
Abstract
Background
Limited access to genetic information has greatly hindered our understanding of the molecular evolution, phylogeny, and differentiation time of subg. Amygdalus. This study reported complete chloroplast (cp) genome sequences of subg. Amygdalus, which further enriched the available valuable resources of complete cp genomes of higher plants and deepened our understanding of the divergence time and phylogenetic relationships of subg. Amygdalus.
Results
The results showed that subg. Amygdalus species exhibited a tetrad structure with sizes ranging from 157,736 bp (P. kansuensis) to 158,971 bp (P. davidiana), a pair of inverted repeat regions (IRa/IRb) that ranged from 26,137–26,467 bp, a large single-copy region that ranged from 85,757–86,608 bp, and a small single-copy region that ranged from 19,020–19,133 bp. The average GC content of the complete cp genomes in the 12 species was 36.80%. We found that the structure of the subg. Amygdalus complete cp genomes was highly conserved, and the 12 subg. Amygdalus species had an rps19 pseudogene. There was not rearrangement of the complete cp genome in the 12 subg. Amygdalus species. All 12 subg. Amygdalus species clustered into one clade based on both Bayesian inference and maximum likelihood. The divergence time analyses based on the complete cp genome sequences showed that subg. Amygdalus species diverged approximately 15.65 Mya.
Conclusion
Our results provide data on the genomic structure of subg. Amygdalus and elucidates their phylogenetic relationships and divergence time.
Similar content being viewed by others


Introduction
Amygdalus L. (Rosaceae), a subgenus of the genus Prunus L. [1,2,3], is a small group within Prunus that includes approximately 24 species. Subg. Amygdalus species are mainly distributed in Iran and eastern Turkey, but a few are distributed in southeastern Europe, the Mediterranean region, Mongolia, and China [4, 5]. Subg. Amygdalus members are shrubs or small trees that mostly grow between 1000 and 2500 m above sea level in mountainous areas [5]. Many studies have elucidated various aspects of subg. Amygdalus members; for example, Maatallaha et al. [6] evaluated mineral nutrients, phenolic and volatile profiles, and antioxidant activities of peach cultivars and assessed their potential for use in cultivar improvement [5, 7]. Moreover, subg. Amygdalus species are valuable fruit trees and can be common ornamental plants, among which P. persica has over one thousand years of cultural history and is one of the five oldest cultivated fruit species with distinct advantages in the world [8].
In recent years, members of subg. Amygdalus have become important subjects of many studies [9,10,11,12]. For example, P. dulcis produces a lot of simple gum exudates that are obtained from its trunk, branches, and fruits [13]. Prunus davidiana var. potaninii Rehd., as an important rootstock of drupe fruit trees in northwestern China, is a wild relative of P. davidiana [14]. Fang et al. [15] constructed regression equations between the ages and base diameters of P. mira by data processing system, and concluded that the populations of P. mira in Linzhi are declining; this species plays an important role in the germplasm improvement of cultivated peach. Prunus mira was also used to facilitate vegetation and rootstock recovery to mitigate land degradation in many areas because of its high tolerance to drought, cold, and barren soil [16]. Prunus kansuensis, which has strong cold resistance and high drought tolerance, can be used as ornamental woody plants. However, the yields of cultivars have recently been seriously affected by diseases and insect pests [17, 18]. Yazbek and Oh [5] reconstructed and analyzed the phylogenetic relationships of subg. Amygdalus by DNA sequencing and morphology, and then evaluated the morphological characteristics used for the classification of subg. Amygdalus. Vafadar et al. [19] identified the pollen morphology of hybrids of subg. Amygdalus, and analyzed common pollen grain features. Several studies have conducted phylogenetic analysis on stone fruits based on internal transcribed spacer (ITS) technology using genetic distance thresholds [20,21,22]. However, a study also showed that accurate identification of species may be impacted by a single threshold [23]. DNA metabarcoding is limited by bias of polymerase chain reaction, resolution of barcoding, universality, and perfect degree of database [24]. The chloroplast (cp) genome of subg. Amygdalus species may be essential for illuminating the evolution of and distinguishing the subg. Amygdalus species. No studies have addressed phylogenetic relationships and estimated divergence time of subg. Amygdalus.
The cp is an important self-replicating organelle that plays a vital role in photosynthesis and energy transformation [25, 26]. Previous studies have shown that the cp genome, which is 115 kb–165 kb in sequence length, consists of a characteristic circular quadripartite structure that includes a large single-copy (LSC) region, a small single-copy (SSC) region, and two inverted repeat (IRa and IRb) regions [27]. Furthermore, compared with the nuclear genome, angiosperm cps are highly conserved in gene composition and genome structure [28], and the structure of plastids, which have unique advantages in phylogenetic reconstruction, is stable, usually uniparental, haploid, and non-recombinant. In this study, complete cp genome sequences of 12 subg. Amygdalus species were compared and analyzed to explore their sequence characteristics and structural differences; these complete cp genome sequences provide additional valuable cp genomic resources of subg. Amygdalus. The aims of the present study were to: (1) explore the complete cp genome sequence of 12 subg. Amygdalus species; (2) clarify the subg. Amygdalus relationships in genus Prunus; and (3) estimate the divergence times of subg. Amygdalus.
Results
Comparative analysis of cp genomes of subg. Amygdalus species
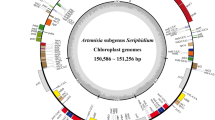
The complete cp genome sequence of 12 subg. Amygdalus species exhibited a circular DNA molecule with a typical quadripartite structure; they have a pair of inverted repeats regions (IRa and IRb), one LSC region, and one SSC region (Fig. 1, Table 1). The complete cp genome sequence of the 12 subg. Amygdalus species ranged from 157,736 bp (P. kansuensis) to 158,971 bp (P. davidiana) in length. The IRa/IRb regions ranged from 26,137–26,467 bp, the LSC region ranged from 85,757–86,608 bp, and the SSC region ranged from 19,020–19,133 bp (Fig. 1, Table 1). The average GC content of the complete cp genome in the 12 species was 36.80%, and the average GC content of the IR regions was 42.60% (Table 1). The cp genomes of the subg. Amygdalus species encoded a total of 131/133 genes, including 86 protein-coding genes (PCGs), 37/39 tRNA genes, and eight rRNA genes. In P. tenella, there were no petB and ycf3 genes. In P. davidiana var. potaninii Rehd., P. kansuensis, P. persica, P. dulcis, P. davidiana, and P. mongolica, there were two rps19 genes and the ycf15 gene was lost. The rps4 gene of P. ferganensis, P. mira, P. tenella, P. pedunculata, P. tangutica, and P. triloba were lost (Table 1, Table 2). There were 18 intron-containing genes; the rps12, clpP, and ycf3 genes contained two introns, and the other genes had a single intron (Table 2).

