying drug targets in viruses (e.g., SARS-CoV-2 [35]), malaria-causing Plasmodium falciparum [36], and additional unicellular pathogens [37]. In addition, several potential antigens have been identified for nematode and platyhelminth parasites of humans, lifestock, and pets, including Ancylostoma duodenale, Ascaris lumbricoides, Brugia malayi, Echinococcus granulosus, Fasciola hepatica, Haemonchus contortus, Necator americanus, Onchocerca volvulus, Ostertagia ostertagi, Schistosoma spp., Strongyloides stercoralis, Taenia solium, Teladorsagia circumcincta, Toxocara canis, and Trichuris trichiura (reviewed in [38]). These examples also reflect the increasing power of omics-guided antigen or target identification, with genomics providing the basis for transcriptomics and proteomics. Consequently, recently proposed strategies in the field increasingly rely on proteomics or a combination of genomics, transcriptomics, and proteomics. Corresponding workflows inherently integrate gene annotations and ontologies [38, 39]. But to the best of our knowledge, there is yet no bioinformatics workflow integrating various omics techniques, annotation, gene ontology analysis, 3D modelling, and virtual ligand screening into the development of novel strategies in parasite control. Here we present a corresponding workflow for the determination of candidate target proteins, the disruption or blocking of which should effectively and specifically kill parasites. We preliminarily characterize the parasite proteins focused, model their 3D structure, and present potential ligands with known properties. To demonstrate its general applicability, we establish the procedure in acanthocephalans.
Acanthocephalans are common parasites in the intestinal tract of many mammals, amphibians, birds, turtles, lizards, snakes, and fishes (e.g., [40]). Depending on the host and the intensity of infection, the worms might penetrate the intestinal wall, which can cause fatal peritonitis [41]. Migrating worms also damage other host organs and mesenteries [42]. Inside the intestine, acanthocephalans injure the intestinal wall with their usually hook-bearing attachment organ, the proboscis [43,44,45]. The resulting lesions reduce the absorptive surface and hence lower the ability of the host to take up nutrients [43]. The gutless worms also absorb minerals and nutrients via their tegument, which they withhold or withdraw from the host [46,47,48]. Intestinal obstruction can also be fatal due to mass infections with up to ~ 1500 thorny-headed worms per individual host in the wild (e.g., [49, 50]). Acanthocephalans additionally parasitize human livestock, including domestic pig (Sus scrofa domestica) and chicken (Gallus gallus domesticus) (e.g., [47, 51]). They are also regular members of the parasitic fauna in marine fish aquaculture (e.g., [11, 52, 53]). Infections with acanthocephalans are further documented for limnocultures of brown trout (Salmo trutta fario), pirarucu (Arapaima gigas), Nile tilapia (Oreochromis niloticus), and tambaqui (Colossoma macropum), amongst others [54,55,56,57]. Here, high intensities can cause reduced growth, weakening, and emaciation of the fish. Deformations and death of infected fish have also been reported [52, 54, 58]. In Brazil, for instance, acanthocephaliasis is regarded as the main obstacle to successful aquaculture [8, 45, 59,60,61,62,63,64].
First genome and transcriptome assemblies for Acanthocephala have lately been published for the Eurasian species Pomphorhynchus laevis (Zoega in Müller, 1776) Monticelli, 1905 (Acanthocephala: Palaeacanthocephala) [65]. In the present investigation, we included worms from common barbel (Barbus barbus), a definitive host in which P. laevis matures and reproduces [66, 67], and European eel (Anguilla anguilla), in which P. laevis survives [68] but usually does not mature and reproduce [69]. For raising effectiveness of any yet-to-be determined agent against acanthocephalans, we searched for transcripts with little variation in abundance at a high level in 20 male and female P. laevis specimens from barbel and eel. To enable enhanced specificity of a future control of acanthocephalans, we screened for transcripts which were unique to or at least highly derived in P. laevis compared to diverse non-acanthocephalan species. Moreover, the ideal candidate target had to be readily accessible to drugs. This criterion was approximated by searching proteome data of P. laevis body walls for high-abundance proteins. To increase transferability of the results, protein sequences were checked for matches in new draft genomes of two additional fish-parasitizing species (Acanthocephala: Eoacanthocephala). These were Neoechinorhynchus agilis (Rudolphi, 1819) Van Cleave, 1916 from Adriatic thinlip mullet (Chelon ramada) and Neoechinorhynchus buttnerae Golvan, 1956 parasitizing above-mentioned tambaqui in South-American limnocultures. Predicted 3D models of the proteins meeting all these requirements were used to screen for drugs that might bind to acanthocephalan target proteins.
Results
Sequencing and mapping
Male and female specimens of P. laevis (Palaeacanthocephala) from two different host species were used for sequencing, resulting in four pairings of worm sex and host species. Each group included five worm specimens, so possible confounding factors in individual samples should not have affected downstream analysis. The resulting 20 RNA-Seq datasets contained 32.6 million single-end reads (75 bp) on average. Quality processing and mapping to an amended version of the reference transcriptome [65] was successfully passed by 95.1% of the sequences.
Candidate target proteins for drug search
Mapping with RSEM revealed that the reads from RNA-Seq spread across transcripts of 18,740 genes. For downstream analysis of transcript abundance (DESeq2) we only considered genes that had at least 50 mapped transcript reads in each of the 20 samples, thus suggesting ubiquitous expression in P. laevis (availability criterion in Fig. 1; Supplementary Fig. S1). As an indication of low regulation up to constitutive expression, the log fold change of transcript abundance was set to ≤1.50 (adjusted p-value < 0.05) in a minimum of two out of four pairs of comparisons: (1) female vs. male worms from barbel, (2) female vs. male worms from eel, (3) male worms from barbel vs. eel, and (4) female worms from barbel vs. eel (Fig. 2; Supplementary Table S1). From the transcripts fulfilling this requirement, we extracted all open reading frames (ORFs) of at least 30 codons. Subsequent BLASTs for translated ORFs reduced the number of candidate targets to 121 (effectiveness & specificity criteria in Fig. 1). The corresponding P. laevis sequences had tblastn hits with E-values <1e-50 in novel genome assemblies of two fish-parasitizing acanthocephalan species from Eoacanthocephala, N. buttnerae and N. agilis (Supplementary Notes S1 & S2). Thus, drugs to be developed against them should be effective not only in P. laevis but fish-parasitizing acanthocephalans in general. The 121 candidates additionally lacked matches in six closer phylogenetic relatives from the Rotifera-Acanthocephala clade (Syndermata or Rotifera sensu lato) and in the SwissProt database at an E-value <1e-50 (tblastn/blastp). Correspondingly, agents tailored to these targets should specifically impair acanthocephalans but no other taxa.

