
Abstract
Background
Sanghuangporus sanghuang is a well-known traditional medicinal mushroom associated with mulberry. Despite the properties of this mushroom being known for many years, the regulatory mechanisms of bioactive compound biosynthesis in this medicinal mushroom are still unclear. Lysine malonylation is a posttranslational modification that has many critical functions in various aspects of cell metabolism. However, at present we do not know its role in S. sanghuang. In this study, a global investigation of the lysine malonylome in S. sanghuang was therefore carried out.
Results
In total, 714 malonyl modification sites were matched to 255 different proteins. The analysis indicated that malonyl modifications were involved in a wide range of cellular functions and displayed a distinct subcellular localization. Bioinformatics analysis indicated that malonylated proteins were engaged in different metabolic pathways, including glyoxylate and dicarboxylate metabolism, glycolysis/gluconeogenesis, and the tricarboxylic acid (TCA) cycle. Notably, a total of 26 enzymes related to triterpene and polysaccharide biosynthesis were found to be malonylated, indicating an indispensable role of lysine malonylation in bioactive compound biosynthesis in S. sanghuang.
Conclusions
These findings suggest that malonylation is associated with many metabolic pathways, particularly the metabolism of the bioactive compounds triterpene and polysaccharide. This paper represents the first comprehensive survey of malonylation in S. sanghuang and provides important data for further study on the physiological function of lysine malonylation in S. sanghuang and other medicinal mushrooms.
Similar content being viewed by others


Background
Sanghuangporus sanghuang (Hymenochaetaceae, Basidiomycota), an herbal mushroom, has been used for more than 2000 years in China. It was previously mistaken for Inonotus linteus or Inonotus baumii for a long time. In 2012, it was identified as a new species Inonotus sanghuang and in 2016, it was renamed S. sanghuang [1, 2].
Extensive work has shown that S. sanghuang has a diverse range of biological activities [3,4,5,6]. The active compounds that play a major role in this medicinal fungus are triterpenoids and polysaccharides. However, the mechanism underlying the regulation of bioactive compound biosynthesis in S. sanghuang is still unclear.
Posttranslational modifications (PTMs) play a pivotal role in modulating different cellular pathways and disease processes, and over 400 distinct forms of PTMs have been found [7, 8]. Lysine malonylation is an evolutionarily conserved PTM. Malonylation has been reported to use malonyl-CoA as a substrate in protein modification [9]. However, we still know little about the enzymes that regulate the malonylation state of proteins [10]. To date, with advances in high-throughput experimental techniques, thousands of malonylated proteins have been discovered. These malonylated proteins have been found to be located in chloroplasts, the mitochondria, the cytoplasm, and the nucleus [10,11,12,13,14,15], suggesting that lysine malonylation is regulated in diverse metabolic processes.
Although malonoyl modifications have been studied in many species, few studies have focused on the mushroom malonylome. Similar to the effects in other organisms, such as mammals, plants, and bacteria [15, 16], we speculated that lysine malonylation may affect various metabolic processes in S. sanghuang. To demonstrate this hypothesis, we conducted a proteomics study of malonylated proteins in S. sanghuang. The results of this study provide a comprehensive view of the regulation of lysine malonylation in a wide range of biological processes, particularly in the biosynthesis of bioactive metabolites and secondary metabolites.
Methods
Fungal strain
The S. sanghuang CGMCC NO.21068 mycelia used in this study were isolated from fruit bodies collected from the mountainous area of Anshun city, Guizhou Province, China. The specimen was deposited in the Mycological Herbarium, Qingdao Agricultural University (HMQAU), Qingdao. In this study, the strain was stored at 4 °C in solid medium slants composed of 20 g/l bran, 30 g/l corn, 30 g/l glucose (catalogue #A501991, Sangon Biotech, China), 1 g/l KH2PO4 (catalogue #A100781, Sangon Biotech, China), 0.5 g/l MgSO4.7H2O (catalogue #A500864, Sangon Biotech, China), 4 g/l yeast extract (catalogue #A100850, Sangon Biotech, China), 3 g/l peptone (catalogue #A505247, Sangon Biotech, China) and 20 g/l agar (catalogue #A100637, Sangon Biotech, China). The strain was incubated on liquid medium (the medium was prepared as the solid medium mentioned-above but without the agar) at 26 °C and 150 rpm for 7 d. Then, the fermentation broth was filtered to collect the mycelia, flash frozen in liquid nitrogen, and stored at − 80 °C to be used for lysine malonylation analysis.
Morphological and molecular identification of the S. sanghuang strain was performed according to a previous study. The microscopic characteristics were studied under a Zeiss/Axioscope A1 microscope at magnifications of up to 1000×. The macroscopic and microscopic morphological characteristics were consistent with previous studies [17,18,19]. Phylogenetic analysis based on ITS sequence also confirmed that the strain was S. sanghuang [19, 20].
