
Abstract
Background
Understanding the genetic structure and local adaptive evolutionary mechanisms of marine organisms is crucial for the management of biological resources. As the ecologically and commercially important small-sized shallow-sea fish, Collichthys lucidus plays a vital role in the structure and functioning of marine ecosystem processes. C. lucidus has been shown to have an obvious population structure. Therefore, it is an ideal candidate for investigating population differentiation and local adaptation under heterogeneous environmental pressure.
Results
A total of 184,708 high-quality single nucleotide polymorphisms (SNPs) were identified and applied to elucidate the fine-scale genetic structure and local thermal adaptation of 8 C. lucidus populations. Population structure analysis based on all SNPs indicated that the northern group and southern group of C. lucidus have a strong differentiation. Moreover, 314 SNPs were found to be significantly associated with temperature variation, and annotations of genes containing temperature-related SNPs suggested that genes were involved in material (protein, lipid, and carbohydrate) metabolism and immune responses.
Conclusion
The high genetic differentiation of 8 C. lucidus populations may have been caused by long-term geographic isolation during the glacial period. Moreover, we suspected that variation in these genes associated with material (protein, lipid, and carbohydrate) metabolism and immune responses was critical for adaptation to spatially heterogeneous temperatures in natural C. lucidus populations. In conclusion, this study could help us determine how C. lucidus populations will respond to future ocean temperature rising.
Similar content being viewed by others

Background
Inferring the genetic diversity, population structure and changing patterns of marine species is critical to the successful management of exploited populations, allowing conservation units to be identified and individuals to be assigned to geographical areas [1, 2]. However, assessing the current genetic structure and population connectivity of marine species remains a major challenge [3] because marine species usually have large population sizes and pelagic eggs. Moreover, the degree of connectivity between populations is often high due to less obvious geographic barriers in the oceans [4]. These biological characteristics may lead to high levels of internal genetic diversity and low levels of genetic differentiation between populations, even in marine species with a large distribution range [5]. Additionally, many marine species may have had insufficient time for divergence since the colonization of postglacial habitats [6]. In conclusion, it may be difficult to evaluate the genetic diversity and population structure of marine species, which may affect the reasonable management of these species.
Population genetics has a great advantage in correctly explaining the population genetic structure of marine species and exploring its influencing factors. Although previous studies have detected genetic differentiation patterns of marine species at a small spatial scale based on a small number of genetic markers [7], the deficiency of hierarchical analysis of variance based on F-statistics (FST) is extremely obvious when it is applied to the genetics of large populations (even for species with weak migration abilities), which is especially obvious when single or limited genetic markers were used [8]. Therefore, it is essential to increase the number of genetic markers to help us understand the population structure of marine species in detail.
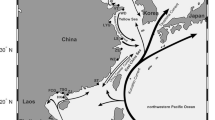
Collichthys lucidus is a short-migratory shallow-sea fish that prefers brackish water in estuaries and has a life cycle including pelagic eggs [9, 10]. With rapid growth, high yield and rich nutrition, C. lucidus has become an important fresh seafood for residents in coastal areas. This species lives in warm water with low salinity and eats shrimp and crabs. As an ecologically and commercially important small-sized fish, C. lucidus is distributed along the inshore waters of China (Fig. 1) and also plays a vital role in the structure and functioning of marine ecosystem processes. Previous population studies based on mitochondrial DNA segments suggested that changes in the sea level during the Pleistocene limited the spread of C. lucidus and promoted the emergence of isolated populations, which ultimately had a significant impact on the systematic geographic pattern of this species [9, 11]. However, the accuracy of population structure research on C. lucidus is limited by the number of genetic markers used, because the hierarchical variance analysis algorithm is not suitable for population genetic studies with a limited number of genetic markers, even for populations with relatively weak migration abilities. Moreover, previous studies restricted to neutral genetic markers have provided limited insights into the local adaptation mechanisms of C. lucidus. Population genomics provides powerful genome-wide genotyping methods and holds great promise for population genetic studies, as it can allow the detection of local adaptation under temperature-driven pressure [12]. This is the case because population genomics may increase the power and resolution of traditional genetic approaches by increasing the number of variable genome-wide genetic markers. Population genomics can also reveal genetic variation in adaptive traits [13].