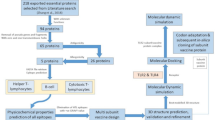
Flowchart of the analysis workflow. Female and male worm specimens were collected from two different hosts and used for mRNA sequencing and mass spectrometry. In-depth analyses ensured target identification (the target sequence is present in acanthocephalans), specificity (the target sequence is absent or has little sequence similarity in non-acanthocephalan species), and availability and accessibility (the target is present as protein in the acanthocephalan body wall). Candidate target protein sequences that fulfill these criteria were passed on to protein modeling and ligand screening

Analysis of transcript abundance. Shown are differential expression values for genes with at least 50 transcript reads in every sample. Each dot represents one gene. Genes with significantly similar transcript abundances (log fold change < 1.5; adjusted p-value < 0.05) are labeled blue, the remaining ones red. Only genes with similar transcript abundances in at least two of the four comparisons were kept for downstream analyses. Up- and downward pointing triangles at the top and bottom margins of the plots represent data points outside of the range depicted. Only a few isolated data points to the right have been omitted for better display
Since transcript abundance does not necessarily correlate with protein abundance (e.g., [70]), we validated the above results in proteome data. Corresponding mass spectrometry (MS) analysis focused on the acanthocephalan tissue promising easiest targetability, i.e., the body wall enclosing the gutless worms (availability criterion in Fig. 1). Based on five pools of P. laevis body-walls, the mass spectra matched 2548 ORFs in the reference transcriptome. Abundances of these proteins (iBAQ values) were significantly positively correlated with transcript abundances according to read counts (coefficient of correlation = 0.51, p-value = 2.4 e-165, Student’s t-test). Thus, higher transcript abundance overall indicated elevated protein abundance. The search for parasitic target molecules was continued with 52 MS-verified proteins (Fig. 3). As to be expected, transcript and protein abundances of these 52 target candidates were even more strongly correlated (coefficient of correlation = 0.81, p-value = 4.4 e-13, Student’s t-test).

Correlation transcript and protein abundances. Each dot represents one of ~ 2500 proteins quantified by mass spectrometry. Candidate target proteins are highlighted in pink. Protein and transcript (RNA) abundances are given as iBAQ values and mean read counts, respectively. The correlation between the two abundances was moderately positive for all proteins (0.51; p-value = 2.4 e-165; Student’s t-test) and strongly positive for the candidate target proteins (0.81; p-value = 4.4 e-13; Student’s t-test). Given the levels of p-values, correction for multiple testing would not have affected the determination of significance
The choice of candidates was supported by the fact that the matching rate between both subsets (52/121 = 0.43) was about three times higher than between the corresponding full lists (2548/18,740 = 0.14). In further support of the validity of the approach, the 52 proteins matched several expected properties. Thus, their mean length (485 amino acid residues) was very close to the average in eukaryotes (472 amino acid residues), and clearly exceeded corresponding averages in unicellular species (ca. 300 amino acid residues) [71]. Furthermore, the amino acid frequencies of the candidates retained was strongly positively correlated with previously reported average values across 614 eukaryotic proteomes (coefficient = 0.88, p = 3.6 e-07, Student’s t-test; Supplementary Table S2) [72]. Of the 52 candidate target proteins, 46 were characterized to be overall hydrophilic (88.5%), and eleven were predicted to have transmembrane helices (21.2%) (Supplementary Table S3). PFAM motifs were found in 46 of the candidate target proteins (88.5%), and Prosite motifs in 34 (65.4%) (Supplementary Table S4).
Protein structure and binding site prediction
The above filtering for dissimilar genes ruled out to use structure models of homologous proteins in non-acanthocephalan species as a starting point. In fact, a database search (NCBI) did not reveal a deposited 3D model of a protein structure for any of the 52 candidate target proteins in P. laevis. This prompted us to perform de novo predictions using AlphaFold2 [31], which in 44 cases succeeded in providing a 3D model of the protein structure. AlphaFold2’s per-residue confidence metric pLDDT (range: 0–100) was 75.7 averaged across all candidates, with mean values ranging from 41.7 to 95.3 for the individual proteins. For estimating the precision of the structure predictions, we employed an additional protein structure prediction program, RoseTTAFold [73] (Supplementary Table S5). Comparison using the Dali Protein Structure Comparison Server [74] revealed high similarity of the models predicted. Thus, Dali’s average confidence or z-score for model comparison was 24.2, which is far beyond the threshold of strong matches (> 2). Likewise, Dali rated the average identity between models from AlphaFold2 and RoseTTAFold predictions as 85.5%, which considerably exceeds the threshold of significant similarities (> 20% [75];).
Virtual ligand screening
Based on the AlphaFold2 models, COACH-D [76] identified putative ligand binding sites in each of the remaining 44 target candidates. In two of the protein models, a secondary binding site was predicted. Although confidence (c) scores varied widely (0.04 to 0.88), we retained all putative binding sites for ligand screening. For each of the 44 candidate targets, virtual screening of clinically tested and approved compounds using AutoDock Vina [77] identified ligands. Supplementary Table S6 provides previous knowledge on indications and molecular targets of these ligands, as extracted from various databases (ChEMBL, ClinicalTrials.gov, DrugBank, PubChem). Applying an arbitrary threshold of binding energy (− 9 kcal/mol), ten compounds remained as strongest-binding ligands to eleven candidate targets (Tables 1, 2). The discrepancy in number reflected that two of the acanthocephalan proteins shared tadalafil as strongest-binding ligand (Fig. 4; Table 1). It is further worthwhile noting that one of the ligands, the nematocidal anthelmintic derquantel, was predicted to bind second strongest to the model of protein 1609, in addition to its strongest binding to protein model 4617. The other eight ligands were each predicted to dock strongly to single acanthocephalan targets only (Table 2).

Three-dimensional structure models of the eleven top candidate target proteins. Shown are de novo models of 3D structures (constructed using AlphaFold2) for eleven proteins which fulfilled all filter criteria. The proteins were each additionally predicted to bind a drug with a free energy of ≤ −9.0 kcal/mol in the virtual screening using AutoDock Vina. The proteins are shown as molecular surfaces colored by AlphaFold2 confidence score (pLDDT; with higher values having greater confidence). Gray markings indicate predicted binding sites (on the surface or within the protein). Values in parentheses below protein identifiers give the average pLDDT of the protein model followed by the percentage identity between this model and one from a second 3D structure prediction program, RoseTTAFold. Both values are on the scale 0–100
Eight of the eleven putative targets in acanthocephalans were likely to have enzymatic activity according to ECPred, and ten of the target molecules contained PFAM motifs (Table 1, Supplementary Tables S3, S4). Furthermore, three of the ten compounds were labelled according to the Globally Harmonized System (GHS). These were etoposide (irritant, health hazard), tadalafil (irritant), and fluazuron (environmental hazard) (Supplementary Table S6). Still, these limitations do not necessarily preclude their repurposing in acanthocephaliasis (see Discussion). Indeed, the usability of all ten agents as drugs was reflected in the fact that most of the parameters giving absorption, distribution, metabolism, excretion, and toxicity (ADMET) were in the optimal range (Table 2). Furthermore, eight of the agents fulfilled the rule of five (RO5), indicating suitability for oral administration, or have already been successfully administered per os in clinical trials (see asterisks in RO5 column of Table 2). This requirement can be considered almost mandatory for the treatment of fish in aquaculture. In sum, a total of five compounds are likely to be most promising for future in vitro testing in acanthocephaliasis. These are pranazepide, piketoprofen, and heliomycin, in addition to above-mentioned derquantel and tadalafil.
Discussion
Present results demonstrate the utility of a workflow for identifying putative ligands to target proteins in parasites. The approach is based on genomic, transcriptomic, and proteomic data, followed by 3D structure and binding site prediction, virtual ligand screening, and ADMET property prediction. Applied to fish-parasitizing acanthocephalans, we identified eleven proteins the blocking of which should enable an effective and specific control of these parasites. Five of the ten identified ligands appear to be particularly promising for further testing in vitro and in fish aquacultures.
