
Abstract
Background
The collection of circRNAs mostly focused on their sequence composition such as protein/miRNA binding motif, and/or regulatory elements such as internal ribosome entry site. However, less attention was paid to subcellular localization. CircVIS aimed to provide a collection of circRNAs with information of subcellular compartments and also integrated the circRNA entries from previous circRNA databases.
Results
A collection of circRNAs from public circRNA databases and de novo identification were annotated according to subcellular localizations including nucleoplasm, chromatin-associated parts, cytoplasm and polyribosome. All circRNAs were aligned to a selected major transcript, and if presence, the circRNA-derived open reading frame with annotation of functional domain were compared to its parental protein. The results showed that distinct circRNAs may exert their molecular and cellular functions in different subcellular compartments. The web service is made freely available at http://lab-x-omics.nchu.edu.tw/circVIS.
Conclusions
CircVIS allows users to visualize the alignment between a given circRNA and its most relevant reference transcript along with information of subcellular localization.
Similar content being viewed by others


Background
Circular RNA (circRNA) is a novel class of single stranded regulatory RNA molecules with covalently enclosed ends by 3’, 5’-phosphodiester bond formed through backsplicing which takes place between a downstream splice donor and an upstream splice acceptor. Recent studies using next generation sequencing and computational analyses have revealed widespread existence of circRNAs in animals and many other organisms [1,2,3].
CircRNAs play various roles such as transcriptional activation, post-transcriptional modulation, translation and protein interaction in different subcellular compartments [4,5,6,7,8]. For those circRNAs regulating gene expression network through interaction with miRNAs [9,10,11,12,13], the majority of these circRNAs resides in cytoplasm to regulate the availability of miRNAs bound to mRNA molecules. For example, more than 75% of circular RNA originated from exon 8–10 of CCDC66, which interacted with miR-33b, 93 and 185, were found in the cytoplasm [11]. In contrast, it was reported that circRNAs modulating transcriptional activation associate with genomic DNA in nuclei. A few intron-retained circRNAs reside in the nuclei and associated with promoter region of target genes [5]. Nevertheless, an exonic circRNA from gene FLI1 modulating DNA methylation in promoter regions also localized in the nuclei [14]. These examples demonstrated that subcellular localizations of a given circRNA may provide clues to their molecular functions. Pioneer studies have made great contribution dissecting and archiving these relationships among miRNAs, circRNAs and associated pathological phenotypes [15,16,17,18]. However, the studies investigating the biological functions of circRNAs are largely limited to the function of miRNA sponge [19,20,21], and thus how to explore alternative molecular functions of circRNA become a critical task.
In this study, we analyzed and categorized circRNAs according to their subcellular localizations, aiming to provide more insight to interpret how circRNAs may exert their biological functions in distinct subcellular compartments. We also integrated potential coding region(s) along with functional domains of circRNA-derived open reading frames in a visual presentation platform.
Implementation
Data retrieval and processing
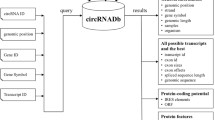
The archived circular RNA coordinates were downloaded from circBase [22] and circRNADb [23] while raw data of RNA sequencing were directly downloaded from SRA and converted to fastq by using SRA tool kit (v 2.9.1). The dataset ‘SRP083953’ was used for ribosomal RNA-depleted cytoplasmic, nucleoplasmic and chromatin-associated RNA [24], while ‘SRP114807’ (all available fractions), ‘SRP139916’ (with cycloheximide treatment) and ‘SRP233220’ were used for polysome fractions [25, 26]. The read sequences were then aligned to reference genome (Homo sapiens GRCh38.92) using Burrows-Wheeler Aligner. For bisulfite treatment-derived samples (SRP233220), reads were aligned to the same reference genome with C converted to T or G converted to A. The resultant Sequence Alignment/Map files were then proceeded to CIRI2 (Fig. 1) [27].