Protein extraction and trypsin digestion
Protein extraction was performed as previously described [7, 10]. Briefly, the tissue samples were ground to powder in a precooled mortar with liquid nitrogen. A fourfold volume of extraction buffer containing 10 mM dithiothreitol (catalogue #D9163-5G, Sigma-Aldrich, USA), 1% protease inhibitor (catalogue #524633-1ML, Calbiochem, Merck, USA), 3 μM trichostatin A (catalogue #58880–19-6, Sigma-Aldrich, USA), and 50 mM nicotinamide (catalogue #N3376-100 g, Sigma-Aldrich, USA) was added and the cells were lysed by sonication using a ultrasonic processor (catalogue #JY92-N, Scientz, Ningbo, China) as previously described [10]. Equivalent volumes of Tris-equilibrated phenol were added. After centrifugation at 5500×g and 4 °C for 10 min (catalogue #5424R, Eppendorf, Germany), the supernatants were collected and sedimented overnight with a 5-fold volume of 0.1 M ammonium acetate (catalogue #73594, Sigma-Aldrich, USA), and the protein precipitate was washed with methanol (catalogue #34860, Sigma-Aldrich, USA) and acetone (catalogue #270725, Sigma-Aldrich, USA). Then, 8 M urea (catalogue #V900119-500G, Sigma-Aldrich, USA) was redissolved for precipitation. The protein concentration was measured using bicinchoninic acid (BCA) kits (catalogue #P0011–1, Beyotime Biotechnology, China). Finally, the extracted proteins were digested by trypsin (catalogue #V5111, Promega, Madison, USA) as according to previously described procedures [7, 10].
HPLC fractionation and affinity enrichment
High-pH reverse HPLC fractionation was used for peptides on an Agilent 300 Extend C18 column (5 μm, 4.6 mm, 250 mm) (Agilent, Santa Clara, USA). The operation was performed as follows: sterilized peptide fractions were isolated in a gradient between 8 and 32% acetonitrile (catalogue #A998–4, Fisher Chemical, USA) (pH 9) for 60 min. They were merged into 4 fractions and freeze-dried under vacuum. The polypeptides were dissolved in IP buffer (100 mM NaCl (catalogue #S3014, Sigma-Aldrich, USA), 1 mM EDTA (catalogue #V900081-500 g, Sigma-Aldrich, USA), 50 mM Tris-HCl (catalogue #V900483-500G, Sigma-Aldrich, USA), and 0.5% NP-40 (catalogue #18896, Sigma-Aldrich, USA), pH 8.0) [10]. The polypeptides were incubated with pan anti-malonyllysine antibody conjugated agarose beads (catalogue #PTM-904, PTM Biotech, China) at 4 °C overnight. Finally, the bound peptides on the agarose beads were eluted three times with 0.1% trifluoroacetic acid (catalogue #58880–19-6, Sigma-Aldrich, USA), followed by desalting using C18 ZipTips (catalogue #Z720046, Merck Millipore, USA) [21,22,23].
LC-MS/MS analysis
The obtained peptides were dissolved in 0.1% formic acid (catalogue #56302-50ML-F, Sigma-Aldrich, USA) and separated by ultrahigh-performance liquid chromatography (UHPLC) using an EASY-nLC 1000 (Thermo Scientific, USA). Mobile phase A consisted of 0.1% formic acid (catalogue #56302-50ML-F, Sigma-Aldrich, USA) and 2% acetonitrile (catalogue #A998–4, Fisher Chemical, USA). Mobile phase B consisted of 0.1% formic acid and 90% acetonitrile. The liquid phase gradient was set as follows: 0–20 min, 7–25% B; 20–34 min, 25–38% B; 34–37 min, 38–80% B; and 37–40 min, 80% B, with a flow rate of 500 nL/min.
After HPLC separation, the peptides were injected into a nanospray ionization (NSI) ion source for ionization and mass spectrometry (MS) analysis by a Q Exactive Plus instrument (Thermo Fisher Scientific, USA) [10]. The ion source voltage was set to 2.2 kV. The primary MS scanning range was 350–1800 m/z, and the secondary MS scanning range was 100.0 m/z. Data collection was performed using the data-dependent acquisition (DDA) procedure. The automatic gain control (AGC) was set to 5e4 [24], The dynamic rejection time was set to 15 s to avoid repeated scanning, the parameter threshold was set to 5e3 ions/s, and the maximum injection time was set to 200 ms.
Database search
The obtained MS/MS data were analysed with MaxQuant software [25]. The S. sanghuang database was used (transcriptome, 23,290 sequences). Reverse libraries were added to calculate the false discovery rate (FDR), and contamination libraries were added to eliminate the effects of contaminating proteins. Trypsin/P was applied as the cleavage enzyme and the number of missed cleavages was set to 4 [10]. The first search and main search primary parent ion mass error tolerance was set to 20 ppm and 5 ppm, respectively. Cysteine alkylation was set as the fixed modification, and the variable modifications were acetylation of the protein N-terminus, deamidation of aspartyl/glutamyl groups, and malonylation of lysine. All the FDRs were set to 1% [26].
