
Abstract
Background
Dendrobium catenatum/D. officinale (here after D. catenatum), a well-known economically important traditional medicinal herb, produces a variety of bioactive metabolites including polysaccharides, alkaloids, and flavonoids with excellent pharmacological and clinical values. Although many genes associated with the biosynthesis of medicinal components have been cloned and characterized, the biosynthetic pathway, especially the downstream and regulatory pathway of major medicinal components in the herb, is far from clear. β-glucosidases (BGLUs) comprise a diverse group of enzymes that widely exist in plants and play essential functions in cell wall modification, defense response, phytohormone signaling, secondary metabolism, herbivore resistance, and scent release by hydrolyzing β-D-glycosidic bond from a carbohydrate moiety. The recent release of the chromosome-level reference genome of D. catenatum enables the characterization of gene families. Although the genome-wide analysis of the BGLU gene family has been successfully conducted in various plants, no systematic analysis is available for the D. catenatum. We previously isolated DcBGLU2 in the BGLU family as a key regulator for polysaccharide biosynthesis in D. catenatum. Yet, the exact number of DcBGLUs in the D. catenatum genome and their possible roles in bioactive compound production deserve more attention.
Results
To investigate the role of BGLUs in active metabolites production, 22 BGLUs (DcBGLU1-22) of the glycoside hydrolase family 1 (GH1) were identified from D. catenatum genome. Protein prediction showed that most of the DcBGLUs were acidic and phylogenetic analysis classified the family into four distinct clusters. The sequence alignments revealed several conserved motifs among the DcBGLU proteins and analyses of the putative signal peptides and N-glycosylation site revealed that the majority of DcBGLU members dually targeted to the vacuole and/or chloroplast. Organ-specific expression profiles and specific responses to MeJA and MF23 were also determined. Furthermore, four DcBGLUs were selected to test their involvement in metabolism regulation. Overexpression of DcBGLU2, 6, 8, and 13 significantly increased contents of flavonoid, reducing-polysaccharide, alkaloid and soluble-polysaccharide, respectively.
Conclusion
The genome-wide systematic analysis identified candidate DcBGLU genes with possible roles in medicinal metabolites production and laid a theoretical foundation for further functional characterization and molecular breeding of D. catenatum.
Similar content being viewed by others

Introduction
Dendrobium, one of the largest genera in the family Orchidaceae with approximately 1800 species worldwide [1], are popular economic plants owing to their beautiful flowers, scientific values and health benefits. Especially, D. catenatum is a highly prized Dendrobium species with a wealth of therapeutic functions in antitumor, anti-angiogenesis, anti-oxidation, anti-inflammation, diabetes alleviation, liver protection, stomach nourishing, body fluids supplementation, and immunity enhancement [2,3,4,5]. The stems of D. catenatum are the principal medicinal part containing large amounts of polysaccharides (> 30% of dry weight) and relatively low levels of ethanol extractives (~ 4.93% of dry weight), such as flavonoids, bibenzyls, phenanthrene and fluorenone [6]. The leaves, comprising approximately half of the total biomass of D. catenatum, are new sources of bioactive molecules, including phenolic compounds [7], flavonoids, polysaccharides, and amino acids [8]. The flowers of D. catenatum are rich with phenolic components (> 30% dry weight), while other substances such as essential and non-essential amino acids, polysaccharides, and volatile components are also found [9].
Many of the secondary metabolites are glucosylated to increase their solubility and stability, and activation of the glucosylated compounds is mediated by enzymes called β-glucosidases (BGLUs) [10]. BGLUs are groups of glycoside hydrolase 1 (GH1) family members found in all domains of living organisms with essential functions in removing nonreducing terminal glucosyl residues from glycosyl esters, oligosaccharides, and glycosides [10]. There are a great many glycosidases in higher plants with extensive redundant functions such as defense, cell wall remodeling, phytohormone activation, scent release, microbe/insect interactions, and secondary metabolism [11, 12]. In the last decades, genome-wide analysis of GH1-BGLUs has been carried out in a few plant species: Arabidopsis thaliana with 47 members (10 subfamilies), Oryza sativa with 40 members (8 subfamilies), Zea mays with 26 members (4 subfamilies), Brassica rapa with 64 members (10 subfamilies), and Medicago truncatula with 51 members (7 subfamilies) [13, 14]. For instance, a specific cytoplasmic BGLU has been demonstrated to hydrolyze the monoterpene alkaloid intermediate strictosidine to produce various monoterpene alkaloids [15]. Although some BGLUs related to metabolite biosynthesis were cloned and characterized from several plant species, limited information is available about BGLU family in orchids. Several BGLU genes expressed in the stems of three Dendrobium species (Dendrobium huoshanese, D. catenatum, and Dendrobium moniliforme) have been identified as hub genes that are possibly involved in polysaccharides biosynthesis [16]. In Tongan vanilla, β-glucosidase has been exogenously applied on green beans to hydrolyze glucovanillin into vanillin [17]. In Cymbidium sinense, BGLU genes are upregulated during ovule development, when β-glucosidase-mediated hydrolysis of cellulose may occur [18]. Fifteen BGLU genes are also significantly downregulated in C. sinense leaves, which may be an important reason for decreased starch content and abnormal sugar metabolism in the chloroplast [19]. In D. catenatum, BGLU genes are significantly upregulated in symbiotically germinated seeds [20], and BGLU genes associated with the glutathione metabolism pathway are upregulated in response to cadmium stress [21]. In Dendrobium crumenatum, a significant increase in β-glucosidase activity is observed during floral bud development [22].
Usually, the accumulation of medicinal metabolites, especially alkaloids, are low across tissues, making them hard to meet the threshold for drug-making [23]. Although genetic engineering and molecular modification are exceedingly helpful in creating improved varieties, the physiological and molecular mechanisms underlying metabolites production remain largely unexplored in Dendrobium plants [24]. Nevertheless, the great achievements in genome sequencing for D. catenatum [25, 26] make it convenient to conduct a genome-wide search for potential genes associated with important traits.