Candidate target identification in parasites
The filtering workflow was designed to converge on druggable targets in fish-parasitizing acanthocephalans (Fig. 1; Supplementary Fig. S1). One requirement was elevated and little varying up to constant transcript abundance in alternative host species providing different physiological environments to the parasite. This suggests targetability in all acanthocephalan specimens. Confirmation of high abundance on the protein level and, especially, in the acanthocephalan body wall should increase their accessibility to orally administered compounds. We expect this to be a requirement for an effective control of gutless acanthocephalans, which take up nutrients and minerals via the surface of the body wall [46,47,48]. A corresponding example is the drug loperamid, which has been shown to enter acanthocephalans via the surface of the tegument making up the outermost part of the body wall upon oral administration to infected pigs [58]. In addition, combating acanthocephalans, so to speak, at the point of entry should be more likely to succeed than targeting proteins in inner organs. In addition, we consider it beneficial for the specificity of acanthocephalan control if target proteins lack homologues in non-acanthocephalan taxa. However, this criterion is not absolute, and we here allowed for the retention of parasite proteins showing high sequence divergence compared to putative homologues in other taxa. Such homology should even be advantageous since corresponding targets could be involved in basic pathways the blocking of which should strongly affect the parasites. On the contrary, we regarded the presence of homologues in two Neoechinorhynchus species as mandatory. This criterion ensured the determination of target proteins that are conserved among fish-parasitizing palaeacanthocephalans and eoacanthocephalans from distant geographic regions (South America and Eurasia). More importantly, one of the species included was N. buttnerae, which is the economically most important acanthocephalan pest species in fish farms. In fact, this species is the major problem in South American limnocultures of fish, decreasing the yield of aquaculture farms by up to 90% [8, 45, 59,60,61,62,63,64]. Thus, confirmation of the candidate targets in N. buttnerae has direct implications for the practical use of the present results.
Drugs against acanthocephalans
We consider present protein structure models as reasonably reliable for the prediction of binding sites and subsequent ligand screening. This is because the 3D models were obtained using the two most accurate de novo modeling tools currently available, Alphafold2 and RoseTTAFold [31, 73, 79]. Among the ten ligands which should bind strongest to our eleven targets in acanthocephalans (Table 1, Table 2), a notable hit was derquantel, which would be a candidate for drug repurposing. The compound is an antagonist of N-acetylcholine receptors in nematodes [80, 81] and as such is contained in a commercial dewormer for sheep, marketed under the label Startect (Zoetis Australia Pty Ltd.; Licence: VPA10387/066/001.2017). Oral administration of Startect was shown to be highly effective (up to 100%) against diverse nematode parasites of the gastrointestinal tract and respiratory system [80, 82]. In virtual ligand screening, derquantel bound most strongly to one of the prime target candidates in acanthocephalans and second most strongly to another one (Table 1). Thus, application of Startect against fish-parasitizing acanthocephalans seems feasible. On the other hand, the second active ingredient of Startect, abamectin, has insecticide and acaricide activity [83, 84]. Thus, dissipation of Startect into the environment might be problematic and the mere administration of derquantel might be the better choice in fish.
Another compound for which high affinity was predicted to two of our eleven candidate targets was tadalafil, a phosphodiesterase (PDE) inhibitor (Table 2). Strikingly, inhibitors of PDE were previously suggested to have potential to control parasitic nematodes due to their disruptive effects on the Caenorhabditis elegans life cycle and nematode-specific active binding sites [85, 86]. In humans, tadalafil competes with the secondary messenger cGMP for binding sites in phosphodiesterase 5 (PDE5), thus relaxing the smooth musculature in several organs [87]. Such a mechanism should adversely affect acanthocephalans, in which the entire musculature is of the smooth type [88]. Also, tadalafil could interfere with acanthocephalan energy metabolism as suggested by PFAM motifs for phosphatidylglycerophosphatase (PGP) activity in both predicted target proteins (Supplementary Table S4). If true, this would be a novel mechanism for tadalafil. Not least, approved use in humans illustrates that the irritant potential of tadalafil according to the GHS is quite manageable.
Anthelmintic potential seems possible for piketoprofen, heliomycin, and pranazepide too, due to their anti-inflammatory, RNA synthesis antagonist, and cholecystokinin receptor type A antagonist activity, respectively [89,90,91]. For two of the remaining five compounds in Table 2, there is evidence for effectiveness against helminths once more. Thus, fluazuron is used in conjunction with above-mentioned abamectin, in the control of the gastrointestinal nematodes infecting cattle [92]. Furthermore, etoposide can induce cell cycle arrest at the G2/M phase and apoptotic cell death in C. elegans [93]. Also, widely constant transcript abundances suggest that the predicted acanthocephalan target proteins should be readily addressable by fluazuron and etoposide (Supplementary Table S1). Yet, both compounds, fluazuron and etoposide, might not be the first choice for acanthocephalan control, given their GHS labels (see Results). Still, all the ten agents in Table 2 result from conservative filtering of their potential target molecules. They all should have potential for use as effective agents against in acanthocephalans. Their application should additionally enable a more specific killing of acanthocephalans than would be achieved with niclosamide or benzimidazole derivates [9, 12, 13].
Conclusions
The development of drugs for parasite control usually takes many years and can easily cause enormous costs for pre-clinical and clinical trials, environmental impact assessment, approval, and the establishment of industrial production. Here, we present a bioinformatics workflow intended to reduce time and cost that is also applicable to non-model parasites for which little functional information is available. The entire workflow includes the identification of candidate targets in parasites and subsequent virtual screening for ligands. Detailed steps are quantitative transcriptome and proteome analyses, prediction of 3D protein structures and binding sites, and virtual database screening for binding compounds. In addition to the novel combination of individual analyses, the approach to the best of our knowledge utilizes for the first time in target identification environmental variation which the parasite is exposed to in definitive and accidental hosts.
Application of the workflow to fish-parasitizing acanthocephalans led to the identification of eleven top-ranked candidate target proteins (Table 1). Compounds predicted to bind to them already exist (Table 2), whereby five appear to be particularly promising according to ADMET, GHS and RO5 classifications: derquantel, tadalafil, pranazepide, piketoprofen, and heliomycin. We take it as confirmation of the usefulness of the present workflow that one of these compounds, derquantel, is an established nematocidal anthelmintic [80, 82]. A second compound, tadalafil, inhibits PDE5 and thus interferes with a metabolic pathway previously suggested to be a promising target for novel nematocidal anthelmintics [85]. Nematocidal effectiveness of two additional compounds, namely fluazuron and etoposide, further corroborates the usefulness of the workflow [92, 93].
Future simulations might shed light on the detailed nature of interaction between the candidate targets and ligands mentioned above. Probably, it will be revealing to examine the extent to which the 3D models determined here represent the active protein structure. Moreover, enabling fit induction by the ligand might uncover hitherto hidden binding sites in acanthocephalan proteins [94, 95]. Such analyses might lead to additional candidate targets but, to our estimation, will unlikely change that the current ones are worthwhile further evaluation, which will also have to include the testing of tolerability, ecotoxicology, specificity, and effectiveness in vitro and in vivo. The candidate targets listed in Table 1 may additionally be used as starting points for screening databases of compounds for which less knowledge is available. Obviously, validation of such compounds would raise costs and take time. But the present filtering of target candidates should increase the probability that any agents enable an effective control of acanthocephalans, whether the drugs will be repurposed or newly developed. Relative to broad-spectrum anthelminthics such as niclosamide and benzimidazole derivatives [9, 12, 13], any novel anti-parasitic strategy developed on the candidate targets in Table 1 should also allow for a specific control of acanthocephalans in fish aquacultures. These efforts might lead to new strategies against acanthocephaliasis, the main current obstacle in establishing successful fish aquaculture in South America (e.g., [61]). Although demonstrated here in acanthocephalans, this novel application can be transferred to a broad range of parasitic taxa [96]. For this purpose, the target determination workflow can be accessed via the Galaxy web server (Supplementary Fig. S1).