Complete chloroplast genome map of 12 subg. Amygdalus species
IR boundary changes and gene rearrangement
The complete cp genome structure of 12 subg. Amygdalus species differed. However, all species had eight genes located at the border of the IR region, i.e., rpl22, rps19, and rpl2 at LSC/IRb; ycf1 and ndhF at IRb/SSC; ycf1 at SSC/IRa; and rpl2 and trnH at IRa/LSC. The border between IRb and SSC extended into the rps19 genes, and the 12 subg. Amygdalus species have similar rps19 pseudogenes. The ycf1 gene of P. mongolica was completely located in the IRb region and was 58 bp away from the IRb/SSC border. The IRb/SSC border extended into the ycf1 genes in the other genomes with a short ycf1 pseudogene of 1–16 bp. The IRa region expanded into trnH (Fig. 2).

Comparison of the borders for LSC, SSC, and IR regions of complete chloroplast genomes of 12 subg. Amygdalus species
The complete cp genome structure and order of the 12 subg. Amygdalus species were relatively conservative; the genomes had high sequence similarity, but there were also some highly variable regions (Fig. 3). These variations mainly existed in non-coding regions. There were substantial differences in intergenic regions in the LSC and SSC regions, including trnH–psbA, trnK–rps16, petN–psbM, rps4–trnT, ndhC–trnV, ycf4–cemA, rps19–rpl2, trnS–trnG, and rpoB–trnC. In addition, there were differences in the coding region of the ycf1 gene and the intron region of the clpP gene.

Sequence alignment of complete chloroplast genomes of 12 subg. Amygdalus specie
The complete cp genome of P. dulcis was considered a reference sequence to compare the remaining subg. Amygdalus complete cp genomes. There was no rearrangement in the complete cp genomes of the 12 subg. Amygdalus species (Fig. 4).