The schematic illustration of circRNA annotation tool. RNA sequencing data compatible with circRNA analysis from distinct subcellular compartments were retrieved from GEO/SRA databases (top-left corner). The coordinates of backsplicing junction were extracted by using CIRI2. The previously archived circRNA information were downloaded from circBase and circRNADb (top-right). Both sources of data were re-annotated by using gene information from Ensembl database. Each backsplicing junction was mapped to a selected representative reference transcripts according to conservation/number of exon/length of transcripts and alignment to exon junctions. CircRNAs will be displayed along with their reference transcript (bottom-left) and/or with the coding region from their parental genes (bottom-right)
Identification of circRNA representative transcript
The information of host genes and transcripts were extracted by comparison between circRNA coordinates and gene annotation (Homo sapiens GRCh38.92). Transcripts matched to circRNA coordinates were further ranked by the presence of Consensus Coding Sequence (CCDS), number of exons, offset to known exon junction and commonness among circRNAs.
Analysis of opening reading frame of circRNAs
The potential ORFs of circRNA sequence were considered beyond its original length. According to the times of ORFs crossing backsplice junction, ORFs were classified to 0-crossing (0C), 1-crossing (toward 5’ or 3’: 1C5’, 1C3’), 2 crossings (2C) and endless crossing (edlsC) (Fig. 2). The zero-crossing ORFs are indistinguishable to ORFs in parental RNAs (Fig. 2, left), while 1C5’ or 1C3’ are ORFs with novel sequences in either N- or C-terminus. In a similar fashion, 2C ORFs would be expected to have novel sequences at both ends (Fig. 2, right). In some cases, endless crossing takes place, generating an ORF with infinite length.

The analysis of ORFs in circular RNAs. The possible ORF organization were illustrated. The ORFs not crossing backsplicing junction were denoted ‘0−crossing’ (left). ORFs crossing backsplicing junctions (once) toward 5’ or 3’ will generate novel N terminal or C terminal sequences. 2C denoted the ORFs crossing backsplicing junction twice. In some cases, circRNA generates ORF crossing backsplicing junction infinite times. Grey circle denotes the absence of a feature (novel sequence or infinite ORF) while green circle denotes the presence of the feature
Data visualization
The circRNA isoforms, corresponding host gene and information of the paired backsplicing exons were integrated and presented by using ‘ggplot2’ [28]. The circRNAs with open reading frame were extracted and aligned with the protein sequence of their parental gene. The functional annotation of the given protein was retrieved and presented by using functions adapted from ‘drawProteins’ with modifications [29].
Results
Genome-wide recollection of circular RNAs with distinct subcellular localizations
The comparison of the records between databases is important for biologists to design their experiments. Due to the incompatibility of accession ID, it is not intuitive to know whether a given circRNA in one database is present in the other. Our annotation pipeline assigned each circRNA a major reference transcript along with a pair of exons for backsplice, making comparison and communication easier. The results of comparison between circBase and circRNADb using our annotation pipeline demonstrated the feasibility of comparison. The pioneer databases of circRNA, circBase and circRNADb, shared 14 thousand circRNA entries, and there are 67 thousand circBase-specific and 17 thousand circRNADb-specific entries respectively. In comparison with circBase and circRNADb, the RNA-seq data we analyzed revealed additional unique 11,858 circRNAs which were not archived previously. Furthermore, we identified circRNAs residing in distinct subcellular localization. Our analyses revealed that the majority of circRNAs resides in cytoplasm based on HeLa and HCT116 cell-lines (Fig. 3B, cytoplasm and polysome, 11,585, 82.48%). Only limited number (524, 3.73%) of circRNAs locates in nuclei and/or associated with chromatin according to data from HeLa cells (Fig. 3B). Of special note, a previous study has found that depletion of particular proteins may impair the nuclear export of circRNAs in a size-dependent manner [30]. It will be worthwhile to further validate the correlation between the cellular distribution of circRNAs and these proteins.