The distribution range of C. lucidus (https://www.fishbase.se/search)
In this study, eight C. lucidus populations were collected from the coastal waters of China (Fig. 2), and RAD-seq was used to identify genome-wide single nucleotide polymorphisms (SNPs) in the species. Genome-wide SNPs were further used to explore the high-resolution population genetic structure and local adaptation mechanism of C. lucidus. This study provides insights into the evolutionary history and genetic diversity of C. lucidus. The management units are defined using the temperature-driven SNPs and provide fundamental information for the management of C. lucidus resources under fishing pressure.

Map of the sampling locations and sample numbers for eight C. lucidus populations
Results
RAD sequencing and genotyping
RAD sequencing of 177 C. lucidus individuals resulted in 2,773,840,982 read pairs, 2,707,124,768 of which were retained after quality filtering (Additional file 1). A total of 2,561,795,216 read pairs were properly mapped to the C. lucidus genome (GCA_004119915.2; https://www.ncbi.nlm.nih.gov/assembly/GCA_004119915.2) for SNP calling (Additional file 1). After filtering out the low-quality SNPs, 184,708 SNPs were retained for subsequent analyses.
Population genetic diversity and structure
These 184,708 SNPs were applied to calculate the genetic diversity of eight C. lucidus populations (Table 1). Estimates of observed heterozygosity (HO), expected heterozygosity (HE), and polymorphism information (Pi) averages over the 184,708 SNPs varied across the eight C. lucidus populations (HO = 0.2353 ~ 0.4147, HE = 0.2279 ~ 0.3486, and Pi = 0.2348 ~ 0.3560). Among the eight C. lucidus populations, the TJ and ZH populations showed the highest and lowest HO, HE, and Pi values, indicating the highest and lowest population genetic diversity, respectively. All eight C. lucidus populations generally had a high percentage of polymorphic loci (73.4023 ~ 99.4640%). However, the genetic diversity in the XM and ZH populations was relatively low according to all indexes. We also found that the inbreeding coefficient (FIS) values of the eight C. lucidus populations were low (− 0.1477 ~ 0.0209), suggesting that each population contained a large number of individuals. Additionally, most of the pairwise fixation index (FST) values between the eight C. lucidus populations were significant (Table 2), ranging from 0.00087 to 0.16222. Across all eight C. lucidus populations except the XM and ZH populations, the Pi and Tajima’s D values of genome-wide SNPs showed slight fluctuation trends (Fig. 3). The XM and ZH populations showed negative Tajima’s D values, while a large proportion of SNPs had positive Tajima’s D values in the other populations.

Genome-wide distribution of Pi and Tajima’s D values across eight C. lucidus populations based on 184,708 SNPs
ADMIXTURE software was first used for clustering analysis of the eight C. lucidus populations. With K values of 2 and 3 (Fig. 4A), the DL, TJ, LYG, NT, ZS, and WZ populations formed a cluster, and the XM and ZH populations formed another cluster (Fig. 4B). Meanwhile, the results of the principal component analysis (PCA) were consistent with the ADMIXTURE results (Fig. 4C), which indicated that all populations formed two distinct clusters. NetView P with K-nearest neighbour (kNN) = 20 was applied to reveal the clustering relationships of all C. lucidus individuals at a fine scale, and the results further supported the previous ADMIXTURE clustering pattern with K = 2 and 3, showing that individuals were grouped into two different clusters, with all individuals from Xiamen and Zhuhai clustered together (Fig. 4D). Additionally, hierarchical AMOVA (Table 3) showed that the FST across the eight populations was 0. 07084, and there was significant genetic differentiation between the two groups (“DL, TJ, LYG, NT, ZS, and WZ” and “XM and ZH”, FCT = 0.13, P = 0.0004).