MAUVE genome alignments of complete chloroplast genome of 12 subg. Amygdalus species. P. dulcis was reference genome
Repeats and simple sequence repeats analysis
Palindromic repeats, dispersed repeats, and tandem repeats were identified in the complete cp genome sequences of 12 subg. Amygdalus species (Fig. 5, Table S1, & Table S2). The numbers and distributions of these three repeats of subg. Amygdalus were similar and conservative. There were 308 dispersed repeats, 259 palindrome repeats, and 199 tandem repeats, which accounted for 40.21, 33.81, and 25.98% of the total repeats, respectively. Prunus mira had the most repeats, including 24 dispersed repeats, 23 palindromic repeats, and 20 tandem repeats. P. triloba had the fewest repeats, including 24 dispersed repeats, 21 palindromic repeats, and 15 tandem repeats. The repeats were concentrated in the region of 24–127 bp; most were distributed in spacers or introns, although a few were distributed in gene regions.

Analysis of repeated sequences of complete chloroplast genomes of 12 subg. Amygdalus species. a Summary of repeat sequences by length. b Numbers of three repeat types. c Distribution of repeat sequences
There were 55–65 SSRs in the complete cp genome sequences of the 12 subg. Amygdalus species (Fig. 6, Table S3). An average of 82.40% of SSRs were located in the non-coding region of LSC/SSC, and 17.60% of SSRs were located in the PCGs (matK, rpoC2, rpoB, atpB, rps18, rpl16, ycf1, ycf3, atpF, ndhE, ndhI, psbE, and psbZ). Prunus mira and P. dulcis have 1 and 2 hexanucleotide repeats, respectively, and mononucleotide, dinucleotide, tetranucleotide, pentanucleotide, and compound nucleotide repeats accounted for averages of 74.69, 6.09, 8.30, 2.35, and 8.16% of all SSRs, respectively. Approximately 90.00% of mononucleotide repeats were A/T repeats. Moreover, there were 4–5 C/G mononucleotide repeats in each genome, and AT/TA was in dinucleotides repeats.

Analysis of SSRs of complete chloroplast genomes of 12 subg. Amygdalus species. a Number and types of SSRs locations. b Summary of SSRs locations. c Distribution of SSRs locations
Chloroplast phylogenetic and divergence time analysis
Phylogenetic trees were constructed based on the complete cp genome sequences of 45 species. The topologies of the maximum likelihood (ML) and Bayesian inference (BI) trees were nearly identical (Fig. 7 & Table S4). All 12 subg. Amygdalus species formed a monophyletic clade that was sister to Maleae and Spiraeae. Colurieae, Rubeae, Roseae, Potentilleae, and Agrimonieae clustered into one clade.

The (a) maximum likelihood (ML) and (b) bayesian inference (BI) phylogenetic tree of 12 subg. Amygdalus species and other 33 species (the NCBI accession numbers see Table S4). a The numbers at nodes correspond to ML bootstrap percentages, (b) the number of the branches represent Bayesian posterior probability (PP)
Divergence time estimates suggested that the 12 subg. Amygdalus species shared a common ancestor around 22.69 Mya (95%HPD: 11.63–35.91 Mya); they diverged into two clades approximately 15.65 Mya (95%HPD: 7.96–24.64 Mya) (P. dulcis, P. davidiana, P. tenella, P. mongolica, P. davidiana var. potaninii Rehd., P. ferganensis, P. kansuensis, and P. mira belonged to one clade; P. tangutica, P. triloba, and P. pedunculata belonged to the other clade). P. dulcis was the oldest species of subg. Amygdalus, and started to independently evolve around 11.86 Mya (95%HPD: 6.07–19.46 Mya). Diversification within subg. Amygdalus occurred over a short period of approximately 0.1 Mya (Fig. 8).