Methods
Sampling and sequencing
Fish were caught and sacrificed by authorized persons immediately prior to excision of acanthocephalans from the guts. As for the processing of the eoacanthocephalans N. agilis and N. buttnerae we refer to Supplementary Notes S1 and S2. Central to present transcriptome analyses was the palaeacanthocephalan P. laevis s.l. [97]. We analyzed N = 20 worms, with 10 specimens (5 males, 5 females) from common barbel and 10 specimens (5 males, 5 females) from European eel. All samples were sequenced as 75 bp single-end reads on an Illumina HiSeq 2500. Raw sequences are available at the EMBL Nucleotide Sequence Database (ENA) repository under accession numbers ERS7302868–87 in project PRJEB47442. Adapter sequences and low-quality parts of the sequences were trimmed with Trimmomatic v0.39 [98]. For more details of sampling and sequencing see [99]. While we aimed to filter out proteins with constant abundance across conditions in the present study, the referenced study demonstrates overall differing proteomic profiles in male and female worms from different hosts. We take this as a confirmation that sample processing did not compromise quantitative analyses of transcriptome data.
Differential gene expression analysis
As reference we used the P. laevis transcriptome published recently under NCBI GenBank accession number GIBA00000000.1 [65]. This transcriptome shotgun assembly was generated by Trinity v2.4.0 [100] from male, female and juvenile specimens. Transcriptome contigs were checked for bacterial contamination by blastn searches in BLAST+ v2.10.0 [101] against 21,820 bacterial reference genomes downloaded from NCBI. All contigs with hits below the E-value cutoff of 1e-20 were removed from the assembly for subsequent analyses. To check for congruence of our RNA-Seq datasets with the reference transcriptome we mapped all datasets with BBMap v38.73 (https://sourceforge.net/projects/bbmap/) to the reference. Since 92–96% of reads mapped successfully under default settings, the transcriptome seems to be quite complete and serves as a useful resource for the analysis.
Transcript quantification was done with the RSEM v1.3.3 [102] software package and the reference transcriptome described above. We applied Bowtie 2 v2.4.1 [103] mapping with settings optimized and implemented for RSEM downstream analysis. The rsem-calculate-expression script was applied with -calc-ci option for calculation of confidence intervals during calculation of relative transcript abundances.
Transcript abundance analyses were carried out with the Bioconductor package DESeq2 v1.28.1 [104] in R v4.0.3 (https://www.gbif.org/tool/81287/r-a-language-and-environment-for-statistical-computing) [105]. Since we were interested in approximating gene expression rather than inferring abundances of single transcripts, we summed up read counts across transcript variants (based on Trinity annotation). Corresponding integers were used as input for DESeq2 analyses between four pairs of comparison: (1) female worms from barbel vs. male worms from barbel, (2) female worms from eel vs. male worms from eel, (3) male worms from barbel vs. male worms from eel, and (4) female worms from barbel vs. female worms from eel. We applied the DESeq2 alternative hypothesis ‘lessAbs’, which tests for genes having transcript read counts within user-defined boundaries.
Identification of candidate targets
As an approximation of targetability, we only kept genes having at least 50 transcript reads in each of the 20 samples. Then, we extracted all significantly unregulated genes, according to likewise transcript read counts in all four DESeq2 pairs of comparisons delineated above. In detail, retention of a gene required a maximum log fold change of 1.50 between at least two of the four groups of comparison (adjusted p-value ≤0.05, each). Corresponding genes were regarded to be expressed independently of sex and host, suggestive of their constitutive expression.
Open reading frames (ORFs) were extracted from candidate target transcripts running getorf within EMBOSS v6.5.7 [106] with default settings. Only ORFs with translated sequences of at least 30 amino acids were kept for subsequent filtering. The resulting protein sequences were further investigated. We especially searched for orthologues of candidate target proteins in newly assembled draft genomes of two additional acanthocephalan species (N. buttnerae, N. agilis) (see Supplementary Notes S1 & S2; Supplementary Tables S7 & S8). Only sequences yielding tblastn hits below an E-value cutoff of 1e-50 were regarded to be conserved within fish-parasitizing acanthocephalans, and hence were kept.
Exclusion of sequences with similar sequences in non-acanthocephalan taxa was accomplished by blastp searches against the SwissProt database. For the same reason, we carried out tblastn searches against the genomes of six species from Bdelloidea, Monogononta, and Seisonidea, i.e., the three other higher-ranked taxa within the Rotifera-Acanthocephala clade commonly referred to as Syndermata or just Rotifera (sensu lato): Adineta vaga (GCA_000513175.1), Adineta ricciae (GCA_900240375.1), Brachionus calyciflorus (GCA_002922825.1), Brachionus koreanus (GCA_009177125.1), Brachionus plicatilis (GCA_010279815.1), and Seison nebaliae (PRJEB43415). All sequences yielding hits with E-values ≤1e-50 were regarded evolutionarily conserved between Acanthocephala and the taxa compared. Corresponding hits were excluded from further analyses.
Protein quantification by mass spectrometry
Protein isolation used the body walls of 192 worms (Supplementary Table S9) freed from the proboscis and emptied from internal organs and body fluid by gentle pressure. Five pools of body walls were boiled in lithium dodecyl sulfate buffer (Life Technologies, Carlsbad, CA, USA). Proteins were separated by polyacrylamide gel electrophoresis on a Novex NuPAGE 4–10% gel (Thermo Fischer Scientific, Waltham, MA, USA). Upon mincing of gel pieces, disulfide bonds were reduced with 10 mM DTT (Sigma-Aldrich, St Louis, MO, USA) at 55 °C, followed by alkylation with 55 mM iodoacetamide (Sigma-Aldrich). In-gel digestion was done with mass spectrometry-grade trypsin (Sigma-Aldrich) at 37 °C overnight. Peptides were eluted from the gel with acetonitrile, which was removed in a concentrator (Eppendorf SE, Hamburg, Germany) prior to loading on an Empore C18 StageTip (3 M Purification Inc., St Paul, MN, USA).
The measurement was performed on an EASY-nLC 1200 HPLC coupled online to an Orbitrap Exploris 480 mass spectrometer (Thermo Fischer Scientific), operated in data-dependent acquisition mode with a top20 method. During the 120 min measurement, peptides were eluted with an optimized 5–40% acetonitrile/water gradient. Raw files were processed with MaxQuant v1.6.5.0 [107, 108] using the settings: digestion = trypsin specific, max missed cleavages = 2, peptide FDR = 0.01, protein FDR = 0.01, variable modifications = oxidation (M) and acetylation (protein N-terminus), fixed modification = carbamidomethylation (C), match between runs = activated, iBAQ quantitation = activated. The search to homologize protein to mRNA sequences was performed against all ORF sequences derived from the transcriptome assembly of P. laevis.