Previously, we sequenced the genome of D. catenatum [25] and reported DcBGLU2 as a key regulator for polysaccharide accumulation in response to phytohormone treatments [27]. However, determining the exact number of DcBGLUs in D. catenatum genome and their corresponding roles in medicinal compounds production deserve more attention. The present study identified 22 candidate GH1 DcBGLUs members through a genome-wide analysis. Then their sequence feature, molecular phylogenetic relationship, conserved motif, gene structure, chromosomal localization, and cis-elements were characterized. Furthermore, the dynamic expression patterns in three more tissues were examined based on our and other published RNA-seq data. These results provide valuable information on the DcBGLU family in D. catenatum and lay a foundation for further exploring its function in plant metabolism.
Materials and methods
Genome-wide identification of GH1 BGLUs in D. catenatum
The genome sequences and protein data of the D. catenatum were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/genome/?term=JSDN00000000) under the accession code JSDN00000000 [25]. To identify D. catenatum BGLU candidates of the GH1 family (DcBGLUs), the hidden Markov model (HMM) profile of Glyco_hydro_1 (PF00232) from the Pfam database (http://pfam.xfam.org/) were downloaded and searched against the BGLU domain in D. catenatum protein sequence data using the HMMER software (version 3.2.1, http://hmmer.org/download.html). After removing repeated or incomplete proteins, the remaining potential candidates were double-checked using the SMART (http://smart.embl-heidelberg.de/) and CDD (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) databases. The gene identifiers used in this study are listed in the Table S1. The molecular weight (MW) and isoelectric point (pI) of each BGLU protein were calculated with the online ProtParam (https://web.expasy.org/protparam/) tool. The subcellular localization of each BGLU protein was predicted using the online ProtComp v. 9.0 server (http://www.softberry.com). The signal sequences were predicted using the SignalP (https://sFervices.healthtech.dtu.dk/service.php?SignalP-5.0) tool, and the N-glycosylation sites were detected by the NetNGlyc 1.0 server (https://services.healthtech.dtu.dk/service.php?NetNGlyc-1.0).
Phylogenetic analysis of DcBGLUs
To classify and investigate phylogenetic relationships of BGLUs, the protein sequences from A. thaliana and D. catenatum were aligned using MEGA X [28]. The phylogenetic tree was constructed according to the neighbor-joining method with the bootstrap set at 1000 replicates, and then visualized and modified using EVOLVIEW (https://www.evolgenius.info/evolview/).
Gene structures and conserved motifs
The exon/intron organization of DcBGLU genes was analyzed using the WebScipio server (https://www.webscipio.org/) [29]. The conserved motifs of DcBGLUs were identified by MEME suite (version 5.4.1) [30]. At the same time, the PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html) was used to identify potential cis-elements in the 2000-bp gene promotors.
RNA isolation and gene expression analysis
Approximately 300 mg of D. catenatum leaves transiently transformed with BGLU-OE plasmids, were homogenized in liquid nitrogen and then subjected to total RNA isolation using TRIzol reagent (Thermo Fisher Scientific) following the manufacturer’s instructions. RNA integrity was checked on a 1.5% agarose gel. Subsequently, 1.0 mg total RNA was reverse transcribed into first strand cDNA with a Prime Script™ reagent Kit with gDNA Eraser (TaKaRa, Japan), according to the manufacturer’s instructions. The cDNAs were subjected to a quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) using SYBR Green Premix Kit (Toyobo, Japan) in the ABI PRISM 7500 Fluorescent Quantitative PCR System (Thermo Fisher Scientific). All the primers used were listed in Table S2, with Actin as the internal control.
Expression profiling of DcBGLUs from Transcriptomic data
Based on the transcriptome data from D. catenatum (GSE155403 and SRP150489) [31, 32], D. huoshanense (SRP122499) [33], and Dendrobium nobile (PRJNA338366) [34], the expressions of DcBGLUs and the corresponding orthologue genes were screened across four more tissues, mainly leaves, stems, roots, and protocorm-like body (PLBs), and under MF23 treatment. All the raw SRA reads were transformed into fastq format, filtered, aligned, assembled, and estimated for expression levels. The results were visualized using heatmap generated from the TBtools [35].
Cloning of DcBGLUs and transient transformation in D. catenatum leaves
The full-length coding sequences of DcBGLU2, DcBGLU6, DcBGLU8, and DcBGLU13 were amplified using D. catenatum cDNA template with the primer sets listed in Table S3. The amplified fragments were cloned into pNC-Cam1304 binary vectors (NC biotech, Hainan, China) for gene overexpression. The corresponding constructs were then infiltrated into 2-year-old D. catenatum leaves following an Agrobacterium-mediated method [36] (Fig. S1).
Measurement of the content of medicinal components
Contents of reducing-polysaccharides were determined following Tonukari et al.’s methods [37]. Contents of soluble-polysaccharides were measured using a plant soluble-polysaccharide assay kit (BC0035, Solarbio, Beijing, China) according to the manufacturer’s instructions. Contents of flavonoids were measured using a flavonoid assay kit (BC1335, Solarbio, Beijing, China) according to the manufacturer’s instructions. Contents of alkaloids were estimated following Wang et al.’s method [38]. Transiently transformed D. catenatum leaves (0.5 g) were harvested, grounded in liquid nitrogen, and used for extraction of compounds. The isolated compounds were determined by using a spectrophotometer (BeckmanCoulter DU730).
Statistical analysis
For qRT-PCR gene expression analysis, Actin from D. catenatum was used as the internal control. The 2-ΔΔCt method was used to calculate relative gene expression. Statistical significance is defined as follows: ** p < 0.01, *** p < 0.001, and **** p < 0.0001 (Student’s t-test).