Population structure analysis. (A). CV plot for the Admixture analysis. (B). Plots of C. lucidus individual ancestry inference for K = 1 to 8 based on 184,708 SNPs. (C). PCA scatter plots with prior population information using first and second components. (D). Individual clustering plot based on NetView P with KNN = 20
Candidate genomic regions under temperature-driven selection
In the present study, we first calculated the average sea surface temperature (ASST), low sea surface temperature (LSST) and high sea surface temperature (HSST) of the eight sea areas (DL, TJ, LYG, NT, ZS, WZ, XM, and ZH) over 68 years (Additional file 1). Then, Bayenv software revealed a total of 314 SNPs associated with temperature variables. Of these SNPs, 255 were associated with ASST, 56 were associated with LSST, and 4 were associated with HSST. There were 314 union sets among SNPs associated with ASST, LSST and HSST and we used the 314 union sets of ASST-related, LSST-related, and HSST-related SNPs as the candidate temperature-selected SNPs. Of the 314 outlier SNPs, 174 were directly located in intergenic region. The others were aligned to gene region, and only 10 SNPs were assigned to exonic region, which might directly affects protein coding. Gene sequences containing 314 SNPs were then used for further annotations, and the results showed that 105 sequences containing temperature-selected SNPs matched homologous protein sequences in the nonredundant protein sequences (Nr) database (Additional file 1). Next, the enrichment of sequences containing temperature-selected SNPs in Gene Ontology (GO) categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was tested, and 30 significantly enriched GO terms (Fig. 5) and 16 significantly enriched KEGG pathways (Additional file 1) were identified.

GO annotation information for gene sequences containing temperature-selected SNPs
Discussion
Habitat heterogeneity has profound effects on the population genetic diversity of most marine species. Genome-level analysis not only provides detailed information on the structure, dynamics, and environmental adaptation processes of different populations but also helps us predict how populations will respond to future temperature increases. We first used RAD-seq to obtain genome-wide SNPs in C. lucidus and then delineated fine population genetic characteristics and local adaptation characteristics of the eight C. lucidus populations at the genomic level. The complete RAD-seq data and hierarchical analysis of variance based on genome-wide SNPs are helpful in providing a reference for population genomics analysis of other marine species and the fine population genetic characteristics based on genome-wide SNPs contributing to the adaptive evolution of C. lucidus, especially in the context of ocean temperature rising.
Genome-wide SNPs delineating the fine population structure of the eight C. lucidus populations
Reports of the genetic differentiation of C. lucidus are quite limited. The most recent population genetic study was based on the mitochondrial control region [11], which identified C. lucidus in offshore China as being divided into a southern group and a northern group, with Zhoushan as the boundary. Song et al. [9] obtained a contradictory conclusion based on a mitochondrial sequence and suggested that the C. lucidus in offshore China could be divided into the East China Sea group and the South China Sea group. Herein, we used 184,708 SNPs to provide a higher-resolution analysis of population structure than the abovementioned studies and suggested that C. lucidus in offshore China could be divided into northern (DL, TJ, LYG, NT, ZS, and WZ populations) and southern (XM and ZH populations) groups (Fig. 4). This distribution pattern is interesting because there are no obvious barriers between Xiamen and Wenzhou. In fact, the same distribution pattern was also found in Chelon haematocheilus [14]. We hypothesize that the reason for the significant genetic differentiation in this distribution pattern as follows: the late Quaternary period (the past one million years) was characterized by a series of large glacial-interglacial changes, and the geographical distributions and abundance of C. lucidus may be acutely affected by cycles of sea level rise-and-fall. We have suspected that the decline in sea levels during the glacial period may have led to long-term geographic isolation between the two C. lucidus clusters [15] and eventually intensified the development of limited dispersal potential, reproductive isolation and local adaptive heterogeneity between the two clusters [9]. During interglacial periods, although rising sea levels enhanced the dispersal of the two clusters [16, 17], the individuals in the two clusters may not have been able to reproduce, or the diffused individuals may have been unable to adapt to the heterogeneous environment, in turn eventually dying. Over time, significant genetic differentiation between the north and south groups developed.