Correlation analyses
Abundances of matched proteins (iBAQ) and transcripts (transcript read counts) was carried out with Excel 2019 (Microsoft).
Protein properties and structure modeling
Sequences of candidate target proteins were screened for PFAM protein motifs and domain features [109] at Kyoto University Bioinformatics Center’s GenomeNet MOTIF search (https://www.genome.jp/tools/motif/; accessed 2021-09-01) and for PROSITE protein domains and functional sites [110] at the Swiss Institute of Bioinformatics’ Resource Portal (https://prosite.expasy.org/; accessed 2021-09-15). Additionally, potential enzyme functions were predicted by ECPred [111].
Protein’s grand average of hydropathy [112] was calculated by the Sequence Manipulation Suite’s GRAVY algorithm implementation (https://www.bioinformatics.org/sms2/protein_gravy.html). N-terminal pre-sequences were predicted by TargetP v2.0 [113] and transmembrane topologies by DeepTMHMM (https://biolib.com/DTU/DeepTMHMM). Prediction of subcellular localization based on sequence information was accomplished by the deep learning algorithm DeepLoc v1.0 [114].
Protein 3D structures were modeled with AlphaFold2 [31] as executed by a Jupyter Notebook [115] on Google Colab (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb) [116]. We applied default settings, including MMseqs2 [117] for sequence alignment. The best out of five calculated models was used for further analysis, based on the ranking by the average predicted Local Distance Difference Test value (pLDDT; see Suppl. Methods of AlphaFold2). For validation, protein 3D structures were additionally modeled with RoseTTAFold [79] applying default settings. Results from both modelers were compared using the University of Helsinki’s Dali protein structure comparison server [74, 118].
Binding site prediction, ligand screening, and docking
Protein-ligand binding site prediction was carried out with COACH-D [76] on protein PDB files as generated by AlphaFold2. Each protein’s best binding site was subjected to ligand screening, plus all secondary sites with confidence scores (C-score) up to 0.3 lower than the site ranked first. High-throughput virtual ligand screening was performed using AutoDock Vina v1.2.0 [77], implemented in MTiOpenScreen [78]. Settings were: Demonstration mode = No, Protein Receptor = PDB, Is lead-like = Yes, Grid calculation = Custom parameters. AlphaFold2-derived PDB files and COACH-D coordinates of the binding site were used as input. We screened against the Drugs-lib compound database that contains 21,276 drugs that are either approved or have been used in clinical trials [78].
Ligands predicted to bind with most favorable free energy to binding sites in the candidate target proteins were further evaluated in the online databases ChEMBL [119], ClinicalTrials.gov (https://clinicaltrials.gov/), DrugBank [120], and PubChem [121]. Special emphasis was given to published indications, known/predicted molecular targets, mode of administration, resp. the fulfilment of the RO5, and annotations within the GHS classification system (Supplementary Table S6). Additionally, physicochemical absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties were predicted using ADMETlab 2.0 [122].
Availability of data and materials
The transcriptome datasets are available in the EMBL Nucleotide Sequence Database (ENA) repository under the accession number PRJEB47442 (ERS7302868–87). The presented workflow is accessible via the Galaxy server under the URL tinyurl.com/yx72rda7. Supplementary Fig. S1 provides a corresponding overview.
References
Selzer PM, Epe C. Antiparasitics in animal health: quo vadis? Trends Parasitol. 2021;37:77–89.
Naylor RL, Hardy RW, Buschmann AH, Bush SR, Cao L, Klinger DH, et al. A 20-year retrospective review of global aquaculture. Nature. 2021;591:551–63.
Food and Agriculture Organization of the United Nations (FAO). The state of world fisheries and aquaculture 2018 - meeting the sustainable development goals. Rome (Italy): FAO; 2018.
Assefa A, Abunna F. Maintenance of fish health in aquaculture: review of epidemiological approaches for prevention and control of infectious disease of fish. Vet Med Int. 2018:5432497.
Rosny HS, Hossain M, Hasan-Uj-Jaman RHS, Aurpa IA, Khondoker S, Biswas C. Dietary supplementation of garlic (Allium sativum) to prevent Acanthocephala infection in aquaculture. Int J Fish Aquat. 2016;4:188–92.
Lal MB. Acanthocephala of trout and anthelmintics: behaviour in vitro. Nature. 1947;159:545–6.
Seixas AT, Umeda Gallani S, Silva Noronha L, Mangabeira Silva JJ, Rizzato Paschoal JA, Kenupp Bastos J, et al. Copaifera oleoresins as a novel natural product against acanthocephalan in aquaculture: insights in the mode of action and toxicity. Aquac Res. 2020;51:4681–8.
Valladão GMR, Gallani SU, Jerônimo GT, Seixas AT. Challenges in the control of acanthocephalosis in aquaculture: special emphasis on Neoechinorhynchus buttnerae. Rev Aquacult. 2020;12:1360–72.
Mehlhorn H. Die Parasiten der Tiere. Heidelberg: Spektrum Akademischer Verlag; 2012.
Woo TK, Buchmann K. Fish parasites - pathology and protection. London: CABI; 2012.
Kayis S, Ozcelep T, Capkin E, Altinok I. Protozoan and metazoan parasites of cultured fish in Turkey and their applied treatments. Isr J Aquacult. 2009;61:93–102.
Calumpang SMF, Medina MJB, Tejada AW, Medina JR. Environmental impact of two molluscicides: niclosamide and metaldehyde in a rice paddy ecosystem. Bull Environ Contam Toxicol. 1995;55:494–501.
Mottier L, Alvarez L, Ceballos L, Lanusse C. Drug transport mechanisms in helminth parasites: passive diffusion of benzimidazole anthelmintics. Exp Parasitol. 2006;113:49–57.
Oh SJ, Park J, Lee MJ, Park SY, Lee J-H, Choi K. Ecological hazard assessment of major veterinary benzimidazoles: acute and chronic toxicities to aquatic microbes and invertebrates. Environ Toxicol Chem. 2006;25:2221–6.
Koschorrek J, Koch C, Rönnefahrt I. Environmental risk assessment of veterinary medicinal products in the EU - a regulatory perspective. Toxicol Lett. 2002;131:117–24.
Raffray M, Cohen GM. Bis (tri-n-butyltin) oxide induces programmed cell death (apoptosis) in immature rat thymocytes. Arch Toxicol. 1991;65:135–9.
Osada T, Chen M, Yang XY, Spasojevic I, Vandeusen JB, Hsu D, et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011;71:4172–82.
Hegde M, Karki SS, Thomas E, Kumar S, Panjamurthy K, Ranganatha SR, et al. Novel levamisole derivative induces extrinsic pathway of apoptosis in cancer cells and inhibits tumor progression in mice. Plos One. 2012;7:e43632.
Wu ZH, Lu M-k, Hu LY, Li X. Praziquantel synergistically enhances paclitaxel efficacy to inhibit cancer cell growth. Plos One. 2012;7:e51721.
Zhang J, Zhao C, Gao Y, Jiang Y, Liang H, Zhao G. Thiabendazole, a well-known antifungal drug, exhibits anti-metastatic melanoma B16F10 activity via inhibiting VEGF expression and inducing apoptosis. Pharmazie. 2013;68:962–8.
Ayyagari VN, Brard L. Bithionol inhibits ovarian cancer cell growth in vitro - studies on mechanism(s) of action. BMC Cancer. 2014;14:61.
