
Abstract
Background
Ziziphus hajarensis is an endemic plant species well-distributed in the Western Hajar mountains of Oman. Despite its potential medicinal uses, little is known regarding its genomic architecture, phylogenetic position, or evolution. Here we sequenced and analyzed the entire chloroplast (cp) genome of Z. hajarensis to understand its genetic organization, structure, and phylogenomic disposition among Rhamnaceae species.
Results
The results revealed the genome of Z. hajarensis cp comprised 162,162 bp and exhibited a typical quadripartite structure, with a large single copy (LSC) region of 895,67 bp, a small single copy (SSC) region of 19,597 bp and an inverted repeat (IR) regions of 26,499 bp. In addition, the cp genome of Z. hajarensis comprises 126 genes, including 82 protein-coding genes, eight rRNA genes, and 36 tRNA genes. Furthermore, the analysis revealed 208 microsatellites, 96.6% of which were mononucleotides. Similarly, a total of 140 repeats were identified, including 11 palindromic, 24 forward, 14 reverse, and 104 tandem repeats. The whole cp genome comparison of Z. hajarensis and nine other species from family Rhamnaceae showed an overall high degree of sequence similarity, with divergence among some intergenic spacers. Comparative phylogenetic analysis based on the complete cp genome, 66 shared genes and matK gene revealed that Z. hajarensis shares a clade with Z. jujuba and that the family Rhamnaceae is the closest family to Barbeyaceae and Elaeagnaceae.
Conclusion
All the genome features such as genome size, GC content, genome organization and gene order were highly conserved compared to the other related genomes. The whole cp genome of Z. hajarensis gives fascinating insights and valuable data that may be used to identify related species and reconstruct the phylogeny of the species.
Similar content being viewed by others


Background
Ziziphus Mill. is one of the medicinally and economically important genera of Family Rhamnaceae. It is disseminated across the sub-tropical and warm-climatic zones of the world. The members of the Ziziphus genus are shrubs (with thorns) and small trees, which vary in numbers as claimed by various reports, like 86 [1, 2], 135 [3] and 170 [4]. The plant list (2013) reports 58 accepted species [5]. Two members Z. mauritiana Lam (ber, Indian jujube) and Z. jujuba Mill (common jujube), are broadly domesticated and grown commercially across the globe. Ziziphus plants hold many medicinally important phytochemicals like phenols, flavonoids, alkaloids, saponins etc., to which may be attributed their medicinal importance and pharmacological activities [6].. Traditionally, the plant cultivars were differentiated based on morphology and pedigree-related information. However, the morphology of a plant is easily affected by environmental fluctuations and hence limits the approach [7]. Furthermore, the Rhamnaceae family has more than 900 species. The available genomic sequences are only of few member plants [8]; because of the diversity in views and limited genomic information available, the intra-generic classification is a difficult problem [9]. The history of the taxonomic relationships of Rhamnaceae was reported by various researchers [10,11,12]. Analyses using the rbcL gene [13] revealed that Elaeagnaceae and Rhamnaceae had a close relationship. Similarly, the relationship between Rhamnaceae and Elaeagnaceae has been established by studies using 18S nuclear ribosomal DNA, atpB, and rbcL sequence data [14, 15].
Z. hajarensis is new and endemic species to Oman. It can be found in Western Hajar (Jabel al Akhdar), which shares the habitat with Juniperux excela subsp. polycarpos, while in Eastern Hajar it can be found in open woodland with Caratonia oreothauma subsp. oreothauma and Prunus arabica. Z. hajarensis is a multi-stemmed shrub or tree with straight, equal-length, uniform spines, juvenile branches with a prominent zig-zag pattern, and dark green leaves with entire margins [16, 17]. Locals consume the fruit and kernels directly from the tree (the fruits persist for several months on the trees). Branch bark is peeled off and used like axes to lower and cut foliage for goat fodder. The identification of this species was performed by molecular studies using chloroplast DNA microsatellites [16, 17]. The identification of Z. hajarensis is in confusion with Z. spina-christi because they share similar morphological characteristics and that is why the taxonomy of Z. hajarensis remains indistinct [16, 17]. Furthermore, the phylogenetic relationships of Ziziphus genus have been controversial and the published data on taxonomic revision is limited. Likewise, little information is available on their genetic structure, especially their chloroplast genomes or precise phylogenetic placement.
The chloroplast is one of the critical cellular organelles in plants, and serves as a reaction site for many vital biochemical reactions. It participates in the biosynthesis of starch, lipid amino acids and pigments [18]. The chloroplast has its own genome, also known as the plastome. The normal chloroplast genome of an angiosperm is a double-helix-circular DNA molecule [19]. The chloroplast genome is quite conserved in gene content, number and organization. The chloroplast genome size may vary from plant to plant, varying from 120 to 160 kb. The chloroplast genome is divided into four parts, a large-single copy of 80 to 90 kb and a small-single copy of 16 to 27 kb, set apart by two inverted repeats of 20–28 kb [20]. It has 110 to 130 unique genes that codes for photosynthesis attributed proteins, transfer RNA and ribosomal RNA [21]. The variation in chloroplast genome size is because of the genes expansion and contraction and the loss of inverted repeats. These characteristics are vital in phylogenetic and evolutionary studies [8].