Bioinformatics analyses
The Gene Ontology (GO) annotations of the proteins were classified into the biological process, cellular component, and molecular function categories [27]. The GO annotations of the malonylated proteins were from the UniProt-Gene Ontology Annotation (GOA) database (http://www.ebi.ac.uk/GOA/) [28]. InterProScan was used to annotate the domain functional domain descriptions of the malonylated proteins [29, 30]. The metabolic pathways associated with the modified proteins were analysed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.genome.jp/kegg/). The subcellular localizations of the identified proteins were annotated using WoLF PSORT [31]. The significance of malonylated protein enrichment was measured by Fisher’s exact (two-sided) test and p value < 0.05 was considered to be significant [32,33,34,35,36]. To investigate the protein-protein interaction (PPI) network, all the modified proteins were searched against the STRING database [37]. Then, the visualization of the PPI network from STRING was presented with the R package “networkD3” [38, 39].
Results
Proteome-scale analysis of malonylated proteins in S. sanghuang
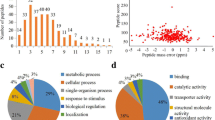
In this project, a range of technologies, such as HPLC, malonylation peptide enrichment and MS-based proteomics technologies, were combined for qualitative proteomics of malonylation in S. sanghuang (Fig. 1a). The results showed that the peptide score was between − 10 and 10 (Fig. 1b). The tolerance of the peptides was in a reasonable range. The distribution of the identified peptide lengths was examined, and the lengths of most peptides were between 7 and 22 (Fig. 1c), meeting the requirements of proteomic analysis. The MS results of malonylated peptides are summarized in Additional file 1: Fig. S1. Finally, 713 malonyl-modified sites matched to 255 different proteins were identified in S. sanghuang (Additional file 2: Table S1). Among them, many were related to triterpene synthesis. Farnesyl pyrophosphate synthases (FPPs), which are are pivotal enzymes in the main pathway of triterpene synthesis, were found to be malonylated, indicating that lysine malonylation was involved in bioactive compound biosynthesis.

Analysis of malonylated sites in S. sanghuang. a Technology roadmap in this study. b Mass distribution of error for all malonyl peptides c Length distribution of modified peptides
Pattern analysis of malonylated sites
To evaluate the distribution of malonylation sites in S. sanghuang, the number of identified modification sites was calculated for each protein. As shown in Fig. 2a, 47% of the proteins had one malonylation site, while only 18, 7, 12, 3, and 13% of the proteins contained 2, 3, 4, 5, and 6 or more modification sites, respectively. It has been documented that modification is prioritized at specific lysine sites (Additional file 2: Table S3) [10]. Therefore, the compositional frequencies of the amino acids surrounding malonyl lysine were examined. As shown in Fig. 2b, lysine (K) had the highest frequencies in the − 10 to + 10 position, whereas arginine (R) and glutamate (E) had the lowest frequencies. Hence, proteins with this group are the preferred substrates for malonyltransferases in S. sanghuang. Consistent with the results of the motif enrichment heatmap (Fig. 2b), only one motif was detected (Fig. 2c,d). To elucidate the secondary structure of proteins and the correlation between modified lysines, the secondary structures of all the malonylated proteins in S. sanghuang were examined (Fig. 2e). More malonylation sites were located more in the coiled-coil regions (p = 0.18) than in the α-helical (p = 0.01) and β-strand (p = 0.48) regions, suggesting that malonylation may favour the disordered structures of S. sanghuang. In addition, we assessed the surface accessibility of malonylated lysine sites and found that 39.62% of the unmodified lysine residues were located on the protein surface, compared to 39.54% of the modified lysine sites (Fig. 2f). As such, the protein’s surface accessibility may be influenced by lysine malonylation.

Characterization of malonylation sites. a Pie chart of the percentage and number of malonylated residues in the protein. b The frequency heat map of amino acid composition around malonylation. c Conservatism of malonylation sites. d Number of malonylation motif peptides. K indicates modified lysine sites and * denotes random amino acid sites. e Predictive analysis of the secondary structure of malonyl proteins. The Y-axis indicates the average probability of secondary structure, and the larger the value of Y, the higher the probability of this secondary structure configuration. f Peptides surface accessibility of malonylation sites
Functional annotation and cellular localization of malonylated proteins in S. sanghuang
For better comprehension of the malonylated proteins in S. sanghuang and their corresponding biological processes and molecular functions, we annotated and classified the identified proteins. GO analysis showed that the malonylated proteins had extensive activity in molecular functions and biological processes in S. sanghuang. The most abundant group of malonylated proteins in the biological process category consisted of enzymes related to metabolism (53%) (Fig. 3a). The majority of the malonylated proteins were associated with organocyclic compound binding (15%), heterocyclic compound binding (15%) and structural constituent of ribosome (10%) within the molecular functional classification (Fig. 3b). Characterization of the subcellular localization of the malonylated proteins showed that the modified proteins were found in the cytoplasm (36%), mitochondria (31%), and nucleus (21%) (Fig. 3c). These observations show that malonylated proteins have multiple functions and are widely present in S. sanghuang.