Divergence times of 12 subg. Amygdalus species obtained from BEAST analysis based on the complete chloroplast genome sequences. Mean divergence time of the nodes were shown next to the nodes while the blue bars correspond to the 95% highest posterior density (HPD)
Discussion
The complete cp genomes of subg. Amygdalus species are typical quadripartite structures with LSC, SSC, and two IR regions. These genomes were similar to the previously reported complete cp genomes of P. mume, P. armeniaca, and P. salicina [29]. The cp genomes of most plants are 120–160 kb in size [30], although those of a few plants are 46–190 kb in size; for example, the cp genomes of Orobanchaceae are 46–190 kb in size [31, 32]. In our study, the cp genomes were conserved and similarly sized, with genomes sizes ranging from 157,736 bp in P. kansuensis to 158,971 bp in P. davidiana. The 12 subg. Amygdalus species encoded 130/133 genes. There were relatively longer LSC and SSC regions than IR regions; the pair of inverted IRa/IRb regions was 26,137–26,467 bp, the LSC region was 85,757–86,608 bp, and the SSC region was 19,020–19,133 bp. There were no differences in the GC contents and composition of subg. Amygdalus species; this indicates that the complete cp genome structure is relatively stable, and that the overall evolution rate is low [33].
In this study, the complete cp genomes of the 12 subg. Amygdalus species ranged from 157,736 bp in P. tenella to 158,971 bp in P. kansuensis. There were differences of only 1235 bp, which indicated that subg. Amygdalus species are highly conservative. Furthermore, there were a total of 130/133 genes present in the complete cp genome of subg. Amygdalus, which included 85/86 PCGs, 37 tRNAs, and 8 rRNAs. The GC contents were similar to those of other plants (such as angiosperms), which showed that they are highly conserved [27]. The average GC content of the complete cp genomes in the 12 species was 36.80%, which is similar to the findings of a previously study that studied Gynostemma species (GC content, 36.9–37.0%) [27]. This high GC content could be caused by the high GC content of the rRNA gene sequences located in IR regions. The function, order, and GC content of these genes are all highly conserved, which was also noted for other angiosperms [27]. The complete cp genomes of the 12 subg. Amygdalus species had an average GC content of 36.80%, which is consistent with the results of Gynostemma species genomes [27], and may be caused by the a high GC content in IR regions [27].
Size increases of plastomes are usually caused by expansion of the IR regions, which are the most conserved regions in the complete cp genome [34]. Our results showed that the cpDNA structure of the 12 subg. Amygdalus species slightly different from each other, although they all have eight genes located at the border of the IR region, i.e., rpl22, rps19, and rpl2 at LSC/IRb; ycf1 and ndhF at IRb/SSC; ycf1 at SSC/IRa; and rpl2 and trnH at IRa/LSC. The border between the IRb and SSC extended into the rps19 genes, and there were rps19 pseudogenes in the 12 subg. Amygdalus species. The expansion of the IR region into the rps19 and ycf1 genes is also present in Cardiocrinum and Amana [35, 36]. These significant differences may contribute to the development of molecular markers and genetic barcodes for subg. Amygdalus species.
SSRs are widely distributed throughout the genome and play important roles in genome recombination and rearrangement; in particular, polymorphic SSRs can be used to study genetic diversity, population structure, and biogeography within and between groups [37]. We identified 55–65 SSRs in the complete cp genomes of the 12 subg. Amygdalus species; on average, 82.40% of SSRs were located in the non-coding LSC or SSC regions, and 17.60% of SSRs were located in the protein-coding region. Furthermore, SSRs are dominated by single nucleotide repeats, and approximately 90% of single nucleotide repeats were A/T repeats in this study. A previous study revealed that the repeated sequences may play a very important role in sequence rearrangement of complete cp genomes [38]. The results of palindromic repeats, dispersed palindromic repeats, and tandem repeats showed that the number and distribution of these repeats in the 12 species of subg. Amygdalus species were similar and conservative. Prunus mira had a maximum of 24 scattered repeats, 23 palindromes, and 20 tandem repeats. P. triloba had the fewest repeats.