Results
Genome-wide identification and characterization of BGLU genes in D. catenatum
The release and continuous update of the complete D. catenatum genome make it easier to conduct genome-wide identification of genes. A total of 22 candidate DcBGLU genes of the GH1 family were identified in the D. catenatum genome after strict HMMER screening and domain confirmation, and they were designated as DcBGLU1 to DcBGLU22 following the LOC accession numbers in the NCBI gene bank (Table S1). Among these 22 listed DcBGLUs, few were functionally characterized. For clarity, information on our previously published DcBGLUs was provided in Table S1 as well.
Physicochemical characteristics of the predicted DcBGLU proteins, including amino acid number, molecular weight, signal peptide, isoelectric point, GRAVY, N-gly site, and possible subcellular localization, are listed in Table 1. Approximately half of the predicted DcBGLU proteins (12/22) were predicted to have signal peptides ranging from 17 to 38 amino acids, targeting them to the secretory pathway. The length of the predicted precursor proteins varied between 245 aa (DcBGLU11) and 1050 aa (DcBGLU18), which correspond to protein molecular weight (MW) varied from 27.87 to 119.28 kDa. Most DcBGLU proteins contain one to ten N-linked glycosylation sites, except four DcBGLUs (DcBGLU11, 12, 16, and 21).
The theoretical isoelectric points (pI) of the predicted proteins varied widely from 5.18 (DcBGLU19) to 8.30 (DcBGLU13). DcBGLU5, 13, and 17 were basic proteins, DcBGLU1 and DcBGLU21 were neutral proteins, and the rest were acidic proteins (Table 1). The GRAVY ranged from − 0.576 to 0.022, suggesting that these DcBGLUs are all hydrophilic proteins. Additionally, most of the DcBGLUs were predicted to be in the vacuole, chloroplast and cytosol (10/22), chloroplast and/or vacuole (7/22). These results showed significant differences among the DcBGLU proteins, reflecting their diversified functions in D. catenatum.
Phylogenetic analysis and classification of DcBGLU proteins
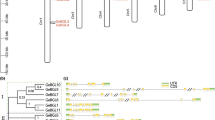
Multiple sequence alignment was conducted to further classify and characterize DcBGLU proteins, and results showed high conservation among the members (Fig. S2). Due to the high sequence similarity, the evolutionary relationship of DcBGLU proteins was investigated. Specifically, a sequence-based neighbor-joining phylogenetic tree was constructed for the proteins, including BGLU proteins both from D. catenatum and A. thaliana, using the MEGA X software (Fig. 1). The results from the phylogenetic tree showed that DcBGLUs could be divided into seven distinct clusters. Among the 22 DcBGLUs, 6, 8, and 2 belong to clusters I, II, and, III, respectively. While cluster IV contains six members only from D. catenatum, clusters AtI (16 members), AtII (6 members), and AtIII (11 members) contain members only from A. thaliana, suggesting that gene deletion could occur during the evolution of D. catenatum.

Phylogenetic relationship of the BGLUs from D. catenatum and A. thaliana. Phylogenetic tree of BGLUs using neighbor-joining (NJ) methods was constructed by MEGA X with 47 AtBGLU and 22 DcBGLU proteins. The subfamilies were marked in different colors. The identified DcBGLUs were highlighted by red circles
Gene structure and conserved motif of DcBGLU proteins
To identify the conserved functional motifs in DcBGLU proteins, we searched using the MEME online tool and identified ten motifs. These conserved motifs possessed 14 to 82 amino acids, and the number of motifs varied from two to ten. The results showed that more than half of the members (13/22) possessed all these ten motifs and that motif two and nine were the two most conserved ones widely present in DcBGLU proteins (Fig. 2a). However, nine DcBGLU proteins lacked the complete combination of the conserved motifs, including five (DcBGLU3, 11, 13, 16 and 19) with less than four motifs, and the other four (DcBGLU17, 20, 21 and 22) have eight motifs. Nevertheless, members within the same cluster tend to share similar compositions of motifs, indicating the highly conservation between these DcBGLU proteins and the validity of cluster classification.

Architecture of conserved motifs and gene structures of DcBGLUs. a The motif composition and distribution of DcBGLU proteins. The colored boxes represent conserved motif and the grey lines indicate non-conserved lines. b Gene structures of DcBGLU genes. Exons and introns were indicated by blue rectangles and orange lines, respectively
In order to better understand the evolutionary relationships of the GH1 family members in D. catenatum, exon/intron structures of all the identified DcBGLU genes were analyzed. The results showed that the exon numbers have varied considerably among the DcBGLU family members, from some having only one exon (DcBGLU11, 13, and 19) to some having up to 27 exons (DcBGLU18), including five with ten or less exons and thirteen with 11-13 exons (Fig. 2b). The exon/intron organization and intron numbers of the most closely related members in the same clusters were very similar.
Promoter analysis and chromosomal distribution of DcBGLU genes
The members of different gene families could display diverse expression patterns due to functional divergence. The regulatory promoter often located upstream of the transcription initiation site of a gene has been recognized as one of the key factors in transcriptional regulation. In order to further investigate the potential regulatory mechanisms of DcBGLU genes in secondary metabolites production in response to environmental stimuli, about 2-kb upstream promoter regions of DcBGLU genes were submitted to the PlantCARE database for scanning the presence of key cis-acting elements. Three types of cis-regulatory elements, i.e., phytohormone-responsive, stress-responsive, and secondary metabolites biosynthesis (Flavonoid) elements, were detected (Fig. 3a). Firstly, five hormone-responsive elements, including MeJA, salicylic acid, abscisic acid, gibberellin, and auxin responses, were commonly presented in the promoter regions of the DcBGLU genes. Secondly, anaerobic, drought, low temperature, and defense and stress responsive elements were detected in these regions. Additionally, many light-responsive regulatory elements and MYB binding sites were widely present in these promoter regions (Supplementary File 1). Among these elements, MeJA responsive elements were the most common type of cis-regulatory elements (16/22) found in the promoters of DcBGLU genes (Fig. S3). This result supported the idea that accumulation of MeJA-induced secondary metabolites might be partially mediated by DcBGLUs. Moreover, flavonoid biosynthesis-associated cis-elements were detected in the promoters of DcBGLU2, 21, and 22, indicating the possible role of DcBGLUs in flavonoid production. The presence of cis-acting elements in the promoters of DcBGLU genes suggested that they might be responsible for adaptation to various stresses and hormone treatments by modulating the production of secondary metabolites.