We also calculated low pairwise FST values within C. lucidus clusters, although the FST values between the two C. lucidus clusters were high (Table 3). This may prove that the genetic differentiation between the northern and southern groups was more obvious. Meanwhile, the FST values between the two C. lucidus clusters probably because that the long-term geographic isolation between the two C. lucidus clusters in the glacial period may have contributed to the emergence of endemic adaptive genotypes, and may have limited gene communication between the two groups (Moody et al. 2015). Additionally, we speculate that the pelagic eggs may be a key driver of the low pairwise FST values within C. lucidus clusters (Table 3). In fact, ocean current transport can enhance the diffusion ability of the pelagic eggs of C. lucidus and therefore significantly increase gene flow within C. lucidus clusters, ultimately contributing to the low pairwise FST values within C. lucidus clusters [18].
Genomic regions of temperature selection in eight C. lucidus populations
With the advantages of identifying adaptive SNPs, RAD-seq can facilitate insights into the genetics of local adaptation in natural populations [19]. The C. lucidus samples in this study were collected from eight different geographical locations and temperature may be one of the important selective forces affecting the genotypic and phenotypic compositions of local populations. In the present study, ASST, LSST and HSST at eight sampling sites were used to identify temperature-selected SNPS and different numbers of candidate SNPs were obtained using different datasets (ASST, LSST, and HSST). We have suspected that those important candidate SNPs could be affected by temperature heterogeneity.
The GO annotation results showed that the sequences containing the temperature-selected SNPs were mainly involved in metabolic and cellular processes, and their functions were mainly in binding and catalytic activity (Fig. 5). This suggests that temperature differences may drive adaptive differentiation in parts of the genome of C. lucidus populations, ultimately leading to differences in physiological regulation. The KEGG annotation results showed that the sequences containing temperature-selected SNPs were mainly associated with material (protein, lipid, and carbohydrate) metabolism and immune responses (Additional file 1). A previous study revealed that the metabolic capacity of organisms was more susceptible to selection under selective force [20]. Moreover, proteins, lipids, and carbohydrates are critical energy materials [21]. Additionally, Lou et al. [22] hypothesized that temperature can affect the priority of metabolic patterns, such as anaerobic carbohydrate metabolism, which is preferred by organisms at low temperatures. Therefore, we speculate that mutations in material metabolism-related genes may cause different C. lucidus populations to change their metabolic patterns in order to maintain a maximum metabolic capacity at different temperatures. Additionally, environmental heterogeneity will inevitably cause variation in environmental pressure among C. lucidus populations. Mutations in genes associated with the immune response may provide evidence of resistance specificity to temperature stress in different geographic populations (Additional file 1). For example, the immune response was shown to be related to local adaptation to different water temperatures in Tylosurus crocodilus crocodilus [23], Trachidermus fasciatus [13], and Larimichthys polyactis [24].
In our outlier study, only RAR-α (Retinoic acid receptor alpha) gene was found to be associated with HSST (Additional file 1). Previous studies have considered that RAR-α gene is involved in the regulation of cell differentiation, proliferation, apoptosis, embryonic development, visual formation, bone formation, metabolism, haematopoiesis and many other pivotal biological processes [25]. Compared with HSST, more SNPs were associated with LSST, which suggests that low temperature pressure may have more serious effects on the spatial distribution of C. lucidus. Poikilotherms have been shown to alter the substance metabolism to maximize ATP production efficiency at ambient temperatures [26]. In our LSST-related outlier study, several genes associated with material metabolic functions were annotated (Additional file 1). For instance, a gene (glucagon-1-like, GLR-1) appears to stimulate insulin production and suppress glucagon production [27]. Therefore, we hypothesize that genes associated with glycometabolism provide energy for cell survival and ultimately increase the ability of different C. lucidus populations to survive in low temperatures. Lower seawater temperatures may enhance oxidative stress of marine organisms. In the present study, we found that some genes were associated with oxidative stress (Additional file 1), such as TLR9 (toll-like receptor 9). TLR9 gene is one of pattern recognition receptors (PRRs). As the sensory receptors of organisms to environmental stress, PRRs has been proved to play an important role in innate immune response [28]. Previous studies have showed that Na+ and Cl− account for a relatively major proportion of the composition of haemolymph in most marine organisms [29]. Our results showed that several LSST-related SNPs are located in ion transport-related genes (SLC6A5; sodium- and chloride-dependent glycine transporter 2), and these genes may influence the sensitivity of different C. lucidus populations to Na+ and Cl− [30]. In fact, the salinity of the low-temperature areas offshore China is higher than that of the high-temperature areas, therefore, we suspected that different C. lucidus populations have evolved different osmotic pressure regulation abilities. Interestingly, one gene (F9; coagulation factor IX) containing LSST-related SNP has been annotated to be involved in the coagulation process. Moderate hypothermia has been used as one of the international anti-coagulant methods [31], the mutations in the F9 gene may has reduces the coagulation rates of C. lucidus populations in the low temperatures.