Lai SR, Castello SA, Robinson AC, Koehler JW. In vitro anti-tubulin effects of mebendazole and fenbendazole on canine glioma cells. Vet Comp Oncol. 2017;15:1445–54.
Zhang Z, Zhao X, Qin X. Potential genotoxic and cytotoxicity of emamectin benzoate in human normal liver cells. Oncotarget. 2017;8:82185–95.
Zhang X, Zhao J, Gao X, Pei D, Gao C. Anthelmintic drug albendazole arrests human gastric cancer cells at the mitotic phase and induces apoptosis. Exp Ther Med. 2017;13:595–603.
Bundschuh M, Hahn T, Ehrlich B, Höltge S, Kreuzig R, Schulz R. Acute toxicity and environmental risks of five veterinary pharmaceuticals for aquatic macroinvertebrates. Bull Environ Contam Toxicol. 2016;96:139–43.
Wagil M, Maszkowska J, Białk-Bielińska A, Stepnowski P, Kumirska J. A comprehensive approach to the determination of two benzimidazoles in environmental samples. Chemosphere. 2015;119:S35–41.
Horvat AJM, Petrović M, Babić S, Pavlović DM, Ašperger D, Pelko S, et al. Analysis, occurrence and fate of anthelmintics and their transformation products in the environment. Trends Anal Chem. 2012;31:61–84.
Reynoldson JA, Behnke JM, Pallant LJ, Macnish MG, Gilbert F, Giles S, et al. Failure of pyrantel in treatment of human hookworm infections (Ancylostoma duodenale) in the Kimberley region of North West Australia. Acta Trop. 1997;68:301–12.
McCarthy J. Is anthelminthic resistance a threat to the program to eliminate lymphatic filariasis? Am J Trop Med Hyg. 2005;73:232–3.
Viana M, Faust CL, Haydon DT, Webster JP, Lamberton PHL. The effects of subcurative praziquantel treatment on life-history traits and trade-offs in drug-resistant Schistosoma mansoni. Evol Appl. 2018;11:488–500.
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9.
Nixon SA, Welz C, Woods DJ, Costa-Junior L, Zamanian M, Martin RJ. Where are all the anthelmintics? Challenges and opportunities on the path to new anthelmintics. Int J Parasitol Drugs Drug Resist. 2020;14:8–16.
Zajíčková M, Nguyen LT, Skálová L, Stuchlíková LR, Matoušková P. Anthelmintics in the future: current trends in the discovery and development of new drugs against gastrointestinal nematodes. Drug Discov Today. 2020;25:430–7.
Geary TG, Sakanari JA, Caffrey CR. Anthelmintic drug discovery: into the future. J Parasitol. 2015;101:125–33.
Barh D, Tiwari S, Weener ME, Azevedo V, Góes-Neto A, Gromiha MM, et al. Multi-omics-based identification of SARS-CoV-2 infection biology and candidate drugs against COVID-19. Comput Biol Med. 2020;126:104051.
Carolino K, Winzeler EA. The antimalarial resistome – finding new drug targets and their modes of action. Curr Opin Microbiol. 2020;57:49–55.
Cowell AN, Winzeler EA. Advances in omics-based methods to identify novel targets for malaria and other parasitic protozoan infections. Genome Med. 2019;11:63.
Daga V, Green E, Ravichandran P, Short M, May M. Multi-omic approaches to vaccine development against helminth diseases. In: Morales-Montor J, Río-Araiza VHD, Bello RH, editors. Parasitic helminths and Zoonoses - from basic to applied research. London: IntechOpen; 2022. p. 80592.
Shivam S, El-Matbouli M, Kumar G. Development of fish parasite vaccines in the OMICs era: progress and opportunities. Vaccines. 2021;9:179.
Herlyn H. Thorny-headed worms (Acanthocephala): jaw-less members of jaw-bearing worms that parasitize jawed arthropods and jawed vertebrates. In: De Baets K, Huntley JW, editors. The evolution and fossil record of parasitism - identification and macroevolution of parasites. Cham: Springer; 2021. p. 273–313.
Weber M, Junge R. Identification and treatment of Moniliformis clarki (Acanthocephala) in cotton-topped tamarins (Saguinus oedipus). J Zoo Wildl Med. 2000;31:503–7.
Choi C-J, Lee H-J, Go J-H, Park Y-K, Chai J-Y, Seo M. Extraintestinal migration of Centrorhynchus sp. (Acanthocephala: Centrorhynchidae) in experimentally infected rats. Korean J Parasitol. 2010;48:139–43.
de Matos LV, de Oliveira MIB, Silva Gomes AL, da Silva GS. Morphological and histochemical changes associated with massive infection by Neoechinorhynchus buttnerae (Acanthocephala: Neoechinorhynchidae) in the farmed freshwater fish Colossoma macropomum Cuvier, 1818 from the Amazon state, Brazil. Parasitol Res. 2017;116:1029–37.
Herlyn H, Taraschewski H. Evolutionary anatomy of the muscular apparatus involved in the anchoring of Acanthocephala to the intestinal wall of their vertebrate hosts. Parasitol Res. 2017;116:1207–25.
Jerônimo GT, de Pádua SB, de Andrade Belo MA, Chagas EC, Taboga SR, Maciel PO, et al. Neoechinorhynchus buttnerae (Acanthocephala) infection in farmed Colossoma macropomum: a pathological approach. Aquaculture. 2017;469:124–7.
Taraschewski H, Mackenstedt U. Autoradiographic and morphological studies on the uptake of the triglyceride [3H]-glyceroltrioleate by acanthocephalans. Parasitol Res. 1991;77:247–54.
Sures B, Franken M, Taraschewski H. Element concentrations in the archiacanthocephalan Macracanthorhynchus hirudinaceus compared with those in the porcine definitive host from a slaughterhouse in La Paz, Bolivia. Int J Parasitol. 2000;30:1071–6.
Sures B. Competition for minerals between Acanthocephalus lucii and its definitive host perch (Perca fluviatilis). Int J Parasitol. 2002;32:1117–22.
Wurmbach H. Zur Krankheitserregenden Wirkung der Acanthocephalen. Die Kratzererkrankung der Barben in der Mosel. Z Fisch Hilfswiss. 1937;35:217–32.
Sanford SE. Mortality in mute swans in southern Ontario associated with infestation with the thorny-headed worm, Polymorphus boschadis. Can Vet J. 1978;19:234–6.
Amin OM, Heckmann RA, Sahara A, Yudhanto S. The finding of Mediorhynchus gallinarum (Acanthocephala: Gigantorhynchidae) in chickens from Indonesia, with expanded description using SEM. Comp Parasitol. 2013;80:39–46.
Yasumoto S, Nagasawa K. Possible life cycle of Longicollum pagrosomi, an acanthocephalan parasite of cultured red sea bream. Fish Pathol. 1996;31:235–6.
Rückert S, Palm HW, Klimpel S. Parasite fauna of seabass (Lates calcarifer) under mariculture conditions in Lampung Bay, Indonesia. J Appl Ichthyol. 2008;24:321–7.
Steinsträsser W. Acanthocephalen als Forellenparasiten. Z Fisch Hilfswiss. 1936;34:177–212.