Genome-wide recollection of circRNAs. A A Venn diagram showed the overlapping unique entries among three datasets. The raw entries from circBase and circRNADb were preprocessed by selection of best representative transcript and paired exons for inter-database comparison. B The numbers of backsplicing events extracted from RNA-seq data using RNA collected from different cellular fractions were shown as a Venn diagram. In addition to the RNA isolated from the cytoplasmic and nuclear fractions, the chromatin fraction was defined as the insoluble parts of the nuclear lysate while the ‘polysome’ came from the heavy fractions of the sucrose gradient for polysome profiling
CircRNAs have great diversity of splicing pattern
To better compare all backsplice junctions from a single gene using minimal number of reference transcript, we integrated coordinates of backsplice junction, representative reference transcript, the pair of exons for backsplice, subcellular location and accession numbers of alternative circRNA databases (Fig. 4A). Furthermore to have better insight to this diversity, a proper visual aid is required to observe an overall exon usage for a given circRNA on a representative transcript. We integrated the annotations of Ensembl transcripts (red track) and circRNAs (blue or green track) in a single transcript plot (Fig. 4B). CircRNAs with predicted open reading frames can be easily identified. From the example of gene ‘PTP4A2’, the transcript ENST00000647444 had 9 pairs of backsplicing exons while ENST00000602725 and ENST00000532001 had 2 and 1 respectively. Obviously, one reference transcript can’t fit to all pairs of backsplicing exons. In addition, potential ORFs were aligned with parental proteins with functional features (Fig. 4C). While the functional domains were shown on the top of the reference protein, the circRNA-derived ORFs were aligned to the parental protein and shown at the bottom. This visual presentation will provide more information for biologists to evaluate what circRNAs may potentially modulate the functions of parental protein.

CircRNA isoform presentation. A The table shows details of the given circRNA including the origin of circRNA, Ref transcript, the pair of exon M/N for backsplice taking place from the representative transcript. The column of subcellular info shows ‘green’ if a given circRNA is identified in corresponding different subcellular compartments. Similarly, if ORF is predicted, the column of ‘cORF’ (circORF) shows green. The last two columns indicate in which database (DB) the given circRNA was archived (cBase: circBase; cRNADb: circRNADb). B Results of circRNA isoform presentation. The representative transcripts are showed in red tracks. A subset of circRNAs are aligned to the upper track (ENST00000647444) while others fit to ENST00000602725 or ENST00000532001. Green track: CircRNAs predicted to have ORF; Blue track: circRNA predicted not to have ORF. C CircRNAs with ORFs aligned to its protein with annotated domains. ORFs from different transcripts are color-coded. Paired exons of backsplicing exons are labeled at the end of gene symbol
Discussion
CircRNA is a class of regulatory molecules with diverse functions. Most of studies focused at its miRNA binding capacity in the cytoplasm. There are a few online resource designing for dissecting this function. For example, both starBase and circAtlas collected the interactions between circRNAs and miRNAs using either CLIP data or bioinformatic prediction while ACT classified the potential sponging activity through common target gene analysis [12, 17, 31]. In addition, predicted IRES and ORF information were archived in circRNADb and circAtlas [23, 31]. Nevertheless, CircInteractome archived the potential RNA-binding proteins associated with circRNA [32]. These annotations are extremely useful to promote functional studies for dissecting the particular downstream genes of circRNAs. However, the fact that overwhelming number of miRNA binding sites and ubiquitous presence of IRES/ORFs in databases of circRNA hinder the precise application of this information in biological researches. Thus, additional information has to be added to facilitate the analysis prior to experimental design.
The regulatory molecules exert their molecular functions in the corresponding subcellular compartments. For example, transcriptional factors such as ‘estrogen receptors’ (ERα and ERβ) or members of ‘signal transducer and activator of transcription’ (e.g. STAT3) have to translocate to nuclei to modulate the transcriptional activity of their target genes. In contrast, the majority of miRNAs and the associated AGO2 protein complexes resides in cytoplasm to target mRNA. Adherent to this concept, we hypothesized that the subcellular compartment where the circRNAs reside may provide extra information to predict or interpret their molecular functions. For example, circZNF609 [4], one of the best studied coding circRNAs was shown in polysome fraction in our analysis while circCCNB1 which modulates CDK1 activity [33] in nuclei was identified in nuclear fraction, suggesting that the information of subcellular localization indeed coincides with molecular functions in some cases. However, the limitation came from the variety of samples analyzed. First, there were limited number of available datasets compatible for circRNA analysis from multiple cell-lines. The majority of RNAseq data were from polyA-enriched samples, and/or oligo-dT-based library construction. Either one renders the circRNA analysis impossible. Second, there were limited number of RNA sequencing datasets available from subcellularly fractionated samples. Thus, the absence of circRNA in certain compartments will require further experimental evaluation.