Functional classification of malonylated proteins in S. sanghuang. Each pie chart shows the percentage of malonylated proteins in each category. The GO annotation classifies proteins according to their biological processes and molecular functions. a Classification of malonylated proteins based on biological process b Classification of malonylated proteins based on molecular function. c Subcellular localization of the malonylated proteins
Functional enrichment analysis of malonylated proteins
To further analyse the proteins and their functions, we performed functional enrichment analysis of the obtained malonylome by GO, KEGG pathway and protein domain analyses (Additional file 2: Table S5, Additional file 2: Table S6). Proteins associated with structural components of the ribosome were highly enriched by functional analysis of GO terms (Additional file 2: Table S4). Based on GO cellular component classification, proteins located in the ribosomal subunit, ribosome, large ribosomal subunit, small ribosomal subunit, and cytosol were more likely to be malonylated (Additional file 1: Fig. S2). Domain enrichment studies indicated that these proteins were the core histone H2A/H2B/H3/H4, proteasome, beta-ketoacyl synthase, 1-cys peroxiredoxin, acyl transferase domain, isocitrate/isopropyl malate dehydrogenase, and oxidoreductase flavin adenine dinucleotide (FAD)-binding domain proteins (Additional file 1: Fig. S3). These enriched domains play a crucial role in glycolysis, polysaccharide synthesis and the tricarboxylic acid (TCA) cycle in S. sanghuang. To probe the process of malonylation regulation, we further performed enrichment analysis of proteins corresponding to malonylation modification sites in KEGG pathways (Fig. 4). Several pathways of the enriched proteins in the ribosome, glucuronide and dicarboxylic acid metabolism, TCA cycle, glycolysis/gluconeogenesis and pyruvate metabolism pathways were enriched. In conclusion, malonylated proteins were enriched in several types of proteins and pathways, suggesting a pivotal role of lysine malonylation in the metabolism of S. sanghuang.

Enrichment bubble plot of S. sanghuang proteins corresponding to modification sites in the KEGG pathways
PPI network of malonylated proteins in S. sanghuang
To determine how the identified proteins were associated with multiple pathways, a PPI network was constructed. Ninety proteins were identified in the PPI database (Fig. 5, Additional file 2: Table S7), presenting a global view of how the identified malonyl proteins are involved in multiple pathways in S. sanghuang. Analysis of the STRING PPI network with Cytoscape identified three strongly correlated clusters of malonylated proteins, including those associated with ribosomes, metabolic pathways, and the biosynthesis of secondary metabolites in S. sanghuang. Overall, we conclude that malonylation is a critical PTM for proteins in S. sanghuang and helps in interactions and coordination with diverse pathways.

Interaction network of the identified proteins in S. sanghuang
Malonylated proteins associated with the biosynthesis of bioactive compounds in S. sanghuang
Malonylated proteins related to ribosomes, glucuronide and dicarboxylic acid metabolism, glycolysis/gluconeogenesis, the TCA cycle, methane metabolism, oxidative phosphorylation, and pyruvate metabolism were greatly enriched (Fig. 4). These findings suggested that the malonylation of lysine may be essential in the biosynthesis of bioactive compounds in S. sanghuang. To further confirm these findings, we analysed malonylated proteins associated with triterpene and polysaccharide biosynthesis in S. sanghuang. Consistent with these hypotheses, a total of 26 enzymes associated with triterpene and polysaccharide biosynthesis were found to be malonylated (Fig. 6, Additional file 2: Table S8). As shown in Fig. 6, a large number of enzymes were affected by malonylation in glycolysis and the TCA cycle, suggesting that malonylation may be associated with multiple levels of intracellular metabolism. Furthermore, our results also showed that 51 malonyl-modified proteins detected on ribosomes, such as ribosomal proteins L24, L13a, and S3, were closely linked to bioactive functions (Fig. 7).

Biosynthesis of triterpenoid and polysaccharide in S. sanghuang. Malonylated proteins are highlighted in red. Additional file 2: Table S2 contains the enzyme annotation

Malonylated modified sites on ribosomal proteins in S. sanghuang. The malonylated modified sites were highlighted in red
Discussion
As a widely used medicinal fungus, S. sanghuang has been known for many years worldwide. It has been reported that S. sanghuang is capable of producing many active substances, such as terpenes, flavonoids, and polysaccharides [5, 40,41,42]. However, the regulatory mechanism of the biosynthesis of these active compounds is still unclear. Lysine malonylation exists widely in eukaryotes and prokaryotes and has many metabolic regulatory functions. To investigate the role of lysine malonylation in the regulation of bioactive compound metabolism, we performed the first proteomic survey of lysine malonylation in S. sanghuang.
The metabolic processes of bioactive substances are related to secondary metabolism. Our malonyl analysis revealed a great number of malonylated proteins participating in secondary metabolism (Fig. 5), demonstrating the essential role of lysine malonylation in all these processes. Other types of PTMs such as acetylation and succinylation also participate in secondary metabolic processes in fungi [7, 43]. Similar to S. sanghuang, Ganoderma lucidum is also a widely used medicinal mushrooms worldwide [43]. It has been well documented that a large number of succinylated proteins are involved in the secondary metabolic process in G. lucidum [43]. Previous studies have shown that the secondary metabolism of Fusarium graminearum is regulated by acetylation [7]. These studies suggest that the secondary metabolic processes associated with the biosynthesis of bioactive substances are regulated by multiple protein modifications.