Cis-elements analysis and chromosomal localization of DcBGLUs. a Cis-elements in promoters of DcBGLU genes. Different colored wedges represented different cis-elements. b Chromosomal localization of DcBGLU genes
The genomic distribution of DcBGLU genes was analyzed to provide an overview of the location on chromosomes. The 16 out of 22 DcBGLU genes were unevenly distributed on 12 of the 19 chromosomes (Fig. 3b). Most of the chromosomes have only one DcBGLU gene on each of them, except chromosome 9 with two members (DcBGLU9 and 12) and chromosome 13 with three members (DcBGLU5, 6, and 22). Interestingly, DcBGLU5, 6 and 22, which belonging to the same subgroup I on the same chromosome 13, tended to cluster together, whereas the other family members were clustered separately.
Organ and stress-specific expression patterns of the DcBGLU genes
Plant BGLUs are well documented to play essential roles in response to developmental and environmental stimuli. Nevertheless, the functions of BGLUs in D. catenatum are far from clear. In the present study, four RNA-seq datasets of D. catenatum (GSE155403 and SRP150489) [31, 32], D. huoshanense (SRP122499) [33], and D. nobile (PRJNA338366) [34] were retrieved from NCBI Web Server, including samples across multiple tissues (root, stem, leaf, and PLB) and samples infected with MF23 (Fig. 4a). Overall, 14 differentially expressed DcBGLUs were identified in D. catenatum, among which DcBGLU8, 16, and 18 were more highly expressed in leaves than in roots. However, DcBGLU2 and 14 were more highly expressed in stems than in roots. The expression analysis indicated that four BGLU orthologue genes corresponding to DcBGLU1, 7, 9, and 20, respectively, in D. nobile were induced by MF23 infection and that the expression patterns of corresponding genes expressed in leaves or stems vs roots in D. catenatum and D. huoshanense are very similar.

Expression profiles showing members of DcBGLUs in varied organs and different treatments. a Expression of DcBGLU genes in stem, leaf, PLB, and under MF23 treatment. The log2(TPM values) of genes were shown by different color dots. Red and blue indicate high and low levels of expression, respectively. Each column indicates a treatment, and each row indicates a DcBGLU gene. b Expression patterns of DcBGLU genes in eight tissues. The expression levels of 17 DcBGLU genes were from the RNA-seq data. The eight samples included the column, sepal, stem, leaf, lip, flower bud, white root and green root tip. The color scale represents the values of log2(TPM value)
Because of the close relevance between gene expression and function, the expression profiles of DcBGLUs in eight tissues (column, sepal, white root, green root tip, stem, leaf, lip, and flower bud) from RNA-seq datasets (PRJNA348403) [39] were analyzed. DcBGLUs showed different transcriptional patterns across various tissues (Fig. 4b). Some DcBGLUs are specifically expressed in floral organs (e.g., DcBGLU9 in the column, DcBGLU1 and 20 in the lip, DcBGLU2, 7, and 14 in the flower buds), suggesting their potential role in pigmentation and scent release. For DcBGLU10, 11, 12, 14, 17, and 21, they tended to be more highly expressed in white roots and green root tips than in other tissues. Additionally, the DcBGLUs classified in the same cluster did not always have the same expression pattern. For example, compared with the members of DcBGLU2, 9, 10 and 14 in cluster IV, DcBGLU8 and 18 from the same clade were more highly expressed in leaves. These results indicated that the expression pattern of DcBGLU genes was diverse and tissue-specific.
Functional validation of DcBGLU genes in medicinal compounds accumulation
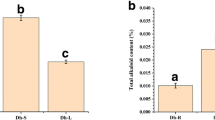
As our previous study suggested that the expression of DcBGLU2 was closely related to reducing-polysaccharide production [27], we thus verified the function by transient overexpression of this gene in D. catenatum leaves. Upregulated expression was detected in the infiltrated leaves 6 hours later (Fig. 5a), with accumulated levels of reducing-polysaccharides, flavonoids, and alkaloids 5 days later. We also overexpressed DcBGLU8, another member in the same cluster as DcBGLU2. Results showed that overexpression of DcBGLU8 had similar promoting roles as DcBGLU2 in flavonoids and alkaloids accumulation but somehow suppressed reducing-polysaccharide production (Fig. 5b). Besides, we also tested the possible functions of the members from the other two clusters, DcBGLU6 from cluster I and DcBGLU13 from cluster II, in medicinal metabolites production. Overexpression of DcBGLU6 greatly induced accumulation of reducing-polysaccharides and flavonoids, and slightly but significantly increased accumulation of alkaloids. Meanwhile, overexpression of DcBGLU13 enhanced the production of soluble-polysaccharides (Fig. 5c) and flavonoids, but slightly reduced alkaloids (Fig. 5d, e). These results reflected the diversified functions of DcBGLUs in regulating medicinal metabolites accumulation in D. catenatum.