The significance of the chloroplast genome, such as its organization and its role in the evolution and phylogenetic studies, has recently gained attention. Thousands of cp genomes have been sequenced and reported in National Centre for Biotechnology Information (NCBI) database. The chloroplast genome is inherited maternally, and this uniparental inheritance is proved to be very convenient in deducing the evolutionary background, phylogeographic and phylogenetic studies of plants [8]. Next-generation sequencing (NGS) is cost-friendly, time-efficient, and high throughput, enabling the chloroplast genomes to be sequenced entirely. In the Ziziphus genus, five chloroplast genomes have been sequenced, including Z. jujuba, Z. acidojujuba, Z. incurva, Z. mauritiana, and Z. spina-christi [9]. To the best of our knowledge, we report the complete chloroplast genome of Z. hajarensis for the first time in the current study. Considering the taxonomic and phylogenetic complications for the genus Ziziphus and lack of concentrated evidence, here, we sequenced and performed a comparative analysis of the complete chloroplast genome of Z. hajarensis and compared it with nine related species from the family Rhamnaceae. We predicted their relationships through a comparative analysis with other Ziziphus species chloroplast genome sequences within phylogenetic clades. These results reshape our understanding of the evolution of the genus Ziziphus and their close relatives.
Results
General features and Organization of Chloroplast Genome
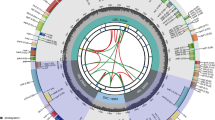
The chloroplast genome of the Z. hajarensis was 162,162 bp: among the largest found among the analyzed genomes (four from Ziziphus genus and five from sub-family members of Rhamnaceae). These genomes thus range in size from 154,962 bp (B. lineata) to 162,162 bp (Z. hajarensis) (Fig. 1, Table 1). The cp genome of Z. hajarensis is a typical circular molecule organized in 4 parts (quadripartite Structure), the two IR of 26,499 bp in size, contributing 16.34% to the genome size, that divides the rest of the genome sequences into a small single copy (SSC) of 19,597 bp and large single copy (LSC) of 89,567 bp, contributing 12.08 and 55.23% respectively. The total number of encoded genes present in the chloroplast genome of Z. hajarensis is 126, including 82 protein-coding, eight rRNA, and 36 tRNA genes (Fig. 1, Table 1). Among the protein-coding genes, 11 genes code for ribosomal proteins of small sub-unit (rps2, 4, 7, 8, 11, 12, 14, 15, 16, 18, and 19) eight genes code for large subunit proteins (rpl14, 16, 2, 20, 23, 32, 33, and 36), five genes codes for the components of photosystem I (psaA, B, C, I and J), 13 genes for photosystem II (psbA, B, C, D, E, F, I, J, K, M, N, Z, and ycf3), and five genes codes for ATP synthase proteins (atpA, B, E, F, H, and I; Table S1). Among these annotated genes there are 17 genes (trnK-UUU, rps16, trnT-CGU, atpF, rpoC1, trnL-UAA, trnC-ACA, accD, rpl2, ndhB, trnE-UUC, trnA-UGC, ndhA, trnA-UGC, trnE-UUC, ndhB and rpl2) contains a single intron while 2 genes (ycf3 and clpP) had 2 introns (Table 2). In the chloroplast genome of Z. hajarensis the protein-coding region is 72,917 bp in size and contributes 44.96%. Furthermore, tRNA and rRNA regions’ size was 2723 bp and 9048 bp and contributed to 1.67 and 5.57%, respectively. The overall GC content in the chloroplast genome of Z. hajarensis was found to be similar to other Ziziphus species (36.2%). In the chloroplast genome of Z. hajarensis, the most commonly occurring codon was ATT (n = 1701) encodes isoleucine followed by TTT (n = 1673) phenylalanine (Table S2). Contrastingly, the least common codon was TGA (n = 112).

Genome Map of the Z. hajarensis Chloroplast genome. The thick lines represent the length of inverted repeat regions (IRs). IRs divide the chloroplast genome into Small single copy (SSC) and Large single copy (LSC) regions. Genes drawn outside the circle are transcribed counter clockwise, while those inside the circle are transcribed clockwise. Functional groups of genes are color-coded. The dark grey in the inner circle corresponds to the GC content, while the light grey corresponds to the AT content
Genome insight, repeats and SSR analysis
Simple-sequence repeats (SSR) works as genetic markers in evolutionary studies and population genetics. SSR or microsatellites are sequences of 1–6 bp repeats. In this study, SSR analysis was performed for the Z. hajarensis chloroplast genome and nine other species from Rhamnaceae. The total identified SSR markers for each species falls between 190 to 223, including mono to hexanucleotides. In Z. hajarensis a total of 208 SSRs were identified, and majority are mono-nucleotides (96.6%), with two di-, four tri-, and one pentanucleotide repeat. Furthermore, the highest and lowest number of SSRs were identified in B. wilsonii (226) and Z. jujuba (183) with 94.6 and 97.3% of mono-nucleotides, respectively (Fig. 2). By exploring all four parts of Z. hajarensis chloroplast genome along with the coding and non-coding regions, the SSRs specified to each part and region have been identified. In the SSC and LSC regions, a total of 36 and 154 repeats have been detected, respectively. It is noteworthy that only one pentanucleotide repeat TTTTC was identified in Z. hajarensis specifically (Fig. 2). Furthermore, nine mono-nucleotide SSRs have been identified in IR regions of Z. hajarensis. Contrastingly, 30 mono-nucleotide SSRs were identified in the protein-coding region. Despite this, most SSRs were identified in intergenic regions in the genome. In the inter-genic spaces of the Z. hajarensis chloroplast genome, a total of 176 repeat sequences have been detected. Still, in other similar species, the number of repeats in inter-genic spaces ranges from 154 to 196 (Fig. S1).