Ferraz de Lima CLB, Lima JAF, Ceccarelli PS. Ocorrência de Acantocéfalos parasitando o Pacu, Piaractus mesopotamicus Holmberg, 1887 (Pisces, Serrassalmidae) em Piscicultura. Bol Tec CEPTA. 1990;2:43–51.
de la Cruz CPP, Paller VGV. Occurrence of Neoechinorhynchus sp. (Acanthocephala: Neoechinorhynchidae) in cultured tilapia, [Oreochromis niloticus (L.), Perciformes: Ciclidae] from Sampaloc Lake, Philippines. Asia Life Sci. 2012;21:287–98.
Marinho RGB, Tavares-Dias M, Dias-Grigório MKR, Neves LR, Yoshioka ETO, Boijink CL, et al. Helminthes and protozoan of farmed pirarucu (Arapaima gigas) in eastern Amazon and host-parasite relationship. Arq Bras Med Vet Zootec. 2013;65:1192–202.
Taraschewski H, Mehlhorn H, Raether W. Loperamid, an efficacious drug against fish-pathogenic acanthocephalans. Parasitol Res. 1990;76:619–23.
Malta JCO, Gomes ALS, Andrade SMS, Varella AMB. Massive infestation by Neoechinorhynchus buttnerae Golvan, 1956 (Eoacanthocephala: Neochinorhynchidae) in young “tambaquis” Colossoma macropomum (Cuvier, 1818) cultured in the Central Amazon. Acta Amazon. 2001;31:133–43.
Martins ML, Moraes FR, Fujimoto RY, Onaka EM, CIF Q. Prevalence and histopathology of Neoechinorhynchus curemai Noronha, 1973 (Acanthocephala: Neoechinorhynchidae) in Prochilodus lineatus Valenciennes, 1836 from Volta Grande Reservoir, MG, Brazil. Braz J Biol. 2001;61:517–22.
Silva Gomes AL, Coelho Filho JG, Viana Silva W, Braga Oliveira MI, Bernardino G, Costa JI. The impact of Neoechinorhynchus buttnerae (Golvan, 1956) (Eoacanthocephala: Neochinorhynchidae) outbreaks on productive and economic performance of the tambaqui Colossoma macropomum (Cuvier, 1818), reared in ponds. Lat Am J Aquat Res. 2017;45:496–500.
de Souza Costa CM, Lima TBC, Cruz MG, Almeida DV, Martins ML, Jerônimo GT. In vitro culture of Neoechinorhynchus buttnerae (Acanthocephala: Neoechinorhynchidae): influence of temperature and culture media. Rev Bras Parasitol Vet. 2018;27:562–9.
Pereira JN, Morey GAM. First record of Neoechinorhynchus buttnerae (Eoacantocephala, Neochinorhynchidae) on Colossoma macropomum (Characidae) in a fish farm in Roraima, Brazil. Acta Amazon. 2018;48:42–5.
Oliveira LCD, Majolo C, Brandão FR, Farias CFS, Oliveira MIB, Santos WB, et al. Avermectins, praziquantel and levamisole have in vitro efficacy against Neoechinorhynchus buttnerae (Neoechinorhynchidae) in Colossoma macropomum: a Serrasalmidae from the Amazon. J Fish Dis. 2019;42:765–72.
Mauer K, Hellmann SL, Groth M, Frobius AC, Zischler H, Hankeln T, et al. The genome, transcriptome, and proteome of the fish parasite Pomphorhynchus laevis (Acanthocephala). Plos One. 2020;15:e0232973.
Dudiňák V, Šnábel V. Comparative analysis of Slovak and Czech populations of Pomphorhynchus laevis (Acanthocephala) using morphological and isoenzyme analyses. Acta Zool Univ Comenianae. 2001;44:41–50.
Cézilly F, Favrat A, Perrot-Minnot MJ. Multidimensionality in parasite-induced phenotypic alterations: ultimate versus proximate aspects. J Exp Biol. 2013;216:27–35.
Bates RM, Kennedy CR. Potential interactions between Acanthocephalus anguillae and Pomphorhynchus laevis in their natural hosts chub, Leuciscus cephalus and the European eel, Anguilla anguilla. Parasitology. 1991;102:289–97.
Moravec F, Scholz T. Observations on the biology of Pomphorhynchus laevis (Zoega in Müller, 1776) (Acanthocephala) in the Rokytná River, Czech and Slovak Federative Republic. Helminthologia. 1991;28:23–9.
Liu Y, Beyer A, Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016;165:535–50.
Tiessen A, Pérez-Rodríguez P, Delaye-Arredondo LJ. Mathematical modeling and comparison of protein size distribution in different plant, animal, fungal and microbial species reveals a negative correlation between protein size and protein number, thus providing insight into the evolution of proteomes. BMC Res Notes. 2012;5:85.
Kozlowski LP. Proteome-pI: proteome isoelectric point database. Nucleic Acids Res. 2016;45(D1):D1112–D16.
Pereira J, Simpkin AJ, Hartmann MD, Rigden DJ, Keegan RM, Lupas AN. High-accuracy protein structure prediction in CASP14. Proteins. 2021;89:1687–99.
Holm L. Using Dali for protein structure comparison. In: Gáspári Z, editor. Structural bioinformatics - methods in molecular biology, vol. 2112. New York: Humana; 2020. p. 29–42.
Holm L, Kääriäinen S, Rosenström P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–1.
Wu Q, Peng Z, Zhang Y, Yang J. COACH-D: improved protein–ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018;46:W438–W42.
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–61.
Labbé CM, Rey J, Lagorce D, Vavruša M, Becot J, Sperandio O, et al. MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res. 2015;43:W448–W54.
Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–6.
Little PR, Hodge A, Watson TG, Seed JA, Maeder SJ. Field efficacy and safety of an oral formulation of the novel combination anthelmintic, derquantel-abamectin, in sheep in New Zealand. N Z Vet J. 2010;58:121–9.
Buxton SK, Charvet CL, Neveu C, Cabaret J, Cortet J, Peineau N, et al. Investigation of acetylcholine receptor diversity in a nematode parasite leads to characterization of tribendimidine- and derquantel-sensitive nAChRs. Plos Path. 2014;10:e1003870.
Little PR, Hodge A, Maeder SJ, Wirtherle NC, Nicholas DR, Cox GG, et al. Efficacy of a combined oral formulation of derquantel-abamectin against the adult and larval stages of nematodes in sheep, including anthelmintic-resistant strains. Vet Parasitol. 2011;181:180–93.
Duchovskienë L. Effects of abamectin on the two-spotted spider mite (Tetranychus urticae Koch.) in greenhouse cucumbers. Scientific Works of the Lithuanian Institute of Horticulture and Lithuanian University of Agriculture. 2007;26:166–75.
Brigante J, Costa JO, Espíndola ELG, Daam MA. Acute toxicity of the insecticide abamectin and the fungicide difenoconazole (individually and in mixture) to the tropical stingless bee Melipona scutellaris. Ecotoxicology. 2021;30:1872–9.
Schuster KD, Cahill KB, Morris K, Thomas WK, Cote RH. PDE inhibitors as potential pesticides targeting parasitic nematodes. FASEB J. 2013;27(S1):1052.6.
Tyagi R, Elfawal MA, Wildman SA, Helander J, Bulman CA, Sakanari J, et al. Identification of small molecule enzyme inhibitors as broad-spectrum anthelmintics. Sci Rep. 2019;9:9085.