Overexpression of DcBGLUs altered major medicinal metabolites accumulation. a qRT-PCR verification of the overexpression status of four selected DcBGLU genes in transiently transformed D. catenatum leaves. Samples transformed with empty vectors were used as the controls (Ctrl). b-e Determination of major medicinal metabolite contents in BGLU-OE leaves. b Reducing-polysaccharide content; c Soluble-polysaccharide content; d Flavonoid content; e Alkaloid content. Leaves transiently transformed with Agrobacterium EHA105 carrying empty vector were used as control. Bars marked with stars indicate significant differences (Student’s t-test, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001)
Discussion
Dendrobium orchids are highly prized medicinal herbs that have been widely used for many years. The plant contains various bioactive components, including polysaccharides, flavonoids, and alkaloids [40]. Several transcriptome profiles have been conducted in Dendrobium species to reveal the putative genes and pathways involved in active metabolites biosynthesis [25, 41, 42]. Moreover, genome-wide characterization of gene families has also begun to shed light on the biosynthesis pathways in several plant species [43]. Glucosidases are multifunctional enzymes that play crucial roles in plant development, secondary metabolites biosynthesis, and responses to biotic and abiotic stresses [11, 12]. Recently, the genome-wide characterization of BGLU members in different plant species has been performed with the rapid advancements of whole-genome sequencing technologies. Compared with the other plant species, the number of CH1-BGLU genes in Orchidaceae (C. sinense, Apostasia shenzhenica, Vanilla shenzhenica, Phalaenopsis equestris, Phalaenopsis aphrodite and D. catenatum) is much smaller, suggesting that there may be gene loss or pseudogenization in the process of evolution [19]. Nevertheless, in D. catenatum, a systematic analysis of BGLU family members has not been available despite being one of the most important medicinal orchids and the importance of BGLU genes in plant secondary metabolism. The recent availability of high-quality annotated reference genome [25] provides valuable resources for studying the BGLU family in D. catenatum. This study, to our knowledge, provides the first report concerning the systematic analysis and functional role of BGLU family genes from D. catenatum on secondary metabolism. This study identified 22 full-length BGLU genes from the D. catenatum genome, 16 of which were unevenly distributed on 12 chromosomes. We also analyzed the phylogenetic relations of DcBGLUs and evaluated gene expression patterns in different tissues and under biotic stress. In addition, correlation analysis between gene expression and metabolites content verified four candidate genes involved in active metabolites biosynthesis. Overall, the current study comprehensively investigated and presented the BGLU genes in D. catenatum. This endeavor will be beneficial for the in-depth exploration of biological functions of the BGLU gene family and provide potential targets for molecular breeding.
The current study identified 22 DcBGLU genes throughout the D. catenatum genome and characterized them using multi-sequence alignment and phylogenetic analysis. The results showed that the DcBGLUs shared high sequence similarity and conserved domain during evolution. DcBGLUs were mainly targeted into cytosol, chloroplast, and vacuole, where they can access the physiological substrates for catalysis. Besides, most DcBGLUs contain at least one predicted N-glycosylation site, corresponding to the estimation that many of DcBGLUs might hydrolyze their substrates through secretory pathways.
Jasmonate (JA) is a plant-specific signaling molecule broadly associated with the biosynthesis of various secondary metabolites. The exogenous application of methyl jasmonate (MeJA) has been frequently used to manipulate the production of polysaccharides [44] and alkaloids in D. catenatum [45]. In the present study (Fig. 3a), we prove that MeJA-responsive cis-elements are widely distributed in most of DcBGLU promoters (16/22), suggesting that DcBGLUs might partially mediate MeJA-induced metabolites accumulation. Salicylic acid (SA) is another stress signaling molecule that can be used as an elicitor to promote the biosynthesis of plant secondary metabolites [46]. Both MeJA and SA have commonly been used in various plant cultures, including PLB, callus, shoot, and root culture systems [47, 48]. The promoters of DcBGLU members, including DcBGLU1, 5, 6, 11, 13, and 18, have SA-responsive elements, implying their possible roles in elicitor-induced active metabolites accumulation.
Even though plant-derived alkaloids are beneficial to human health, for plants, alkaloids are essential for defensive responses to environmental stresses. For example, binary stress can increase indole alkaloid levels in Catharanthus roseus [49], drought stresses can increase the accumulation of alkaloid in roots of motherwort (Leonurus japonicas) [50], Ceratocystis fimbriata infection can enhance alkaloids production in mango (cultivar Ubá) [51], and MF23 infection can increase alkaloids accumulation in D. nobile [34]. Accordingly, many defense and stress responsive elements presented in the promoter regions of DcBGLUs. MF23 induced BGLU1, 7, 19, and 20 expressions (Fig. 4a) with increased dendrobine alkaloid levels [34]. Anthocyanin flavonoids, normally present as glycosides, are the main determinants of flowers colors [52]. Tea made from D. catenatum flowers has been consumed for many years, in which flavonoids might be one of the major beneficial ingredients. DcBGLU1, 2, 7, 9, 14, and 20 were highly expressed in D. catenatum floral organs (Fig. 4b), indicating their possible functions in flavonoid biosynthesis.
Stem-specific expressions of AtBGLU45 and AtBGLU46 in A. thaliana are responsible for hydrolysis of lignin precursors in response to various stresses [53]. Grouped in the same cluster (i.e., cluster I), however, DcBGLU16 was mainly expressed in leaves of D. catenatum (Fig. 4b). Root-specific AtBGLU42 has been known to modulate rhizobacteria-induced systematic resistance in A. thaliana [54]. Even though DcBGLU12 in the same clade (i.e., clade II) expressed in roots, it failed to respond to MF23 infection as revealed by the RNA-seq analysis (Fig. 4a), indicating the diversified functions between AtBGLUs and DcBGLUs.