Analysis of the simple sequence repeats (SSRs) in the chloroplast genome of Z. hajarensis and compared cp genomes of related species; A Total number of SSR repeats in genomes; B Frequency of the simple sequence repeat motif in the chloroplast genome of Z. hajarensis and compared cp genomes of related species; C Mono-nucleotides SSRs; D Di-nucleotides SSRs; E Tri-nucleotides SSRs; F Tetra-nucleotides SSRs; G Penta-nucleotides SSRs (H) Hexa-nucleotides SSRs
A total of 140 repeats were identified in the Z. hajarensis cp genome, including 11 palindromic, 24 forward, 14 reverse, and 104 tandem repeat sequences. Similarly, the lowest number of palindromic repeats and the highest number of reverse repeats were noted in the Z. hajarensis cp genome, 11 and 14, respectively (Fig. 3). In the chloroplast genome of Z. hajarensis, the lengthwise distribution of palindromic, forward, reverse. Tandem repeats were analysed in which the highest number (5 repeats each) of palindromic and forward repeats were recorded in size range of 21–40 bp, whereas the most number (9) of reverse repeats were identified in 41–60 bp size range. Similarly, for all other compared nine species, the most significant number of palindromic, forward, and reverse repeat sequences were identified in size range of 21–40 bp, as shown in Fig. 3. Furthermore, the maximum number (74) of tandem repeats were identified in range of 11–20 bp in Z. hajarensis and, similar results were observed in related species cp genomes (Fig. 3).

Analysis of the repetitive sequences in chloroplast genome of Z. hajarensis and related species. A A total number of repetitive sequences in cp genomes; B Lengthwise frequency of palindromic repeats (C) Lengthwise frequency of forward repeats (D) Lengthwise frequency of reverse repeats (E) Lengthwise frequency of tandem repeats
Sequence divergence analysis
A Z. hajarensis cp genome comparison with related species showed sequence variation in various regions. Z. hajarensis was selected as reference genome. The results showed high sequence similarities among these cp genomes, especially in protein-coding and IR regions. Variations were observed in the intergenic regions such as psbI-atpA, atpH-atpI, psbM-psbD, ycf3-rps4, ndhC-atpE, ndhF-rpl32, psbF-petG, rps15-trnN and rpl32-ccsA. In addition to these regions, some divergence was also observed in protein-coding genes such as matK, atpF, rpoC2, ycf3, rbcL, clpP, petB, ndhH, ycf2, and psaB (Fig. 4). The average pairwise sequence divergence was determined among the Z. hajarensis and other related chloroplast genomes (Table S3). The highest divergence 0.134 was found with B. lineata, while the lowest was found with Z. jujuba (0.0040). Moreover, among all the compared genes, the most divergent genes were atpF, ccsA, clpP, matK, ndhF, ndhH, petN, rbcL, rpl36 and rpoC2. The highest average pairwise divergence 1.408 was recorded for rbcL gene and followed by petN gene (0.427). Furthermore, the highest divergence 1.022 was observed among the Ziziphus species in rbcL genes. However, in Berchemia species, this rbcL gene showed the highest divergence (4.3) among all analyzed species cp genomes. (Table S4).

Visual alignment of chloroplast genomes of Z. hajarensis vs. previously reported chloroplast genomes of related species. VISTA-based identity plot showing sequence identities among 10 species, using Z. hajarensis as a reference. Genome regions are color-coded as protein coding, rRNA coding, tRNA coding, or conserved noncoding sequences (CNS). The x-axis represents the coordinate in the chloroplast genome. Annotated genes are displayed along the top. The sequences similarity of the aligned regions is shown as horizontal bars indicating the average percent identity between 50 and 100%
Expansion and contraction of IR regions
In angiosperms, the variations in chloroplast genome size result from the expansion or contraction of the IR/LSC or IR/SSC regions. In this study, an extensive evaluation and comparison of all the four IRa/LSC (JLA), IRb/LSC (JLB), IRa/SSC (JSA) and IRb/SSC (JSB) junctions of the chloroplast genomes of Z. hajarensis, with related species cp genomes were performed. The rps19 gene is located at JLB i.e., 172 bp in LSC and 107 bp in IRb (Fig. 5). This gene is also present at the exact location in Z. jujuba and Z. incurva, while a slight variation in Z. spina-christi and Z. mauritiana, 62 bp in LSC and 217 bp in IRb at JLB in both species. The ndhF gene is located 69 bp away from the JSB in SSC region. The trnN gene is located at 1461 bp from JSA in the IRa region.

Distances between adjacent genes and junctions of the small single-copy (SSC), large single-copy (LSC), and two inverted repeat (IR) regions among chloroplast genomes of Z. hajarensis and related species within the Rhamnaceae family. Boxes above and below the primary line indicate the adjacent border genes. The figure is not to scale regarding sequence length and only shows relative changes at or near the IR/SC borders
Furthermore, the trnH gene is located 3 bp away from JLA in the LSC region compared to Z. spina-christi and Z. mauritiana, which is 87 bp away from JLA in the LSC region. Like IR length variation was observed in genes present on IR borders with B. lineata cp genome. Similarly, the rpl22 gene is present in IRb region about 74 bp away from JLB junction. Similarly, the ndhF gene is present in SSC region about 12 bp away from JSB junction.
Phylogenetic relationships
Here, the phylogenetic position of Z. hajarensis within the order Rosales was established by multiple alignment analysis of the complete cp genome, 66 shared protein coding genes sequences and the matK gene of Rosales members representing 7 families and 15 genera (Fig. 6 and Fig. S2). Phylogenetic analysis was executed using four different methods i.e., ML (maximum likelihood), MP (maximum parsimony), NJ (Neighbour Joining) and BI (Bayesian inference). The phylogenetic trees constructed based on complete cp genomes, 66 shared genes (both nucleotides and proteins sequences) and matK gene of Z. hajarensis formed a clade with Z. jujuba via bootstrap and BI support. The phylogenetic analysis revealed that Z. hajarensis shares the monophyletic clade with Z. jujuba with high bootstrap values within the phylogenetic tree based on all of the above methods. Nonetheless, the Z. hajarensis and Z. jujuba shared a sub-clade with Z. 8ncurve. Additionally, based on the current findings, Ziziphus species were in monophyletic clade with Hovenia species, i.e. H. dulcis, H. trichocarpa and H. acerba. In the analyzed data sets, Barbeyaceae and Elaeagnaceae were found the nearest families with Rhamnaceae based on the complete cp genome, 66 shared genes and matK gene (Fig. 6 and Fig. S2).