Huang Y-Y, Li Z, Cai Y-H, Feng L-J, Wu Y, Li X, et al. The molecular basis for the selectivity of tadalafil toward phosphodiesterase 5 and 6: a modeling study. J Chem Inf Model. 2013;53:3044–53.
Schmidt-Rhaesa A. The evolution of organ systems. Oxford: Oxford University Press; 2007.
Arora SK. Molecular structure of heliomycin, an inhibitor of RNA synthesis. J Antibiot. 1985;38:113–5.
Burgos A, Pérez Busquier M, Gómez Reino J, Ferreiro JL, Navarro F, Valverde J, et al. Double-blind, double-dummy comparative study of local action transcutaneous flurbiprofen (flurbiprofen LAT) versus piketoprofen cream in the treatment of extra-articular rheumatism. Clin Drug Invest. 2001;21:95–102.
Berna MJ, Jensen RT. Role of CCK/gastrin receptors in gastrointestinal/metabolic diseases and results of human studies using gastrin/CCK receptor agonists/antagonists in these diseases. Curr Top Med Chem. 2007;7:1211–31.
Coelho CN, Oliveira GF, Coumendouros K, Moraes PA, Medeiros MT, Borges DA, et al. Anthelmintic efficacy of abamectin with fluazuron association in control of the gastrointestinal nematodes of cattle. Braz. J Vet Med. 2015;37:100–5.
Lee SY, Kim JY, Jung Y-J, Kang K. Toxicological evaluation of the topoisomerase inhibitor, etoposide, in the model animal Caenorhabditis elegans and 3T3-L1 normal murine cells. Environ Toxicol. 2017;32:1836–43.
Sotriffer CA. Accounting for induced-fit effects in docking: what is possible and what is not? Curr Top Med Chem. 2011;11:179–91.
Zhao Q, Capelli R, Carloni P, Lüscher B, Li J, Rossetti G. Enhanced sampling approach to the induced-fit docking problem in protein-ligand binding: the case of mono-ADP-ribosylation hydrolase inhibitors. J Chem Theory Comput. 2021;17:7899–911. https://doi.org/10.1021/acs.jctc.1c00649 Epub 2021 Nov 23. PMID: 34813698.
Goater TM, Goater CP, Esch GW. Parasitism: the diversity and ecology of animal parasites. New York: Cambridge University Press; 2013.
Perrot-Minnot M-J, Bollache L, Lagrue C. Distribution of Pomphorhynchus laevis s.l. (Acanthocephala) among fish species at a local scale: importance of fish biomass density. J Helminthol. 2019;94:e99.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Schmidt H, Mauer K, Hankeln T, Herlyn H. Host-dependent impairment of parasite development and reproduction in the acanthocephalan model. Cell Biosci. 2022;12:75.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314.
Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–7.
Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72.
Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11:2301–19.
Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 2020;49(D1):D412–D19.
de Castro E, Sigrist CJA, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, et al. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34:W362–W65.
Dalkiran A, Rifaioglu AS, Martin MJ, Cetin-Atalay R, Atalay V, Doğan T. ECPred: a tool for the prediction of the enzymatic functions of protein sequences based on the EC nomenclature. BMC Bioinform. 2018;19:334.
Kyte J, Doolittle R. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–32.
Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–16.
Armenteros JJA, Sønderby CK, Sønderby SK, Nielsen H, Winther O. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics. 2017;33:3387–95.
Kluyver T, Ragan-Kelley B, Pérez F, Granger B, Bussonnier M, Frederic J, et al. Jupyter notebooks – a publishing format for reproducible computational workflows. In: Loizides F, Schmidt B, editors. Positioning and power in academic publishing: players, agents and agendas. Amsterdam: IOS Press; 2016. p. 87–90.
Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold - making protein folding accessible to all. Nat Methods. 2022;19:679–82.
Steinegger M, Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017;35:1026–8.
Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W49.
Davies M, Nowotka M, Papadatos G, Dedman N, Gaulton A, Atkinson F, et al. ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015;43(W1):W612–W20.
Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D72.
Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37:W623–W33.
Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49:W5–W14.
Acknowledgements
We gratefully acknowledge support by the holders of fishing licenses, Sven Fischer (Mainz) and Hartmut Roessel† (Gieselwerder). We are further indebted to three anonymous reviewers for constructive comments on previous manuscript drafts.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by funds awarded to HH in collaboration with TH from the Deutsche Forschungsgemeinschaft (HE 3487/5–1). MG and RCW gratefully acknowledge the support of the Klaus Tschira Foundation. ALSG acknowledges funding by the Acanthocephalan Control Project in Fish Farming – CAP, Amazonas Strategic - FAPEAM 004/2018.
Author information
Authors and Affiliations
Contributions
H.S. contributed to data analysis and interpretation, software handling, bioinformatics analyses, figure and table generation, and manuscript writing and editing. K.M. contributed to data curation and analysis, and manuscript editing. M.G. contributed to methodology and software handling. B.S.D. contributed resources and in manuscript editing. S.L.H. contributed to data curation and methodology. A.L.S.G. contributed resources and in manuscript editing. F.B. contributed to carrying out mass spectrometry and manuscript editing. R.C.W. contributed to the conception of the study, funding acquisition, data analysis, and manuscript editing. T.H. contributed to the conception of the study, funding acquisition, data analysis, and manuscript editing. H.H. contributed to the conception of the study, funding acquisition, project administration, data analysis, and manuscript writing and editing. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Animal usage followed the guidelines of the European Union (Directive 2010/63/EU). All parasites used in our study were excised from fishes which had been caught and sacrificed by authorized and experienced persons: (a) Neoechinorhynchus buttnerae: withdrawal of fish from aquaculture under the umbrella of the Brazilian government project: Control Project in Fish Farming – CAP, Amazonas Strategic - FAPEAM 004/2018, (b) Pomphorhynchus laevis from eel: license no. 16692 issued by VDSF Verbands Deutscher Sportfischer e.V., LV Rheinland-Pfalz e.V., Germany, (c) Neoechinorhynchus agilis: fishes provided by Po Delta Park Administration, Italy, (d) Pomphorhynchus laevis from barbel: fishing rights with fishing cooperative Hannoversch Münden, Germany. Without exception, material (intestines) from natural infections was used that would otherwise have been discarded. For carrying out sequencing of acanthocephalan DNA and RNA no approval by an ethics committee was required. The study was conducted in accordance with the ARRIVE guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Note S1.
Assembly of Neoechinorhynchus buttnerae draft genome. Supplementary Note S2. Assembly of Neoechinorhynchus agilis draft genome. Supplementary Table S1. Transcript abundance differences of candidates. Supplementary Table S2. Amino acid composition of candidate target proteins. Supplementary Table S3. Properties of target proteins. Supplementary Table S4. PFAM motifs of target proteins. Supplementary Table S5. 3D structure prediction of target proteins. Supplementary Table S6. Virtual ligand screening results. Supplementary Table S7. Bacterial genome assemblies used for data decontamination. Supplementary Table S8. Annotation of the mitochondrial genome of N. buttnerae. Supplementary Table S9. Specimens used for mass spectrometry. Supplementary Table S10. Standard InChI keys and 2D structures of candidate ligands. Supplementary Figure S1. Target identification workflow at Galaxy.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Schmidt, H., Mauer, K., Glaser, M. et al. Identification of antiparasitic drug targets using a multi-omics workflow in the acanthocephalan model. BMC Genomics 23, 677 (2022). https://doi.org/10.1186/s12864-022-08882-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08882-1