Polysaccharides are among the main bioactive substances produced by medicinal mushrooms [43]. In G. lucidum, 9 kinds of enzymes associated with polysaccharide biosynthesis have been found [44]. Among them, phosphoglucomutase (PGM) and UDP-glucose 6-dehydrogenase (UGDH) are succinyl-modified proteins in G. lucidum [43]. As shown in Fig. 6, PGM and UDP-glucosepyrophosphorylase (UGPG) were malonylated in S. sanghuang. To date, more than 700 kinds of medicinal mushrooms have been shown to produce bioactive polysaccharides [45]. These observations suggest that multiple PTMs, including malonylation and succinylation, participate in the regulation of polysaccharide biosynthesis in medicinal mushrooms.
Another major bioactive substance produced by S. sanghuang was triterpenoids. Previous studies have proven that triterpenoids are biosynthesized by the mevalonic acid (MVA) pathway [44]. As shown in Fig. 6, the first enzyme in the MVA pathway is acetyl-CoA acetyltransferase (ACAT), which converts acetyl-CoA to acetoacetyl-CoA. FPPs are crucial enzymes in the MVA pathway of triterpene metabolism [46]. Further modification of terpenes involves the introduction of acyl, aryl, or glycosyl groups, usually starting with oxidation catalysed by cytochrome P450 monooxygenases (P450s, also known as CYPs). P450s are ubiquitous in nature and are involved in fundamental biological pathways such as terpene biosynthesis [47,48,49,50]. All these key enzymes were detected by malonyl enrichment (Fig. 6). Thus, lysine malonylation plays a multilevel regulatory role in the biosynthesis of secondary metabolism enzymes.
Furthermore, different types of ribosomal proteins may have different biological activities. Ribosomal protein S5 (RPS5) is closely associated with liver fibrosis in Sprague-Dawley rats [51]. RPS13a plays a role in plant defence against Verticillium dahliae infection [52]. RPS3 protected cells in the substantia nigra against MPTP-induced oxidative stress in a mouse model of Parkinson’s disease [53] and RPL24 had time and dose-dependent effects on HepG-2 cell growth inhibition [54]. In addition, ribosomal synthesis and posttranslationally modified peptides (RiPPs) are an important family of bioactive products [55]. As shown in Fig. 7, a total of 51 ribosomal proteins were modified by malonylation. These findings all support the irreplaceable role of protein malonylation in the synthesis of bioactive substances.
Conclusions
In this study, we found 714 lysine malonyl-modified residues in 255 proteins in S. sanghuang.
Malonylated proteins are involved in a variety of biological processes, especially in secondary metabolic pathways. Further analysis showed that a large number of enzymes involved in the biosynthesis of polysaccharides and triterpenoids were modified by malonylation. This research widens the scope of protein malonylation and provides a rich resource for exploring the physiological regulation of protein malonylation in S. sanghuang.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files. The datasets supporting the results of this article are included within the article and additional files. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD025835.
References
Zhou LW, Ghobad-Nejhad M, Tian XM, Wang YF, Wu F. Current status of 'Sanghuang' as a group of medicinal mushrooms and their perspective in industry development. Food Rev Int. 2020. https://doi.org/10.1080/87559129.2020.1740245.
Tian XM, Dai YC, Song AR, Xu K, Ng LT. Optimization of liquid fermentation medium for production of Inonotus Sanghuang (higher basidiomycetes) mycelia and evaluation of their mycochemical contents and antioxidant activities. Int J Med Mushrooms. 2015;17(7):681–91. https://doi.org/10.1615/IntJMedMushrooms.v17.i7.80.
Cheng J, Song J, Liu Y, Lu N, Wang Y, Hu C, et al. Conformational properties and biological activities of α-D-mannan from Sanghuangporus Sanghuang in liquid culture. Int J Biol Macromol. 2020;164:3568–79. https://doi.org/10.1016/j.ijbiomac.2020.08.112.
Lin W, Deng J, Huang S, Wu S, Chen C, Lin W, et al. Anti-inflammatory activity of Sanghuangporus Sanghuang mycelium. Int J Mol Sci. 2017;18(2):347. https://doi.org/10.3390/ijms18020347.
Ma X, She X, Peterson EC, Wang YZ, Zheng P, Ma H, et al. A newly characterized exopolysaccharide from Sanghuangporus Sanghuang. J Microbiol. 2019;57(9):812–20. https://doi.org/10.1007/s12275-019-9036-4.
Sun Y, Zhong S, Yu J, Zhu J, Ji D, Hu G, et al. The aqueous extract of phellinus igniarius (SH) ameliorates dextran sodium sulfate-induced colitis in C57BL/6 mice. PLoS ONE. 2018;13(10):1–13. https://doi.org/10.1371/journal.pone.0205007.