Phylogenetic trees were constructed for 36 species from seven families representing 15 genera using different methods. The entire genome dataset was analysed using four different methods: Bayesian inference (BI), maximum parsimony (MP), neighbour joining (NJ) and maximum likelihood (ML). The branches above represent bootstrap values in the ML, NJ and MP, and posterior probabilities in the BI trees. Star represents the position of Z. hajarensis
Discussion
In this study, the chloroplast genome of Z. hajarensis was sequenced using Ion Torrent S5 sequencing methods and compared with the available chloroplast genomes from family Rhamnaceae. The Z. hajarensis cp genome shared a typical structure (quadripartite) arranged circularly, with one large and one small single copy (LSC, SSC) as well as two inverted repeats (IR) regions as reported previously in angiosperms [22, 23]. The cp genomes studied here were highly conserved as reported in other angiosperms [24]. The size of Z. hajarensis cp genome is in agreement with the already sequenced chloroplast genomes of Z. jujuba (160,920 bp) and other subfamily members [8]. Similarly, the IR size of Z. hajarensis is 26,499 bp which falls in the typical angiosperm size range (20–28 kb) of chloroplast genomes [25]. Considering the Similar sizes of IR, it is proposed that the contributing factor to the difference of genome sizes could be the variations in LSC region as evident by [23, 26]. Furthermore, in the chloroplast genome of Z. hajarensis, 19 genes were identified with introns (11 protein-coding genes and 8 tRNA genes). Similar to other angiosperm cp genomes, in Z. hajarensis these protein coding genes have two genes (ycf3 and clpP) with two introns (Table 2).
Sequences of repetitive nature have a crucial role in rearranging and providing stability to the chloroplast genome sequences and determines the variation in copy number in different and same species. Because of the variable copy number and variation in length, the SSRs have gained considerable importance in various studies like biogeographic and population genetics [27,28,29]. A total of 140 repeats were identified in the chloroplast genome of Z. hajarensis compared to 146, 151 and 154 repeats in Z. incurve, Z. jujuba and Z. mauritiana. These repeats in cp genomes play a pivotal role in genomic expansions or rearrangements and structural variation and stability [30,31,32]. In all the compared chloroplast genomes the highest and lowest number of repeat sequences were found in H. dulcis (165) and R. taquetii (85), respectively (Fig. 2). The number of forward and reverse sequences repeats found in Z. hajarensis was 11 and 14 respectively. Similarly, the forward sequence repeats were the lowest and reverse sequence repeats were the highest for Z. hajarensis compared to the other related species. The highest number of forward repeats found were noted in Z. mauritiana (25), while the lowest reverse repeats were found in Z. jujuba and R. taquetii (3). Similarly, the number of palindromic sequences found in Z. hajarensis were 11. The number of tandem repeat sequences recorded for Z. hajarensis was 104. The highest value of tandem repeats was noted for H. dulcis (116), followed by Z. spina-christi (107) and Z. jujuba (105). In contrast, the least tandem repeats were recorded for R. tanquetii (36). These repeats exhibit a similar pattern as reported previously [8, 9, 33, 34]. The complex repeats and high numbers are key components in studying the chloroplast genome evolution [35, 36].
SSRs (simple sequence repeats) hold several essential characteristics, including haploid nature, relative recombination, and maternal inheritance. Because of these features, SSRs are considered one of the valuable markers. They have been diversely employed in various studies like estimating genetic variation, gene flow analysis, and exploring animal and plant populations [37,38,39]. The significance and applicability of SSRs markers have been reported in various other Ziziphus species [9, 40, 41]. In the current study, the distribution and type of SSRs markers have been analysed in Z. hajarensis and related cp genomes. The number of SSRs identified in Z. hajarensis, Z. incurve, Z. jujuba and Z. mauritiana were 208, 98, 183, and 190, respectively. Mono-nucleotide chloroplast genome SSRs in a single copy region is responsible for the intra-species chloroplast genome variations [37]. The current study’s findings agree with previously reported findings. The SSRs in chloroplast genomes combine polyadenine or polythymine repeats containing tandem guanine or cytosine repeats [42], resulting in AT-rich chloroplast genomes [43, 44].
Like typical angiosperm cp genomes, the Z. hajarensis shared a high sequence with all analyzed species. However, some regions showed lower sequence similarity in these cp genomes. As reported previously, the sequence divergence recorded in the IR region was lower than LSC and SSC regions [25]. These results agree with previous reports that showed a higher sequence divergence because of copy correction for gene conservation in IR regions [45, 46]. The Z. hajarensis cp genome showed high sequencing divergence in various intergenic regions and genes like psbI-atpA, atpH-atpI, psbM-psbD, ycf3-rps4, matK, atpF, rpoC2, ycf3, rbcL and clpP (Fig. 4). The average pairwise sequence divergence was calculated and Z. hajarensis showed an average pairwise sequence divergence of 0.041with related species cp genomes. The highest divergence was observed with B. lineata. Similarly,the rbcL gene was found with the highest pairwise sequence divergence followed by petN gene. Comparable findings for these genes have previously been reported [26, 47, 48], and our results are supported by Yang et al. [49] suggesting that similar variations exist among different coding regions. These findings are also supported by earlier report that these divergent genes are primarily found in LSC regions and are evolving rapidly [47]. Extensive IRs play a significant role in maintaining the conserved structure and stability of chloroplast genome [50, 51]. Variations in length among the cp genomes were observed because of the expansion or contraction of IR regions [52, 53]. An IR copy was lost in the plastomes of tribes in the legume subfamily Papilionoideae [54] during the evolution of the angiosperms, and cp genome rearrangements are more common in these species compared with species possessing typical IRs [55]. We evaluated and compared all four IRa/LSC (JLA), IRb/LSC (JLB), IRa/SSC (JSA) and IRb/SSC (JSB) junctions of the cp genome of Z. hajarensis, with related species. The rps19 gene in Z. hajarensis, Z. jujuba and Z. incurva was found the same, which is 172 bp in LSC and 107 bp in IRb (JLB). In contrast, the same gene location is slightly different in Z. spina-christi and Z. mauritiana (Fig. 5). Similarly, variations were also noted with B. lineata in the location of rpl22 gene present on IR borders. Previous studies have revealed that there is an expansion of the IR and LSC regions in angiosperm plastomes during evolution [23, 56, 57].