Conclusions
Our circRNA annotation platform not only provides a unique information about the subcellular location, but also a straightforward presentation and nomenclature. The integrative information is much improved compared to these pioneer databases (Table 1), and will serve as an alternative hub for circRNA studies.
Availability and requirements
Project name: circVIS
Project home page: http://lab-x-omics.nchu.edu.tw/circVIS
Operating system(s): Platform independent (Web-based service)
Programming language: Perl 5 and R 3.5.0
Other requirements: Not applicable
License: GNU GPL; non-academic user: license needed
Datasets used
circBase:http://www.circbase.org/
circRNADb:http://reprod.njmu.edu.cn/cgi-bin/circrnadb/index.php
SRA datasets:SRP083953, SRP114807, SRP139916, SRP233220
Availability of data and materials
Web service is made freely available at http://lab-x-omics.nchu.edu.tw/circVIS.
Abbreviations
- circVIS:
-
CircRNA visual presentation
- circRNA:
-
Circular RNA
References
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE. 2012;7(2):e30733.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–8.
Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15(7):409.
Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M, et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol Cell. 2017; 66(1):22–37e29.
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22(3):256–64.
Holdt LM, Stahringer A, Sass K, Pichler G, Kulak NA, Wilfert W, Kohlmaier A, Herbst A, Northoff BH, Nicolaou A, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. 2016;7:12429.
Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016;44(6):2846–58.
Hsiao KY, Sun HS, Tsai SJ. Circular RNA - New member of noncoding RNA with novel functions. Exp Biol Med (Maywood). 2017;242(11):1136–41.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–8.
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215.
Hsiao KY, Lin YC, Gupta SK, Chang N, Yen L, Sun HS, Tsai SJ. Noncoding Effects of Circular RNA CCDC66 Promote Colon Cancer Growth and Metastasis. Cancer Res. 2017;77(9):2339–50.
Lin YC, Lee YC, Chang KL, Hsiao KY. Analysis of common targets for circular RNAs. BMC Bioinformatics. 2019;20(1):372.
Lin YC, Yu YS, Lin HH, Hsiao KY. Oxaliplatin-induced DHX9 phosphorylation promotes oncogenic circular RNA CCDC66 expression and development of chemoresistance. Cancers. 2020;12(3):697.
Chen N, Zhao G, Yan X, Lv Z, Yin H, Zhang S, Song W, Li X, Li L, Du Z, et al. A novel FLI1 exonic circular RNA promotes metastasis in breast cancer by coordinately regulating TET1 and DNMT1. Genome Biol. 2018;19(1):218.
Liu YC, Hong HC, Yang CD, Lee WH, Huang HT, Huang HD. Ouroboros resembling competitive endogenous loop (ORCEL) in circular RNAs revealed through transcriptome sequencing dataset analysis. BMC Genomics. 2018;19(Suppl 2):171.
Liu YC, Li JR, Sun CH, Andrews E, Chao RF, Lin FM, Weng SL, Hsu SD, Huang CC, Cheng C, et al. CircNet: a database of circular RNAs derived from transcriptome sequencing data. Nucleic Acids Res. 2016;44(D1):D209–215.
Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(Database issue):D92–97.
Ghosal S, Das S, Sen R, Basak P, Chakrabarti J. Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet. 2013;4:283.
Zheng J, Liu X, Xue Y, Gong W, Ma J, Xi Z, Que Z, Liu Y. TTBK2 circular RNA promotes glioma malignancy by regulating miR-217/HNF1beta/Derlin-1 pathway. J Hematol Oncol. 2017;10(1):52.