Zhou S, Yang Q, Yin C, Liu L, Liang W. Systematic analysis of the lysine acetylome in fusarium graminearum. BMC Genomics. 2016;17(1):1019. https://doi.org/10.1186/s12864-016-3361-3.
Ma YY, Yang MK, Lin XH, Liu X, Huang H, Ge F. Malonylome analysis reveals the involvement of lysine malonylation in metabolism and photosynthesis in cyanobacteria. J Proteome Res. 2017;16(5):2030–43. https://doi.org/10.1021/acs.jproteome.7b00017.
Zhou BD, Du YP, Xue YJ, Miao GB, Wei TB, Zhang P. Identification of malonylation, succinylation, and glutarylation in serum proteins of acute myocardial infarction patients. Proteomics Clin Appl. 2020;14(1):1–6. https://doi.org/10.1002/prca.201900103.
Liu J, Wang G, Lin Q, Liang W, Gao Z, Mu P, et al. Systematic analysis of the lysine malonylome in common wheat. BMC Genomics. 2018;19(1):209. https://doi.org/10.1186/s12864-018-4535-y.
Bowman CE, Wolfgang MJ. Role of the malonyl-CoA synthetase ACSF3 in mitochondrial metabolism. Adv Biol Regul. 2019;71:34–40. https://doi.org/10.1016/j.jbior.2018.09.002.
Bowman CE, Rodriguez S, Alpergin ESS, Acoba MG, Zhao L, Hartung T, et al. The mammalian malonyl-CoA synthetase ACSF3 is required for mitochondrial protein malonylation and metabolic efficiency. Cell Chem Biol. 2017;24(6):673–84. https://doi.org/10.1016/j.chembiol.2017.04.009.
Sophie T, Claudia DL, Nathaniel WS, Kathryn EW. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol Metab. 2019;38:100941.
Shi Y, Zhu J, Xu Y, Tang X, Yang Z, Huang A. Malonyl-proteome profiles of Staphylococcus aureus reveal lysine malonylation modification in enzymes involved in energy metabolism. Proteome Sci. 2021;19(1):1–13. https://doi.org/10.1186/s12953-020-00169-1.
Fan B, Li Y, Li L. Malonylome analysis of rhizobacterium Bacillus amyloliquefaciens FZB42 reveals involvement of lysine malonylation in polyketide synthesis and plant-bacteria interactions. J Proteome. 2017;154:1–12. https://doi.org/10.1016/j.jprot.2016.11.022.
Chung CR, Chang YP, Hsu YL, Chen SY, Wu LC, Horng JT, et al. Incorporating hybrid models into lysine malonylation sites prediction on mammalian and plant proteins. Sci Rep. 2020;10(1):1–12. https://doi.org/10.1038/s41598-020-67384-w.
Chang HY, Shih SY, Yu TW, Wu SH, Hattori T, Parmasto E, et al. Species clarification for the medicinally valuable 'Sanghuang' mushroom. Bot Stud. 2012;53(1):135–49.
Tian XM, Yu HY, Zhou LW, Decock C, Vlasak J, Dai YC. Phylogeny and taxonomy of the Inonotus linteus complex. Fungal Divers. 2013;58(1):159–69. https://doi.org/10.1007/s13225-012-0202-9.
Zhou LW, Vlasak J, Decock C. Global diversity and taxonomy of the Inonotus linteus complex (Hymenochaetales, Basidiomycota): Sanghuangporus gen. Nov. Tropicoporus excentrodendri and T.guanacastensis gen. Et spp. nov. and 17 new combinations. Fungal Divers. 2016;77(1):335–47. https://doi.org/10.1007/s13225-015-0335-8.
Shen S, Liu SL, Jiang JH, Zhou LW. Addressing widespread misidentifications of traditional medicinal mushrooms in Sanghuangporus (Basidiomycota) through ITS barcoding and designation of reference sequences. IMA Fungus. 2021;12(1):1–21. https://doi.org/10.1186/s43008-021-00059-x.
Schmelter C, Funke S, Treml J. Comparison of two solid-phase extraction (SPE) methods for the identification and quantification of porcine retinal protein markers by LC-MS/MS. Adv Tech Biol Med. 2018;6(3):9. https://doi.org/10.4172/2379-1764.1000262.
Timothy WR, Jared RW, Lopez N, Morré JT, Bradford CS, Joseph SB. Using theoretical protein isotopic distributions to parse small-mass-difference post-translational modifications via mass spectrometry. J Am Soc Mass Spectrom. 2013;24(1):115–24. https://doi.org/10.1007/s13361-012-0500-1.
Yang Z, Shen X, Chen D, Sun L. Microscale reversed-phase liquid chromatography/ capillary zone electrophoresis-tandem mass spectrometry for deep and highly sensitive bottom- up proteomics: identification of 7500 proteins with five micrograms of an MCF7 proteome digest. Anal Chem. 2018;90(17):10479–86. https://doi.org/10.1021/acs.analchem.8b02466.