In summary, the temperature-selected SNPs analysis revealed a number of potential genes that help different C. lucidus populations adapt to local temperatures. However, it is undeniable that some temperature-selected SNPs could be false positives. Meanwhile, future studies will also need to conduct experiments (i.e. RNA-seq, gene knockdown or genome editing) to investigate the exact function and expression levels of these candidate genes.
Conclusion
In the present study, RAD-seq was applied to develop the genome-wide SNPs of C. lucidus. All SNPs were further used to reveal the population genetic structure and genomic regions under temperature-driven selection in eight C. lucidus populations. Genetic structure analysis showed that there was significant genetic differentiation between the northerm (DL, TJ, LYG, NT, ZS, and WZ populations) and southern (XM and ZH populations) groups. Combined with the temperature data of eight sea areas (DL, TJ, LYG, NT, ZS, WZ, XM, and ZH) over 68 years, 314 SNPs associated with temperature variables were identified and genes that contained temperature-selected SNPs were involved in material (protein, lipid, and carbohydrate) metabolism and immune responses. The present research could help us determine how different C. lucidus populations respond to local temperatures.
Methods
Specimen collection and RAD sequencing
All C. lucidus samples were collected from eight locations in China, namely, Dalian (DL), Tianjin (TJ), Lianyungang (LYG), Nantong (NT), Wenzhou (WZ), Zhoushan (ZS), Xiamen (XM), and Zhuhai (ZH) (Fig. 2 and Additional file 1). Muscles were extracted from each individual using sterilized scissors and forceps. All muscle tissues were separately preserved in 95% ethanol and stored at − 80 °C prior to the subsequent experiments. Genomic DNA was extracted following the standard phenol-chloroform extraction method. After assessing the quality of the genomic DNA, we constructed a paired-end library with high-quality genomic DNA following the protocol described by Etter et al. [32] and then sequenced the library on the Illumina HiSeq 2500 sequencing platform.
RAD data processing and SNP filtering
All raw reads in FASTQ format were filtered using Trimmomatic software (version 0.36 [33];) based on the following criteria: (I) raw reads with sequencing adaptors; (II) a ratio of unidentified nucleotides in the raw reads ≥8%; and (III) raw reads that had more than 50% of base calls with a low quality score (Q < 30). After filtering, we downloaded the whole-genome sequence of C. lucidus [34] and used it as a reference sequence for subsequent SNP filtering. The whole-genome sequence was first constructed into an index file using BWA software (version 0.7.12 [35];). The clean reads of each sample were then aligned to the whole-genome sequence using the “bwa-mem” algorithm in BWA software (version 0.7.12 [35];) with default parameters. SNP calling was subsequently performed using SAMtools software (version 1.3.1 [36];) with the following parameters: -q 1 -C 50 -t AD, ADF, ADR, DP, SP -m 2 -F 0.002. The generated SNPs were sorted in a variant call format (VCF) file. Furthermore, we removed the low-quality SNPs using SAMtools software (version 1.3.1 [36];) with the following parameters: --maf 0.01 --max-missing 0.1 --min-meanDP 150 --min-alleles 2 --max-alleles 2 --minGQ 98 --minQ 30 --remove-indels --hwe 0.05.
Population genetic diversity and differentiation
The genome-wide patterns of genetic variation, including nucleotide diversity (Pi) and Tajima’s D at each SNP, were estimated using TASSEL software (version 5.2.31 [37];). The results were visualized using Circos software [38]. To calculate the genetic diversity within populations, nucleotide diversity (Pi), observed heterozygosity (HO), expected heterozygosity (HE), and the inbreeding coefficient (FIS) were calculated using the “populations” module in Stacks software (version 1.34 [39];. Pairwise genetic differentiation (FST) values between populations and their significance were calculated using Arlequin software (version 3.5 [40]; with 10,000 permutations.