Zhang J, Liu H, Hou L, Wang G, Zhang R, Huang Y, Chen X, Zhu J. Circular RNA_LARP4 inhibits cell proliferation and invasion of gastric cancer by sponging miR-424-5p and regulating LATS1 expression. Mol Cancer. 2017;16(1):151.
Chen G, Shi Y, Liu M, Sun J. circHIPK3 regulates cell proliferation and migration by sponging miR-124 and regulating AQP3 expression in hepatocellular carcinoma. Cell Death Dis. 2018;9(2):175.
Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20(11):1666–70.
Chen X, Han P, Zhou T, Guo X, Song X, Li Y. circRNADb: A comprehensive database for human circular RNAs with protein-coding annotations. Sci Rep. 2016;6:34985.
Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vagbo CB, Geula S, Hanna JH, Black DL, Darnell JE Jr, Darnell RB. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017;31(10):990–1006.
Schumann U, Zhang HN, Sibbritt T, Pan A, Horvath A, Gross S, Clark SJ, Yang L, Preiss T. Multiple links between 5-methylcytosine content of mRNA and translation. BMC Biol. 2020;18(1):40.
Chen TM, Lai MC, Li YH, Chan YL, Wu CH, Wang YM, Chien CW, Huang SY, Sun HS, Tsai SJ. hnRNPM induces translation switch under hypoxia to promote colon cancer development. EBioMedicine. 2019;41:299–309.
Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform. 2018;19(5):803–10.
Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag; 2016.
Brennan P. drawProteins: a Bioconductor/R package for reproducible and programmatic generation of protein schematics. F1000Res. 2018;7:1105.
Huang C, Liang D, Tatomer DC, Wilusz JE. A length-dependent evolutionarily conserved pathway controls nuclear export of circular RNAs. Genes Dev. 2018;32(9–10):639–44.
Wu W, Ji P, Zhao F. CircAtlas: an integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020;21(1):101.
Dudekula DB, Panda AC, Grammatikakis I, De S, Abdelmohsen K, Gorospe M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016;13(1):34–42.
Fang L, Du WW, Awan FM, Dong J, Yang BB. The circular RNA circ-Ccnb1 dissociates Ccnb1/Cdk1 complex suppressing cell invasion and tumorigenesis. Cancer Lett. 2019;459:216–26.
Acknowledgements
The authors would like to show their great appreciation to Mr. Yu-Cheng Chen for his technical support on establishing the website. We also deeply appreciate the full support of the Bioinformatics Center at National Cheng Kung University. In addition, HH Lin carried out her thesis research under the auspices of the Ph.D. program in Tissue Engineering and Regenerative Medicine, National Chung Hsing University and National Health Research Institutes in Taiwan.
About this supplement
This article has been published as part of BMC Genomics Volume 22 Supplement 5 2021: Selected articles from the 19th Asia Pacific Bioinformatics Conference (APBC 2021): genomics The full contents of the supplement are available at https://bmcgenomics.biomedcentral.com/articles/supplements/volume-22-supplement-5.
Funding
This research was funded by Ministry of Science and Technology of Taiwan (MOST 109–2320-B-005–008-MY3) to KY Hsiao, and Hsing Chung inter-institutional project (NCHU-CSMU-10805, NCHU-CSMU-10906 and NCHU-CSMU-11007) to KY Hsiao and YC Lee. Publication costs were funded by Ministry of Science and Technology of Taiwan (MOST 109–2320-B-005–008-MY3). None of the funding agencies were involved in the design of the study, analysis, interpretation of data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YC Lin, YC Lee and KL Chang developed the concept and wrote the codes. YC Wang and YC Tai optimized the codes and implemented the visual outputs. YC Lin, YC Lee, YC Wang and HH Lin analyzed and evaluated the results. HH Lin curated the datasets. YC Lin and YC Lee drafted the manuscript. KY Hsiao revised the manuscript and supervised the project. All authors have read and approved the final version of the manuscript for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, YC., Wang, YC., Lee, YC. et al. CircVIS: a platform for circRNA visual presentation. BMC Genomics 22 (Suppl 5), 921 (2021). https://doi.org/10.1186/s12864-022-08650-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08650-1