Ameli A, Hooshyar A, Yazdavar AH, El-Saadany EF, Youssef A. Attack detection for load frequency control systems using stochastic unknown input estimators. IEEE Trans Inf Forensics Secur. 2018;13(10):2575–90. https://doi.org/10.1109/TIFS.2018.2824253.
Tyanova S, Temu T, Cox J. The maxquant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11(12):2301–19. https://doi.org/10.1038/nprot.2016.136.
An Z, Zhai L, Ying W, Qian X, Gong F. PTMiner: localization and quality control of protein modifications detected in an open search and its application to comprehensive post-translational modification characterization in human proteome. Mol Cell Proteomics. 2019;18(2):391–405. https://doi.org/10.1074/mcp.RA118.000812.
Wang R, Wang C, Sun L, Liu G. A seed-extended algorithm for detecting protein complexes based on density and modularity with topological structure and GO annotations. BMC Genomics. 2019;20(1):1–28. https://doi.org/10.1186/s12864-019-5956-y.
Wei X, Zhang C, Freddolino PL, Zhang Y. Detecting gene ontology misannotations using taxon-specific rate ratio comparisons. Bioinforma (Oxford, England). 2020;10:btaa548.
Shah A. Joseph, Schielzeth H. transcriptome assembly for a colour-polymorphic grasshopper (Gomphocerus sibiricus) with a very large genome size. BMC Genomics. 2019;20(1):1–10. https://doi.org/10.1186/s12864-019-5756-4.
Jiao X, Sherman BT, Huang DW, Stephens R, Baseler MW, Lane HC, et al. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics. 2012;28(13):1805–6. https://doi.org/10.1093/bioinformatics/bts251.
Kanehisa M, Sato Y. KEGG mapper for inferring cellular functions from protein sequences. Protein Sci. 2020;29(1):28–35. https://doi.org/10.1002/pro.3711.
Bradley NL, Wiseman SM. Papillary thyroid microcarcinoma: the significance of high risk features. BMC Cancer. 2017;17(1):142. https://doi.org/10.1186/s12885-017-3120-0.
Gao Y, Li SP, Lai ZY, Zhou ZH, Wu F, Huang YZ, et al. Analysis of long non-coding RNA and mRNA expression profiling in immature and mature bovine (Bos taurus) testes. Front Genet. 2019;10:646. https://doi.org/10.3389/fgene.2019.00646.
Wang Y, Wu CY, Zhang F, Zhang YN, Ren ZW, Guo X. Screening for differentially expressed circular RNAs in the cartilage of osteoarthritis patients for their diagnostic value. Genet Test Mol Biomark. 2019;23(10):706–16. https://doi.org/10.1089/gtmb.2019.0108.
Lotfan M, Ali SA, Yadav ML, Choudhary S, Jena MK, Kumar S, et al. Genome-widegene expression analysis of 45 days pregnant fetal cotyledons Vis-à-Vis non-pregnant caruncles in buffalo (Bubalus bubalis). Gene. 2018;654:127–37. https://doi.org/10.1016/j.gene.2018.02.038.
Yan M, Jing X, Liu Y, Cui X. Screening and identification of key biomarkers in bladder carcinoma: evidence from bioinformatics analysis. Oncol Lett. 2018;16(3):3092–100. https://doi.org/10.3892/ol.2018.9002.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–8. https://doi.org/10.1093/nar/gkw937.
Qi JP, Rui Z. The effects of plumbagin on pancreatic cancer: a mechanistic network pharmacology approach. Med Sci Monit Int Med J Exp Clin Res. 2019;25:4648–54. https://doi.org/10.12659/MSM.917240.
Sween D, Vandana S, Pawan K, Ajit K. Protein-protein interaction network analyses of human WNT proteins involved in neural development. Bioinformation. 2019;15(5):307–14. https://doi.org/10.6026/97320630015307.
Ge Q, Mao JW, Zhang AQ, Wang YJ. Sun Pl. purification, chemical characterization, and antioxidant activity of a polysaccharide from the fruiting bodies of Sanghuang mushroom (Phellinus baumii Pilát). Food Sci Biotechnol. 2013;22(2):301–7. https://doi.org/10.1007/s10068-013-0081-1.
Ran L, Zou Y, Cheng J, Lu F. Silver nanoparticles in situ synthesized by polysaccharids from Sanghuangporus Sanghuang and composites with chitosan to prepare scaffolds for the regeneration of infected full-thickness skin defects. Int J Biol Macromol. 2019;125:392–403. https://doi.org/10.1016/j.ijbiomac.2018.12.052.
Wen Y, Wan YZ, Qiao CX, Xu XF, Wang J, Shen Y. Immunoregenerative effects of the bionically cultured Sanghuang mushrooms (Inonotus sanghuagn) on the immunodeficient mice. J Ethnopharmacol. 2019;245:1–9. https://doi.org/10.1016/j.jep.2019.112047.
Guang YW, Xu LL, Yu H, Gao J, Guo LZ. Systematic analysis of the lysine succinylome in the model medicinal mushroom ganoderma lucidum. BMC Genomics. 2019;20(1):585. https://doi.org/10.1186/s12864-019-5962-0.