Plant BGLUs are groups of GH1 enzymes that remove the nonreducing terminal β-D-glucosyl residue from glucoconjugates. BGLU enzymes contribute to various biological functions in plants, including cell wall remodeling, defense and stress response, scent release, phytohormone activation, and secondary metabolism [55]. BGLUs can hydrolyze metabolic intermediates to release glucosyl blocking groups and allow further metabolization to various natural products, many of which are medicinally relevant compounds. For example, the monoterpene alkaloid strictosidine is hydrolyzed by a specific cytoplasmic β-glucosidase to produce various monoterpene alkaloids [15, 56]. In addition to hydrolysis activity, many BGLUs also showed transglucosidase activities for synthesizing glucoconjugates, such as anthocyanin biosynthesis in Agapanthus africanus and A. thaliana [57, 58]. This glycosylation and deglycosylation process perfectly maintained the homeostasis of plant metabolites. In a previous study, we found a close association between the expression of DcBGLU2 and polysaccharide content in D. catenatum [27], suggesting that the DcBGLU2 might work as a transglucosidase in polysaccharide biosynthesis. Consistent with these findings, overexpression of DcBGLU2 increased the reducing-polysaccharides, flavonoids, and alkaloids in D. catenatum (Fig. 5). Moreover, SENSITIVE TO FREEZING 2 (SFR2) encoded enzymes from carnation (Dianthus caryophyllus), delphinium (Delphinium grandiflorum), and A. taliana can transglycosylate monogalactosyl diacylglyceride to di-, tri-, and tetra-galactosyl diacylglycerides [59, 60]. The present study also identified DcBGLU3 and DcBGLU16 as BGLU-like SFR2 genes in cluster I (Table S1), indicating similar functions between them. Overexpression of exogenous β-glucosidase in tobacco chloroplasts increased phytohormone levels and thus the resistance to white flies and aphids [61]. Likewise, the transformation of the same BGLU into Artemisia annua vacuole increased trichome numbers and artemisinin production [62]. These works showed the potential application of manipulating β-glucosidase in plants to prompt medicinal metabolites production.
Conclusion
The present study has investigated the classification and expression profile of 22 GH1 DcBGLUs in different tissues and under stress conditions. Although some family members of AtBGLUs and DcBGLUs shared high sequence similarities, the tissue and stress responsiveness were diversified. Therefore, the present study paves the way for further dissection of the distinct role of DcBGLUs in secondary metabolism and other functions.
Availability of data and materials
The genome sequences and protein data of the D. catenatum were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/genome/?term=JSDN00000000) under the accession code JSDN00000000 [25]. Transcriptome data from D. catenatum (GSE155403 and SRP150489) [31, 32], D. huoshanense (SRP122499) [33], and D. nobile (PRJNA338366) [34] were used for BGLU expression profiling.
References
Wood HP. The dendrobiums. Koenigstein: ARG Gantner Verlag; 2006.
Bulpitt CJ, Li Y, Bulpitt PF, Wang JG. The use of orchids in Chinese medicine. J Roy Soc Med. 2007;100(12):558–63.
Wei W, Feng L, Bao WR, Ma DL, Leung CH, Nie SP, et al. Structure characterization and immunomodulating effects of polysaccharides isolated from Dendrobium officinale. J Agric Food Chem. 2016;64(4):881–9.
Tang H, Zhao T, Sheng Y, Zheng T, Fu L, Zhang Y. Dendrobium officinale Kimura et Migo: a review on its ethnopharmacology, phytochemistry, pharmacology, and industrialization. Evid Based Compl Alt. 2017. https://doi.org/10.1155/2017/7436259.
Teixeira da Silva JA, Ng TB. The medicinal and pharmaceutical importance of Dendrobium species. Appl Microbiol Biotechnol. 2017;101(6):2227–39.
Tang H, Zhao T, Sheng Y, Zheng T, Fu L, Zhang Y. Dendrobium officinale Kimura et Migo: a review on its ethnopharmacology, phytochemistry, pharmacology, and industrialization. Evid Based Complement Alternat Med. 2017;2017:7436259.
Zhang Y, Zhang L, Liu J, Liang J, Si J, Wu S. Dendrobium officinale leaves as a new antioxidant source. J Funct Foods. 2017;37:400–15.
Liu JJ, Liu ZP, Zhang XF, Si JP. Effects of various processing methods on the metabolic profile and antioxidant activity of Dendrobium catenatum Lindley leaves. Metabolites. 2021;11(6):351.
Zhang X, Zhang S, Gao B, Qian Z, Liu J, Wu S, et al. Identification and quantitative analysis of phenolic glycosides with antioxidant activity in methanolic extract of Dendrobium catenatum flowers and selection of quality control herb-markers. Food Res Int. 2019;123:732–45.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–5.
Ketudat Cairns JR, Esen A. β-Glucosidases. Cell Mol Life Sci. 2010;67(20):3389–405.
Opassiri R, Pomthong B, Onkoksoong T, Akiyama T, Esen A, Ketudat Cairns JR. Analysis of rice glycosyl hydrolase family 1 and expression of Os4bglu12 β-glucosidase. BMC Plant Biol. 2006;6(1):1–19.
Dong X, Jiang Y, Hur Y. Genome-wide analysis of glycoside hydrolase family 1 β-glucosidase genes in Brassica rapa and their potential role in pollen development. Int J Mol Sci. 2019;20(7):1663.
Yang J, Ma L, Jiang W, Yao Y, Tang Y, Pang Y. Comprehensive identification and characterization of abiotic stress and hormone responsive glycosyl hydrolase family 1 genes in Medicago truncatula. Plant Physiol Biochem. 2021;158:21–33.
Stöckigt J, Zenk MH. Strictosidine (isovincoside): the key intermediate in the biosynthesis of monoterpenoid indole alkaloids. J Chem Soc Chem Commun. 1977;18:646–8.
Yuan Y, Zhang B, Tang X, Zhang J, Lin J. Comparative transcriptome analysis of different Dendrobium species reveals active ingredients-related genes and pathways. Int J Mol Sci. 2020;21(3):861.
Perera CO, Owen E. Effect of tissue disruption by different methods followed by incubation with hydrolyzing enzymes on the production of vanillin from Tongan Vanilla beans. Food Bioprocess Technol. 2008;3(1):49.
Zeng D, Que C, Teixeira da Silva JA, Xu S, Li D. Comparative transcriptomic and metabolic analyses reveal the molecular mechanism of ovule development in the orchid, Cymbidium sinense. Front Plant Sci. 2022;12:814275.
Yang FX, Gao J, Wei YL, Ren R, Zhang GQ, Lu CQ, et al. The genome of Cymbidium sinense revealed the evolution of orchid traits. Plant Biotechnol J. 2021;19(12):2501–16.