Population genetic structure based on all SNPs was analysed using five methods. (I) The Bayesian model-based clustering program ADMIXTURE (version 1.3.0 [41];) was used to investigate individual ancestries, with five replicates of coancestry cluster (K) values ranging from 1 to 8. The optimal K value corresponded to the lowest cross-validation error. (II) We conducted a principal component analysis (PCA) using the “adegenet” package [42] implemented in R software to infer population structure. (III) We calculated the allele-sharing distance using PLINK software [43]. The NetView pipeline (version 0.7.1 [44];) with a KNN step ranging from 1 to 45 was then used to construct the fine-scale relationships between all individuals, and the networks were visualized using Cytoscape software [45]. (IV) On the basis of the optimal value of K, we divided all the groups into two groups (group 1: DL, TJ, LYG, NT, ZS, and WZ; group 2: XM and ZH) and then performed an analysis of molecular variance (AMOVA) in Arlequin software (version 3.5 [40];) to estimate the differentiation among groups (FCT) and the differentiation among populations within groups (FSC).
Outlier SNP detection and annotation
The genotype-environment association method implemented in Bayenv (version 2.0 [46];) was applied to detect putative SNPs correlated with temperature variations. First, we obtained the high-resolution mean LSST, mean ASST and mean HSST data of eight sea areas (DL, TJ, LYG, NT, ZS, WZ, XM, and ZH) over 68 years (from 1950 to 2017) by combining data from the Japan Meteorological Agency (JMA; http://www.jma.go.jp/jma/index.html), Advanced Very High Resolution Radiometer (AVHRR; http://oceanwatch.pifsc.noaa.gov/thredds/catalog.html) and Geostationary Operational Environmental Satellites (GOES; http://oceanwatch.pifsc.noaa.gov/thredds/catalog.html). We first converted SNPs files and environmental data files into matrix files needed for software operation and the matrix was evaluated according to the software specification, the parameters were set as follows: -k 100,000 -r 63,479. Then, Bayenv tests were applied to identify putative SNPs correlated with temperature variations using “calc_bfs.sh” script in the Bayenv2 software, and a Bayes factor (BF) value higher than 10 was set as the filtering condition for putative SNPs. We repeated the Bayenv analysis four times to avoid false positives, and only the SNPs that were continuously screened were used for subsequent analysis. Thereafter, we used the union set of ASST-related, LSST-related, and HSST-related SNPs as the candidate temperature-selected SNPs. To determine the genetic mechanisms underlying temperature-related adaptive differentiation between C. lucidus populations, gene sequences containing these SNPs were then annotated using Blast2GO software along with KEGG database [47, 48]. Meanwhile, for identified the outlier SNPs in genome, we used ANNOVAR software [49] to annotate them based on genome annotation information of C. lucidus (GCA_004119915.2).
Availability of data and materials
Sequences are available from GenBank with the Bioproject accession number PRJNA679902 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA679902).
Abbreviations
- H O :
-
Observed heterozygosity
- H E :
-
Expected heterozygosity
- Pi:
-
Polymorphism information;
- F IS :
-
The inbreeding coefficient
- F ST :
-
Fixation index
- PCA:
-
Principle component analysis
- kNN:
-
K-nearest neighbor
- ASST:
-
Average sea surface temperature
- LSST:
-
Low sea surface temperature
- HSST:
-
High sea surface temperature
References
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW. Harnessing genomics for delineating conservation units. Trends Ecol Evol. 2012;27:489–96.
Nielsen EE, Cariani A, Mac Aoidh E, Maes GE, Milano I, Ogden R, et al. Gene-associated markers provide tools for tackling illegal fishing and false eco-certification. Nat Commun. 2012;3:851.