Chloroplast genomes have been played a significant role in molecular, evolutionary and phylogenetic studies. Analyses based on complete chloroplast genome sequence comparison have solved numerous phylogenetic problems at the deep node level. They have contributed to understanding less known evolutionary associations between angiosperms [26, 58]. Several phylogenetic studies have been conducted on the subfamily Rhmnaceae and intra-generic within Ziziphus but could not address the problems related to the classification of Z. hajarensis based on ITS regions, SSRs regions, rbcL gene etc. [59,60,61]. On the other hand, complete genome sequencing provides more in-depth information [43, 45, 62]. The complete cp genome sequence of Z. hajarensis has been overlooked in this respect; the new dataset will give more detailed insights into the role of different genes, allowing for a better knowledge of the plant’s history. Chloroplast genomes have proven to be helpful in phylogenetic analyses and molecular and evolutionary systematics. In recent years, various studies based on the entire cp genome and compared with many protein-coding genes have been undertaken at deep nodes to solve phylogenetic problems [63, 64]. This approach allows a better understanding of the complex evolutionary links among angiosperms [58]. Therefore, in this study, the phylogenetic position of Z. hajarensis within Rhamnaceae and Rosales was established by utilizing the complete cp genomes, 66 shared genes proteins coding genes and matK gene among the members of 7 families representing 15 genera Four different methods, which are ML (maximum likelihood), MP (maximum parsimony), NJ (neighbor-joining) and BI (Bayesian inference), were used for phylogenetic analysis. The results revealed that complete cp sequences (Fig. 6), 66 shared genes (Fig. S2 A and matK gene (Fig. S2 B) from all the analysed species generated a phylogenetic tree with the same topology. In these phylogenetic trees (Fig. 6 & S2) constructed by employing ML, MP, NJ, and BI methods, Z. hajarensis formed a single clade with Z.jujuba with high bootstrap (100%) and BI support.
Furthermore, Barbeyaceae and Elaeagnaceae were the nearest families to the Rhamnaceae. Similar results were reported previously based on chloroplast rbcL and atpB genes where Elaeagnaceae and Rhamnaceae showed a close relationship [13]. Similarly, phylogenetic study based on plastid non-coding region revealed family Barbeyaceae and Dirachmaceaein in a close relationship with Rhamnaceae [14, 15]. These findings also suggest that Rhamnaceae germplasm-related genetic resources are important and valuable for Rhamnaceae species identification, phylogenetic inference, and taxonomy clarification. Furthermore, if plastid genomes are made accessible, phylogenetic inferences within Rosales and Rhamnaceae might be improved, potentially offering hundreds of useful molecular markers for future studies.
Conclusion
For the first time, the current findings provide comprehensive insights into the entire cp genome of Z. hajarensis. The structure and gene content of the Z. hajarensis cp genome was determined to be in synergy with similar Rhamnaceae species. We retrieved important genetic characteristics such as repetitive sequences, SSRs, codon use, IR contraction and expansion, sequence divergence, and phylogenomic position using thorough bioinformatic analysis and comparative assessments. Repetitive sequences like tandem repeats and SSRs were examined within these cp genomes. Overall, there was a significant sequence similarity amongst these cp genomes. However, these cp genomes had several divergent genes and intergenic regions, including psbI-atpA, atpH-atpI, psbM-psbD, matK, atpF rpoC2, ycf3, and rbcL. The current work presents a valuable set of complete chloroplast genome analyses of Z. hajarensis and related species, which might aid in species identification and biology, genetic diversity, and phylogenetic studies.
Methods
Sample collection
The fresh juvenile leaves were collected from Z. hajarensis plant growing in natural habitat of Jabal Al-Akhdar, Oman (23° 6′ 11.110″ N; 57° 22′ 47.14″ E). The climate of natural habitat is high in temperature and low in precipitation, with an average temperature of 25–46 °C. Permission (6210/10/73) to collect plants for research purposes was obtained from Ministry of Environment & Climate Affairs, Muscat Oman. The voucher specimen (UoN-ZH1) was deposited in the Herbarium Centre, University of Nizwa as plant was identified by lead taxonomist (Saif Al-Hathmi) at Oman Botanic Garden, Muscat Oman. Collected leaves were put in zipper bags, immediately kept in liquid nitrogen, and stored at − 80 °C for DNA extraction.