Zhao MM, Zhang G, Zhang DW, Hsiao YY, Guo SX. ESTs analysis reveals putative genes involved in symbiotic seed germination in Dendrobium officinale. PLoS One. 2013;8(8):e72705.
Jiang W, Wu Z, Wang T, Mantri N, Huang H, Li H, et al. Physiological and transcriptomic analyses of cadmium stress response in Dendrobium officinale seedling. Plant Physiol Biochem. 2020;148:152–65.
Yap YM, Loh CS, Ong BL. Regulation of flower development in Dendrobium crumenatum by changes in carbohydrate contents, water status and cell wall metabolism. Sci Hortic. 2008;119(1):59–66.
Hussain MS, Fareed S, Saba Ansari M, Rahman A, Ahmad IZ, Saeed M. Current approaches toward production of secondary plant metabolites. J Pharm Bioallied Sci. 2012;4(1):10–20.
Adejobi OI, Guan J, Yang L, Hu JM, Yu A, Muraguri S, et al. Transcriptomic analyses shed light on critical genes associated with bibenzyl biosynthesis in Dendrobium officinale. Plants. 2021;10(4):633.
Zhang GQ, Xu Q, Bian C, Tsai WC, Yeh CM, Liu KW, et al. The Dendrobium catenatum Lindl. Genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci Rep. 2016;6:19029.
Niu Z, Zhu F, Fan Y, Li C, Zhang B, Zhu S, et al. The chromosome-level reference genome assembly for Dendrobium officinale and its utility of functional genomics research and molecular breeding study. Acta Pharm Sin B. 2021;11(7):2080–92.
Wang ZC, Zhao ML, Zhang XJ, Zhang ZL, Li SZ, Li J, et al. Phytohormone-triggered transcriptional changes revealed β-glucosidase as a key player for polysaccharide metabolism in Dendrobium officinale. Prog Biochem Biophys. 2022;1-12. https://doi.org/10.16476/j.pibb.2021.0352.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547.
Odronitz F, Pillmann H, Keller O, Waack S, Kollmar M. WebScipio: an online tool for the determination of gene structures using protein sequences. BMC Genomics. 2008;9:422.
Zhang T, Cui Z, Li Y, Kang Y, Song X, Wang J, et al. Genome-wide identification and expression analysis of MYB transcription factor superfamily in Dendrobium catenatum. Front Genet. 2021;12:714696.
Wang Z, Jiang W, Liu Y, Meng X, Su X, Cao M, et al. Putative genes in alkaloid biosynthesis identified in Dendrobium officinale by correlating the contents of major bioactive metabolites with genes expression between protocorm-like bodies and leaves. BMC Genomics. 2021;22(1):579.
Yuan Y, Zhang J, Liu X, Meng M, Wang J, Lin J. Tissue-specific transcriptome for Dendrobium officinale reveals genes involved in flavonoid biosynthesis. Genomics. 2020;112(2):1781–94.
Yuan Y, Yu M, Jia Z, Song X, Liang Y, Zhang J. Analysis of Dendrobium huoshanense transcriptome unveils putative genes associated with active ingredients synthesis. BMC Genomics. 2018;19(1):978.
Li Q, Ding G, Li B, Guo SX. Transcriptome analysis of genes involved in dendrobine biosynthesis in Dendrobium nobile Lindl. Infected with mycorrhizal fungus MF23 (Mycena sp.). Sci Rep. 2017;7(1):316.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Moon H, Park HJ, Jeong AR, Han SW, Park CJ. Isolation and identification of Burkholderia gladioli on Cymbidium orchids in Korea. Biotechnol Biotec Eq. 2017;31(2):280–8.
Tonukari NJ, Avwioroko OJ, Ezedom T, Anigboro AA. Effect of preservation on two different varieties of Vernonia amygdalina Del.(bitter) leaves. Food Nutr Sci. 2015;6(07):623.
Wang HQ, Jin MY, Paek KY, Piao XC, Lian ML. An efficient strategy for enhancement of bioactive compounds by protocorm-like body culture of Dendrobium candidum. Ind Crop Prod. 2016;84:121–30.
Zhang GQ, Liu KW, Li Z, Lohaus R, Hsiao YY, Niu SC, et al. The Apostasia genome and the evolution of orchids. Nature. 2017;549(7672):379–83.
Ng TB, Liu J, Wong JH, Ye X, Wing Sze SC, Tong Y, et al. Review of research on Dendrobium, a prized folk medicine. Appl Microbiol Biotechnol. 2012;93(5):1795–803.
Guo X, Li Y, Li C, Luo H, Wang L, Qian J, et al. Analysis of the Dendrobium officinale transcriptome reveals putative alkaloid biosynthetic genes and genetic markers. Gene. 2013;527(1):131–8.
Shen C, Guo H, Chen H, Shi Y, Meng Y, Lu J, et al. Identification and analysis of genes associated with the synthesis of bioactive constituents in Dendrobium officinale using RNA-Seq. Sci Rep. 2017;7(1):187.
Wu B, Gao L, Gao J, Xu Y, Liu H, Cao X, et al. Genome-wide identification, expression patterns, and functional analysis of UDP glycosyltransferase family in peach (Prunus persica L. Batsch). Front Plant Sci. 2017;8:389.
Yuan Z, Zhang J, Liu T. Enhancement of polysaccharides accumulation in Dendrobium officinale by exogenously applied methyl jasmonate. Biol Plantarum. 2017;61(3):438–44.
Jiao C, Song C, Zheng S, Zhu Y, Jin Q, Cai Y, et al. Metabolic profiling of Dendrobium officinale in response to precursors and methyl jasmonate. Int J Mol Sci. 2018;19(3):728.
Manorma S, Archana S, Ashwani K, Basu SK. Enhancement of secondary metabolites in cultured plant cells through stress stimulus. Amer J Plant Physiol. 2011;6(2):50–71.
Largia MJV, Pothiraj G, Shilpha J, Ramesh M. Methyl jasmonate and salicylic acid synergism enhances bacoside a content in shoot cultures of Bacopa monnieri (L.). Plant Cell Tiss Org. 2015;122(1):9–20.