Miller AD, van Rooyen A, Rašić G, Ierodiaconou DA, Gorfine HK, Day R, et al. Contrasting patterns of population connectivity between regions in a commercially important mollusc Haliotis rubra: integrating population genetics, genomics and marine LiDAR data. Mol Ecol. 2016;25:3845–64.
Allendorf FW, Hohenlohe PA, Luikart G. Genomics and the future of conservation genetics. Nat Rev Genet. 2010;11:697–709.
DeWoody JA, Avise JC. Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. J Fish Biol. 2000;56:461–73.
Hauser L, Carvalho GR. Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish Fish. 2008;9:333–62.
Teske PR, Sandoval-Castillo J, van Sebille E, Waters J, Beheregaray LB. On-shelf larval retention limits population connectivity in a coastal broadcast spawner. Mar Ecol Prog Ser. 2015;532:1–12.
Xu S, Song N, Zhao L, Cai S, Han Z, Gao T. Genomic evidence for local adaptation in the ovoviviparous marine fish Sebastiscus marmoratus with a background of population homogeneity. Sci Rep. 2017;7:1562.
Song N, Ma G, Zhang X, Gao T, Sun D. Genetic structure and historical demography of Collichthys lucidus inferred from mtDNA sequence analysis. Environ Biol Fish. 2014;97:69–77.
Song W, Jiang KJ, Zhang FY, Zhao M, Ma LB. Molecular cloning and gene expression analysis of cystatin C-like proteins in spinyhead croaker Collichthys lucidus. Genet Mol Res. 2016;15. https://doi.org/10.4238/gmr.15017417.
Liang SZ, Song W, Ma CY, Jiang KJ, Zhang FY, Zhao M, et al. Genetic structure of Collichthys lucidus populations from China coastal waters based on mtDNA control region. Mar Fish. 2019;41:138–48.
Lozier JD, Zayed A. Bee conservation in the age of genomics. Conserv Genet. 2017;18:713–29.
Li YL, Xue DX, Zhang BD, Liu JX. Population genomic signatures of genetic structure and environmental selection in the catadromous roughskin sculpin trachidermus fasciatus. Genome Biol Evol. 2019;11:1751–64.
Liu JX, Gao TX, Wu SF, Zhang YP. Pleistocene isolation in the northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus (Temminck & Schlegel, 1845). Mol Ecol. 2007;16:275–88.
Palumbi SR. Genetic divergence, reproductive isolation, and marine speciation. Annu Rev Ecol Syst. 1994;25:547–72.
Shen KN, Jamandre BW, Hsu CC, Tzeng WN, Durand JD. Plio-Pleistocene Sea level and temperature fluctuations in the northwestern Pacific promoted speciation in the globally distributed flathead mullet Mugil cephalus. BMC Evol Biol. 2011;11:83.
Xu J, Chan TY, Tsang LM, Chu KH. Phylogeography of the mitten crab Eriocheir sensu stricto in East Asia: pleistocene isolation, population expansion and secondary contact. Mol Phylogenet Evol. 2009;52:45–56.
Hu ZM, Zhang J, Lopez-Bautista J, Duan DL. Asymmetric genetic exchange in the brown seaweed Sargassum fusiforme (Phaeophyceae) driven by oceanic currents. Mar Biol. 2013;160:1407–14.
Catchen JM, Hohenlohe PA, Bernatchez L, Funk WC, Andrews KR, Allendorf FW. Unbroken: RADseq remains a powerful tool for understanding the genetics of adaptation in natural populations. Mol Ecol Resour. 2017;17:362–5.
Priede IG. Metabolic scope in fishes. In: Tytler P, Calow P, editors. Fish energetics: new perspective. London: Croom-Helm; 1985.
Wang J. Advances in studies on the ecology and reproductive biology of Trachidermus fasciatus Heckel. Acta Hydrobiol Sin. 1999;23:729–34.
Lou F, Han Z, Gao T. Transcriptomic responses of two ecologically divergent populations of Japanese Mantis Shrimp (Oratosquilla oratoria) under thermal stress. Animals (Basel). 2019;9:399.
Flanagan SP, Rose E, Jones AG. Population genomics reveals multiple drivers of population differentiation in a sex-role-reversed pipefish. Mol Ecol. 2016;25:5043–72.