DNA extraction and sequencing
The chloroplast DNA of Z. hajarensis was extracted from its finely powdered leaves according to the protocol [65] with brief modifications. Genomic libraries were prepared according to the provided instructions (Life Technologies USA, Eugene, OR, USA). Ion Shear™ Plus Reagents kit and Ion Xpress™ Plus, gDNA Fragment Library kit, were used to arrange the chloroplast DNA into the 400 bp fragments enzymatically and construct libraries. The quantification of prepared libraries was performed by Qubit 3.0 fluorometer and bioanalyzer (Agilent 2100 Bioanalyzer system, Life Technologies USA) and followed by the amplification of template using Ion OneTouch™ 2. By Ion OneTouch™ ES enrichment system, the templates (amplified) were enriched using Ion 530 & 520 OT2 Reagents. For sequencing, Ion sample loading on S5 530 Chip was performed according to the Ion S5 protocol.
Genome assembly and annotation
A total of 1,315,423 raw reads were obtained for Z. hajarensis cp genome. The Z. jujuba was used as a reference genome for mapping the produced reads by Bowtie2 (v.2.2.3) [66] in Geneious Pro (v.10.2.3) software [67]. Assembly means coverage of Z. hajarensis. The annotation of the chloroplast genome of Z. hajarensis was performed by using CpGAVAS [68] and DOGMA [69] (http://dogma.ccbb.utexas.edu/, China). TRNAs can-SE (v.1.21) [70] software was utilized to detect tRNA genes. Furthermore, Geneious Pro (v.10.2.3) [67] and tRNAs can-SE (v.1.21) [70] were used for manual alteration and comparison of the genome (Z. hajarensis) with previously published Z. jujuba genome and intron boundaries. These tools were also utilized to adjust the start and stop codons manually. Illustration of chloroplast genome OGDRAW [71] was used for structural features. Moreover, genome divergence was determined by mVISTA [72] in shuffle-LAGAN mode, while Z. hajarensis chloroplast genome was chosen as the reference genome.
Repeat identification
Forward reverse and palindromic repeats were identified on the REPuter online tool [73] with a minimum repeat size of eight bp and maximum computed repeats of 50. Similarly, SSRs were identified using MISA Software [74] with requirements set to ≥10 repeat units for one bp repeats; ≥8 repeat units for two bp repeats; ≥4 repeat units for 3 and 4 bp repeats, and ≥ three repeat units for 5 and 6 bp repeats. Tandem Repeats Finder v.4.09 [75] online tool was used to identify the tandem repeats.
Genome divergence and phylogenetic analysis
The sequence divergence in the complete chloroplast genome and shared genes were determined among the Ziziphus and closely related species. After multiple sequence alignment, a comparative analysis strategy was used to compare gene order to identify the ambiguous and missing gene annotation. MAFFT version 7.222 [76] was used to align complete chloroplast genomes using default settings. Pairwise sequence divergence was determined using Kimura’s two parameter model (K2P) [76]. The phylogenetic position of Z. hajarensis within the order Rosales and sub-family Rhamnaceae was inferred using 36 chloroplast genome sequences retrieved from the NCBI database. First the complete chloroplast genomes were aligned based on chloroplast genome structures and conserved gene orders [77]. The phylogenetic tree was inferred on four different methods: Bayesian inference (BI), implemented in Mr. Bayes 3.1.2 [78], maximum parsimony (MP) using PAUP 4.0 [79], and maximum likelihood (ML) using MEGA 6 [80], using already described settings [25, 45]. The GTR + G substitution model was tested via jModel Test version v2.1.02 [81] as per AIC (Akaike information criterion) for Bayesian posterior probabilities in BI analysis. The Markov Chain Monte Carlo (MCMC) method was run using four incrementally heated chains across 1,000,000 generations, starting from random trees and sampling 1 out of every 100 generations. The first 30% values were considered as burn-in and discarded. In Maximum Parsimony run heuristic search was used with random addition of 1000 sequence replicates with the tree-bisection-reconnection (TBR) branch-swapping tree search criterion to estimate the posterior probabilities.
Furthermore, for ML run, the parameters were optimized by BIONJ tree [82] as the starting tree with 1000 bootstrap replicates by employing the Kimura 2-parameter model with invariant sites gamma-distributed rate heterogeneity. A set of 66 shared genes and matK gene from the plastome genomes of 36 Rosales species were aligned using MAFFT version 7.22294 under default parameters and made various manual adjustments to preserve and improve reading frames in the second and third tiers of phylogenetic analysis. The above four phylogenetic inference models (ML, MP, NJ, and BI) were used to construct trees utilizing 66 concatenated genes, matK gene, and 66 concatenated protein sequences as reported previously [83].
Availability of data and materials
All data generated or analysed during this study are included in this article. The Ziziphus hajarensis genome deposited to NCBI (MZ475300), https://www.ncbi.nlm.nih.gov/nuccore/MZ475300.
References
Evreinoff V. Notes sur le Jujubier (Zizyphus sativa G.). J Agricult Trad Botan Appliquée. 1964;11(5):177–87.
Polhill RM, et al. Flora of tropical east Africa. London: CRC Press; 2020.
Bhansali A. Monographic study of the family Rhamnaceae of India. Jodhpur: University of Jodhpur; 1975.
Liu M, Cheng C. A taxonomic study on the genus Ziziphus. in Internat. Symposium on Medicinal and Aromatic Plants 390; 1994.
El Maaiden E, et al. Genus Ziziphus: a comprehensive review on ethnopharmacological, phytochemical and pharmacological properties. J Ethnopharmacol. 2020;259:112950.
Medan D, Schirarend C. Rhamnaceae. In: Flowering plants· Dicotyledons. London: Springer; 2004. p. 320–38.
Xu C, et al. Identifying the genetic diversity, genetic structure and a core collection of Ziziphus jujuba Mill. Var. jujuba accessions using microsatellite markers. Sci Rep. 2016;6(1):1–11.
Ma Q, et al. Complete chloroplast genome sequence of a major economic species, Ziziphus jujuba (Rhamnaceae). Curr Genet. 2017;63(1):117–29.