Chen S, Xu J, Liu C, Zhu Y, Nelson DR, Zhou S. Genome sequence of the model medicinal mushroom ganoderma lucidum. Nat Commun. 2012;3(1):913. https://doi.org/10.1038/ncomms1923.
Wasser SP. Medicinal mushroom science: current perspectives, advances, evidences, and challenges. Biom J. 2014;37(6):345–56. https://doi.org/10.4103/2319-4170.138318.
Qi Q, Li R, Gai Y, Jiang XN. Cloning and functional identification of farnesyl diphosphate synthase from Pinus massoniana lamb. J Plant Biochem Biotechnol. 2017;26(2):132–40. https://doi.org/10.1007/s13562-016-0373-7.
Mnguni FC, Padayachee T, Chen W, Gront D, Yu JH, Nelson DR, et al. More p450s are involved in secondary metabolite biosynthesis in Streptomyces compared to Bacillus, Cyanobacteria, and Mycobacterium. Int J Mol Sci. 2020;21(13):4814. https://doi.org/10.3390/ijms21134814.
Karunanithi PS, Berrios DI, Wang S, Davis J, Shen T, Fiehn O, et al. The foxtail millet (Setaria italica) terpene synthase gene family. Plant J. 2020;103(2):781–800. https://doi.org/10.1111/tpj.14771.
Lemire CA, Mafu S. Elucidation of terpenoid biosynthesis in sclerotinia. FASEB J. 2020;34(1):1. https://doi.org/10.1096/fasebj.2020.34.s1.03053.
Rudolf JD, Chang CY. Terpene synthases in disguise: enzymology, structure, and opportunities of non-canonical terpene synthases. Nat Prod Rep. 2020;37(3):425–63. https://doi.org/10.1039/C9NP00051H.
Xu WH, Hu HG, Tian Y. Bioactive compound reveals a novel function for ribosomal protein S5 in hepatic stellate cell activation and hepatic fibrosis. Hepatol (Baltimore, Md). 2014;60(2):648–60.
Yang L, Xie C, Li W, Zhang R, Jue D, Yang Q. Expression of a wild eggplant ribosomal protein L13a in potato enhances resistance to Verticillium dahliae. Plant Cell Tissue Organ Cult. 2013;115(3):329–40. https://doi.org/10.1007/s11240-013-0365-4.
Ahn EH, Kim DW, Shin MJ, Kim YN, Kim HR, Woo SJ, et al. PEP-1-ribosomal protein S3 protects dopaminergic neurons in MPTP-induced parkinson's disease mouse model. Free Radic Biol Med. 2012;55:36–45. https://doi.org/10.1016/j.freeradbiomed.2012.11.008.
Hou YL, Ding X, Hou W. Overexpression, purification, and pharmacologic evaluation of anticancer activity of ribosomal protein L24 from the giant panda (Ailuropoda melanoleuca). Genet Mol Res. 2013;12(4):4735–50. https://doi.org/10.4238/2013.October.18.11.
Cao L, Gurevich A, Alexander KL, Naman CB, Leão T, Glukhov E, et al. MetaMiner: a scalable peptidogenomics approach for discovery of ribosomal peptide natural products with blind modifications from microbial communities. Cell Syst. 2019;9(6):600–608e604. https://doi.org/10.1016/j.cels.2019.09.004.
Acknowledgements
Not applicable.
Funding
The research was supported by the National Natural Science Foundation of China (Project No. 31772376, 31970014, 31301827); University Youth Innovation and Technology Program of Shandong Province, China (Project No. 2019KJE003); Research Foundation of Qingdao Agricultural University, China (Project No. 631445).
Author information
Authors and Affiliations
Contributions
TW conceived and designed the study and drafted the manuscript. GW participated in the design and discussion of the research and helped to carefully revise English editing. GZ participated in the design and provided technical implementation assistance. RH participated in the design and provided technical implementation assistance. LZ participated in the discussion of the research and revised English editing. XT participated in its design and coordination and exercised general supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Fig. S1. The MS/MS spectra of examples of malonyl peptides. Fig. S2. GO-based enrichment analysis. Fig. S3. Domain enrichment analysis of the malonylproteins.
Additional file 2:
Table S1. The identified malonylated sites in S. sanghuang. Table S2. Protein annotation analysis. Table S3. Analysis the sequence motifs of the malonylpeptides. Table S4. GO functional annotation of the malonylproteins. Table S5. The modified proteins based on KEGG pathway enrichment analysis. Table S6. The malonylated proteins based on domain enrichment analysis. Table S7. The proteins obtained from PPI network analysis. Table S8. The 26 enzymes associated with triterpene and polysaccharide biosynthesis were found to be malonylated. Table S9. BH correction (FDR) of the GO enrichment. Table S10. BH correction (FDR) of the KEGG enrichment. Table S11. BH correction (FDR) of the protein domain enrichment.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, T., Wang, G., Zhang, G. et al. Systematic analysis of the lysine malonylome in Sanghuangporus sanghuang. BMC Genomics 22, 840 (2021). https://doi.org/10.1186/s12864-021-08120-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-08120-0