Zhang BD, Xue DX, Li YL, Liu JX. RAD genotyping reveals fine-scale population structure and provides evidence for adaptive divergence in a commercially important fish from the northwestern pacific ocean. PeerJ. 2019;7:e7242.
Zhang GL, Yan GR, Fu ZL, Wu YH, Wu F, Zheng Z, et al. Loss of retinoic acid receptor-related receptor alpha (Rorα) promotes the progression of UV-induced cSCC. Cell Death Dis. 2021;12:247.
Takeuchi T, Masaoka T, Aoki H, Koyanagi R, Fujie M, Satoh N. Divergent northern and southern populations and demographic history of the pearl oyster in the western Pacific revealed with genomic SNPs. Evol Appl. 2020;13:837–53.
Ma J, Stevens JE, Cukier K, AF Maddox AF, Wishart JM, Jones KL, Clifton PM, Horowitz M, Rayner CK. Effects of a protein preload on gastric emptying, glycemia, and gut hormones after a carbohydrated meal in diet-controlled type 2 diabetes. Diabetes Care. 2009;32:1600–2.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20.
Velotta JP, Wegrzyn JL, Ginzburg S, Kang L, Cezsny S, O'Neill RJ, et al. Transcriptomic imprints of adaptation to fresh water: parallel evolution of osmoregulatory gene expression in the alewife. Mol Ecol. 2017;26:831–48.
Lou FR, Gao TX, Han ZQ. Effect of salinity fluctuation on the transcriptome of the Japanese mantis shrimp Oratosquilla oratoria. Int J Biol Macromol. 2019;140:1202–13.
Yi X, Liu MX, Luo Q, Zhuo HL, Cao H, Wang JX, et al. Toxic effects of dimethyl sulfoxide on red blood cells, platelets, and vascular endothelial cells in vitro. FEBS Open Bio. 2017;7:485–94.
Etter PD, Preston JL, Bassham S, Cresko WA, Johnson EA. Local de novo assembly of RAD paired-end contigs using short sequencing reads. PLoS One. 2011;6:e18561.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Cai M, Zou Y, Xiao S, Li W, Han Z, Han F, et al. Chromosome assembly of Collichthys lucidus, a fish of Sciaenidae with a multiple sex chromosome system. Sci Data. 2019;6:132.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22:3124–40.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour. 2010;10:564–7.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64.
Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010;11:94.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Steinig EJ, Neuditschko M, Khatkar MS, Raadsma HW, Zenger KR. Netview p: a network visualization tool to unravel complex population structure using genome-wide SNPs. Mol Ecol Resour. 2016;16:216–27.
Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303.
Coop G, Witonsky D, Di Rienzo A, Pritchard JK. Using environmental correlations to identify loci underlying local adaptation. Genetics. 2010;185:1411–23.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:545–51.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010;38:163–9.
Acknowledgements
We would like to thank Fangrui Lou for his technical support and Binbin Shan for collecting samples.
Funding
This work was supported by the National Natural Science Foundation of China (41706187).
Author information
Authors and Affiliations
Contributions
Z.Z., Z.L. and G.T. conceived and designed the experiments. Q.F. and S.N. performed the sample DNA extraction. Z.L. and H.Z. performed the computational analyses. Z.L. drafted the manuscript. All authors read and approved the final manuscript.
Authors’ information
1 First Institute of Oceanography, Ministry of Natural Resources, Qingdao, Shandong, 266100, China.
2 Fishery College, Ocean University of China, Qingdao, Shandong, 266061, China.
3 School of Fishery, Zhejiang Ocean University, Zhoushan, Zhejiang, 316022, China.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The samples are not privately owned or protected, and no specific permission is required for sample collection. This study was approved by Institutional Animal Care and Use Committee of First Institute of Oceanography, Ministry of Natural Resources. All experimental protocols and procedures were performed in strict accordance with the Laboratory Animal Management Principles of China.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, L., Qu, F., Song, N. et al. Population genomics provides insights into the population structure and temperature-driven adaptation of Collichthys lucidus. BMC Genomics 22, 729 (2021). https://doi.org/10.1186/s12864-021-08045-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-08045-8