Huang J, Chen R, Li X. Comparative analysis of the complete chloroplast genome of four known Ziziphus species. Genes. 2017;8(12):340.
Takhtajan AL. Outline of the classification of flowering plants (Magnoliophyta). Bot Rev. 1980;46(3):225–359.
Cronquist A. The evolution and classification of flowering plants. The evolution and classification of flowering plants; 1968.
Thorne RF. Classification and geography of the flowering plants. Bot Rev. 1992;58(3):225–327.
Soltis DE, et al. Chloroplast gene sequence data suggest a single origin of the predisposition for symbiotic nitrogen fixation in angiosperms. Proc Natl Acad Sci. 1995;92(7):2647–51.
Soltis DE, et al. Angiosperm phylogeny inferred from 18S ribosomal DNA sequences. Ann Mo Bot Gard. 1997;84 (1):1–49.
Savolainen V, et al. Phylogenetics of flowering plants based on combined analysis of plastid atpB and rbcL gene sequences. Syst Biol. 2000;49(2):306–62.
Duling DW, Ghazanfar SA, Prendergast HD. A new species of Ziziphus mill.(Rhamnaceae) from Oman. Kew Bull. 1998;53(3):733–9.
Rohlf FJ. NTSYSpc numerical taxonomy and multivariate analysis system version 2.0 user guide; 1998.
Kirchhoff H. Chloroplast ultrastructure in plants. New Phytol. 2019;223(2):565–74.
Cheng Y, et al. Complete chloroplast genome sequence of Hibiscus cannabinus and comparative analysis of the Malvaceae family. Front Genet. 2020;11:227.
Ozeki H, et al. The chloroplast genome of plants: a unique origin. Genome. 1989;31(1):169–74.
Wang W, Lanfear R. Long-reads reveal that the chloroplast genome exists in two distinct versions in most plants. Genome Biol Evol. 2019;11(12):3372–81.
Khan A, et al. Comparative chloroplast genomics of endangered Euphorbia species: insights into hotspot divergence, repetitive sequence variation, and phylogeny. Plants. 2020;9(2):199.
Khan A, et al. First complete chloroplast genomics and comparative phylogenetic analysis of Commiphora gileadensis and C. foliacea: myrrh producing trees. PLoS One. 2019;14(1):e0208511.
Chumley TW, et al. The complete chloroplast genome sequence of Pelargonium× hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol. 2006;23(11):2175–90.
Asaf S, et al. Unraveling the chloroplast genomes of two prosopis species to identify its genomic information, comparative analyses and phylogenetic relationship. Int J Mol Sci. 2020;21(9):3280.
Asaf S, et al. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front Plant Sci. 2016;7:843.
do Nascimento Vieira L, et al. The complete chloroplast genome sequence of Podocarpus lambertii: genome structure, evolutionary aspects, gene content and SSR detection. PLoS One. 2014;9(3):e90618.
Piovani P, et al. Conservation genetics of small relic populations of silver fir (Abies alba mill.) in the northern Apennines. Plant Biosystems. 2010;144(3):683–91.
Asaf S, et al. Complete chloroplast genomes of Vachellia nilotica and Senegalia senegal: comparative genomics and Phylogenomic placement in a new generic system. PLoS One. 2019;14(11):e0225469.
Cho K-S, et al. Complete chloroplast genome sequences of Solanum commersonii and its application to chloroplast genotype in somatic hybrids with Solanum tuberosum. Plant Cell Rep. 2016;35(10):2113–23.
Williams AV, et al. Integration of complete chloroplast genome sequences with small amplicon datasets improves phylogenetic resolution in Acacia. Mol Phylogenet Evol. 2016;96:1–8.
Khan A, et al. Complete chloroplast genomes of medicinally important Teucrium species and comparative analyses with related species from Lamiaceae. PeerJ. 2019;7:e7260.
Wang Y, et al. The complete chloroplast genome sequence of Ziziphus incurva. Mitochondrial DNA Part B. 2019;4(2):3465–6.
Zhang L, et al. Characterization of the complete chloroplast genome of Hovenia acerba (Rhamnaceae). Mitochondrial DNA Part B. 2020;5(1):934–5.
Milligan BG, Hampton JN, Palmer JD. Dispersed repeats and structural reorganization in subclover chloroplast DNA. Mol Biol Evol. 1989;6(4):355–68.
Asaf S, et al. Complete chloroplast genome characterization of oxalis Corniculata and its comparison with related species from family Oxalidaceae. Plants. 2020;9(8):928.
Ebert D, Peakall R. Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol Ecol Resour. 2009;9(3):673–90.
Addisalem A, et al. Genomic sequencing and microsatellite marker development for Boswellia papyrifera, an economically important but threatened tree native to dry tropical forests. AoB Plants. 2015;7:plu086.
Kalia RK, et al. Microsatellite markers: an overview of the recent progress in plants. Euphytica. 2011;177(3):309–34.
Zhang Y, et al. Chloroplast genome sequence of the wild Ziziphus jujuba Mill. Var. spinosa from North China. Mitochondrial DNA Part B. 2021;6(2):666–7.
Huang J, et al. Development of chloroplast microsatellite markers and analysis of chloroplast diversity in Chinese jujube (Ziziphus jujuba mill.) and wild jujube (Ziziphus acidojujuba mill.). PLoS One. 2015;10(9):e0134519.
Yi X, et al. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol Evol. 2013;5(4):688–98.
Asaf S, et al. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front Plant Sci. 2017;8:304.
Kuang D-Y, et al. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome. 2011;54(8):663–73.
Asaf S, et al. Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: structures and comparative analysis. Sci Rep. 2017;7(1):1–15.
Qian J, et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One. 2013;8(2):e57607.