Sivanandhan G, Kapil Dev G, Jeyaraj M, Rajesh M, Arjunan A, Muthuselvam M, et al. Increased production of withanolide a, withanone, and withaferin a in hairy root cultures of Withania somnifera (L.) dunal elicited with methyl jasmonate and salicylic acid. Plant Cell Tiss Org. 2013;114(1):121–9.
Zhu W, Yang B, Komatsu S, Lu X, Li X, Tian J. Binary stress induces an increase in indole alkaloid biosynthesis in Catharanthus roseus. Front Plant Sci. 2015;6:582.
Wei H, Li L, Yan X, Wang Y. Effects of soil drought stress on the accumulation of alkaloids and flavonoids in motherwort. Adv Inf Sci Serv Sci. 2013;5(6):795.
Araujo L, Bispo WMS, Rios JA, Fernandes SA, Rodrigues FÁ. Alkaloids and phenolics biosynthesis increases mango resistance to infection by Ceratocystis fimbriata. Bragantia. 2016;75:199–211.
Xiao T, Guo Z, Sun B, Zhao Y. Identification of anthocyanins from four kinds of berries and their inhibition activity to α-glycosidase and protein tyrosine phosphatase 1B by HPLC–FT-ICR MS/MS. J Agric Food Chem. 2017;65(30):6211–21.
Chapelle A, Morreel K, Vanholme R, Le-Bris P, Morin H, Lapierre C, et al. Impact of the absence of stem-specific β-glucosidases on lignin and monolignols. Plant Physiol. 2012;160(3):1204–17.
Zamioudis C, Hanson J, Pieterse CM. β-Glucosidase BGLU 42 is a MYB 72-dependent key regulator of rhizobacteria-induced systemic resistance and modulates iron deficiency responses in Arabidopsis roots. New Phytol. 2014;204(2):368–79.
Ketudat Cairns JR, Mahong B, Baiya S, Jeon JS. β-Glucosidases: multitasking, moonlighting or simply misunderstood? Plant Sci. 2015;241:246–59.
Warzecha H, Gerasimenko I, Kutchan TM, Stöckigt J. Molecular cloning and functional bacterial expression of a plant glucosidase specifically involved in alkaloid biosynthesis. Phytochemistry. 2000;54(7):657–66.
Miyahara T, Takahashi M, Ozeki Y, Sasaki N. Isolation of an acyl-glucose-dependent anthocyanin 7-O-glucosyltransferase from the monocot Agapanthus africanus. J Plant Physiol. 2012;169(13):1321–6.
Miyahara T, Sakiyama R, Ozeki Y, Sasaki N. Acyl-glucose-dependent glucosyltransferase catalyzes the final step of anthocyanin formation in Arabidopsis. J Plant Physiol. 2013;170(6):619–24.
Matsuba Y, Sasaki N, Tera M, Okamura M, Abe Y, Okamoto E, et al. A novel glucosylation reaction on anthocyanins catalyzed by acyl-glucose–dependent glucosyltransferase in the petals of carnation and delphinium. Plant Cell. 2010;22(10):3374–89.
Moellering ER, Muthan B, Benning C. Freezing tolerance in plants requires lipid remodeling at the outer chloroplast membrane. Science. 2010;330(6001):226–8.
Jin S, Kanagaraj A, Verma D, Lange T, Daniell H. Release of hormones from conjugates: chloroplast expression of β-glucosidase results in elevated phytohormone levels associated with significant increase in biomass and protection from aphids or whiteflies conferred by sucrose esters. Plant Physiol. 2011;155(1):222–35.
Singh ND, Kumar S, Daniell H. Expression of β-glucosidase increases trichome density and artemisinin content in transgenic Artemisia annua plants. Plant Biotechnol J. 2016;14(3):1034–45.
Acknowledgments
The authors are grateful to Ms. Yu Wang from Orchid Conservation and Research Center of Shenzhen for her help caring for the D. catenatum plants, and to Dr. Guoqiang Zhang for his help in chromatin localization.
Funding
This work was supported by grants from The Science, Technology and Innovation Commission of Shenzhen Municipality (JCYJ20210324123005016) and The Research Grants for Postdocs Settle in Shenzhen from The Human Resources and Social Security Bureau of Shenzhen to Dr. Zhicai Wang.
Author information
Authors and Affiliations
Contributions
Z.W. and M.W. conceived the study. Z.W. designed the experiments, analyzed the data, and wrote the manuscript. X.Z. and X.D. cloned the DcBGLU genes and constructed the overexpression vectors, M.Z. and X.D. performed Agrobacterium-mediated gene transformation, qRT-PCR, and measured the metabolites content. Z.W., J.L. and M.W. revised the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study is conducted with plant material complies with relevant institutional, national, and international guidelines and legislation. Also, the study did not use any endangered or protected species. The D. catenatum seedlings are widely planted and commercially available in China. No stable transgenic lines were generated. The 2-year-old D. catenatum seedlings used in this study were grown in a greenhouse at the Orchid Conservation and Research Center of Shenzhen, Shenzhen, China, at 25 ± 2 °C with a 12/12 h light/dark cycle (40 μmol/m 2 /S) and 60-70% relative humidity.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 2: Fig. S1.
Transient expression of DcBGLUs in leaves of D. catenatum. Fig. S2. Amino acid sequence alignment of 22 DcBGLU enzymes in D. catenatum. Fig. S3. Analysis of the numbers and types of cis-acting elements in DcBGLU genes. Table S1. Accession numbers for proteins used in the phylogenetic tree. Table S2. Primers used for qRT-PCR validation. Table S3. Primers used for gene cloning. Table S4. Abbreviations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Z., Zhao, M., Zhang, X. et al. Genome-wide identification and characterization of active ingredients related β-Glucosidases in Dendrobium catenatum. BMC Genomics 23, 612 (2022). https://doi.org/10.1186/s12864-022-08840-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08840-x