Chen J, et al. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front Plant Sci. 2015;6:447.
Asaf S, et al. Mangrove tree (Avicennia marina): insight into chloroplast genome evolutionary divergence and its comparison with related species from family Acanthaceae. Sci Rep. 2021;11(1):1–15.
Yang M, et al. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS One. 2010;5(9):e12762.
Palmer JD, Thompson WF. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell. 1982;29(2):537–50.
Zhang Y, et al. The complete chloroplast genome sequence of Taxus chinensis var. mairei (Taxaceae): loss of an inverted repeat region and comparative analysis with related species. Gene. 2014;540(2):201–9.
Raubeson LA, et al. Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics. 2007;8(1):1–27.
Yao X, et al. The first complete chloroplast genome sequences in Actinidiaceae: genome structure and comparative analysis. PLoS One. 2015;10(6):e0129347.
Lavin M, Doyle JJ, Palmer JD. Evolutionary significance of the loss of the chloroplast-DNA inverted repeat in the Leguminosae subfamily Papilionoideae. Evolution. 1990;44(2):390–402.
Hirao T, et al. Complete nucleotide sequence of the Cryptomeria japonica D. Don. Chloroplast genome and comparative chloroplast genomics: diversified genomic structure of coniferous species. BMC Plant Biol. 2008;8(1):1–20.
Zhu A, et al. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016;209(4):1747–56.
Saeed S, et al. Phylogenetics of selected Plantago species on the basis of rps14 chloroplast gene. J Med Plant Res. 2011;5(19):4888–91.
Hohmann N, et al. Taming the wild: resolving the gene pools of non-model Arabidopsis lineages. BMC Evol Biol. 2014;14(1):1–21.
Islam MB, Simmons MP. A thorny dilemma: testing alternative intrageneric classifications within Ziziphus (Rhamnaceae). Syst Bot. 2006;31(4):826–42.
Kellermann J, Udovicic F, Ladiges PY. Phylogenetic analysis and generic limits of the tribe Pomaderreae (Rhamnaceae) using internal transcribed spacer DNA sequences. Taxon. 2005;54(3):619–31.
Richardson JE, et al. A phylogenetic analysis of Rhamnaceae using rbcL and trnL-F plastid DNA sequences. Am J Bot. 2000;87(9):1309–24.
Wambugu PW, et al. Relationships of wild and domesticated Rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 2015;5(1):1–9.
Goremykin VV, et al. The chloroplast genome of Nymphaea alba: whole-genome analyses and the problem of identifying the most basal angiosperm. Mol Biol Evol. 2004;21(7):1445–54.
Moore MJ, et al. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc Natl Acad Sci. 2010;107(10):4623–8.
Shi C, et al. An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS One. 2012;7(2):e31468.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357.
Kearse M, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–9.
Liu C, et al. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics. 2012;13(1):1–7.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–5.
Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33(suppl_2):W686–9.
Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52(5–6):267–74.
Frazer KA, et al. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32(suppl_2):W273–9.
Kurtz S, et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29(22):4633–42.
Beier S, et al. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33(16):2583–5.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80.
Katoh K, Toh H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics. 2010;26(15):1899–900.
Wicke S, et al. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3):273–97.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–4.
Swofford DL. Phylogenetic analysis using parsimony (* and other methods); 2002.
Kumar S, et al. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008;9(4):299–306.
Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25(7):1253–6.
Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14(7):685–95.
Asaf S, et al. Expanded inverted repeat region with large scale inversion in the first complete plastid genome sequence of Plantago ovata. Sci Rep. 2020;10(1):1–16.
Acknowledgments
The authors are thankful to OAPGRC (Oman’s Animal and Plant Genetic Resource Centre) for their support and Mr. Arif Khan’s initial experimental work.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
‘ALK’ and ‘SA’ performed experiments; ‘ALK’, ‘SA’ and ‘WA’ wrote the original draft and Bioinformatics analysis: ALK and AH supervision and arranging resources. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The plants (Z. hajarensis) samples were collected from the study area through official Permission (6210/10/73) from Ministry of Environment & Climate Affairs, Muscat-Oman. A voucher specimen of Z. hajarensis (UoN-ZH1) was also deposited in the Herbarium Centre at University of Nizwa.
Consent for publication
Not applicable.
Competing interests
The authors have declared that no competing interests exist.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Analysis of the simple sequence repeats (SSRs) in the chloroplast genome of Z. hajarensis and compared cp genomes of related species. (A) Frequency of SSRs in coding and intergenic regions (B) Frequency of SSRs in inverted repeats (IR) small single copy (SSC) and large single copy (LSC) regions. Figure S2. Phylogenetic trees were constructed for 36 species from seven families representing 15 genera using different methods and the tree is shown for 66 protein coding shared genes (A) and matK (B) data sets. These sequences data sets were used with four different methods: Bayesian inference (BI), maximum parsimony (MP), neighbour joining (NJ) and maximum likelihood (ML). The branches above represent bootstrap values in the ML, NJ and MP, and posterior probabilities in the BI trees. Table S1. Gene composition in Z. hajarensis chloroplast genome.
Additional file 2: Table S2.
Codon Usage in Z. hajarensis chloroplast genome.
Additional file 3: Table S3.
Average pairwise distance of complete chloroplast sequence from Z. hajarensis with related species.
Additional file 4: Table S4.
Average pairwise distance of chloroplast shared genes from Z. hajarensis with related species.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Asaf, S., Ahmad, W., Al-Harrasi, A. et al. Uncovering the first complete plastome genomics, comparative analyses, and phylogenetic dispositions of endemic medicinal plant Ziziphus hajarensis (Rhamnaceae). BMC Genomics 23, 83 (2022). https://doi.org/10.1186/s12864-022-08320-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08320-2