Phylogenetic relationships in Rosaceae have long been problematic because of frequent hybridization, apomixis, presumed rapid radiation, and historical diversification [29]. Development of the cp phylogeny and time estimation provides new evidence for future comparative evolutionary studies [29], and there have been an increasing number of studies using complete cp genome sequences to assess phylogenetic relationships among angiosperms [27, 39]. Our phylogenetic tree was based on complete cp genome data. The ML and BI methods were used to conduct phylogenetic analysis on the 12 subg. Amygdalus species. The phylogenetic trees indicated that the 12 subg. Amygdalus species were clearly closely related with high bootstrap support and posterior probabilities (Fig. 7). The results showed that the 12 subg. Amygdalus species were divided into one subclade, which consistent with results of Yazbek et al. [5]. In addition, the results are consistent with the traditional classification system; this indicated that the current classification of the 12 subg. Amygdalus species is reasonable, such as the classification used in the Flora of China (www.iplant.cn/frps). In morphological cladistic analysis, the appearance, shape, and other characteristics of the 12 subg. Amygdalus species were similar. However, Vafadar et al. [40] suggested that P. mira Koehne, P. davidiana (Carriere) Franch., P. triloba Ltdl., and P. tenella L. should be excluded from Amygdalus, and this may require further study. There are differences in the morphological tree structure and molecular phylogeny of the Rosaceae family and relationships among various genera. Therefore, these cp genome sequences will provide genetic information that may help elucidate the evolution of these species.
The divergence time of the 12 subg. Amygdalus was estimated. The results showed that the 12 subg. Amygdalus species shared a common ancestor around 22.69 Mya, and the two clades diverged approximately 15.65 Mya (95%HPD: 7.96–24.64 Mya). According to fossil evidence from southwest China, peach (P. persica) was present in the late Pliocene (ca. 2.6 Mya) [41]; however, we found that P. persica diverged from P. ferganensis approximately 0.1 Mya. In addition, Liu et al. [42] showed that P. persica diverged approximately 10.0 Mya based on plastid ndhF, rps16, and rpl16 sequence data, which may be based on different data. Moreover, our results showed that Amygdaleae diverged approximately 36.33 Mya. The Rosaceae patterns indicate that hybridization and polyploidy may have played a pivotal role in the early evolution of the family in the Eocene [43], which indicated that the Rosaceae was different about Eocene (53–36.5 Mya), they are similar to the results of our study.
Conclusion
This study reported the complete cp genome sequences of subg. Amygdalus species, which further enriches the availability of valuable complete cp genomes of subg. Amygdalus species. The results showed that subg. Amygdalus species exhibited a tetrad structure, with sizes ranging from 157,736 bp in P. kansuensis to 158,971 bp in P. davidiana; the pair of inverted IRa/IRb regions ranged from 26,137–26,467 bp, the LSC region ranged from 85,757–86,608 bp, and the SSC region ranged from 19,020–19,133 bp. The average GC content of the complete cp genome in the 12 species was 36.80%. In addition, it was found that the structure of the subg. Amygdalus complete cp genome was highly conserved, and all 12 subg. Amygdalus species had an rps19 pseudogene. There was no rearrangement in the complete cp of the 12 subg. Amygdalus species. All 12 subg. Amygdalus species clustered into one clade based on both the BI and ML methods. The divergence time analyses based on the complete cp genome sequences showed that subg. Amygdalus species events occurred approximately 15.65 Mya. Our study provides data on the phylogenetic structure of subg. Amygdalus and its phylogeny position with Prunus, and offers a reference for the divergence time of subg. Amygdalus.
Materials and methods
Plant material sampling and chloroplast genomic DNA extraction
Fresh and healthy leaves were collected from adult P. pedunculate, P. mira, P. ferganensis, P. tangutica, P. tenella, P. triloba, P. mongolica, P. dulcis, and P. davidiana var. potaninii Rehd. plants in the field in northwest China (Table 3). The voucher specimens were placed in the herbarium of the School of Life Science, Yulin University, and the spare materials were placed in an ultra-low temperature refrigerator at − 80 °C (for accession numbers, see Table 3; all nine subg. Amygdalus specimens were identified and sorted by Yizhong Duan). Prunus persica, P. davidiana, and P. kansuensis sequences were collected from the GenBank database (Table 1). The modified hexadecyl trimethyl ammonium bromide method [44, 45] was used to extract the total genomic DNA of the 9 species. The extracted DNA was subjected to 0.5% agarose gel electrophoresis and ultraviolet spectrophotometer to check the quality. After passing the total genomic DNA test, fragment it with ultrasound; then, fragment purification and end repair were performed, A was added at the 3′ end, and the sequencing adapter was connected. Subsequently, we used agarose gel electrophoresis to select the size of the fragment and performed polymerase chain reaction for sequencing library preparation. The built library was first subjected to library quality inspection. The library that passed quality inspection was sequenced by Beijing Biomax Biotechnology Co., Ltd. (http://www.biomarker.com.cn/) using the Illumina HiSeq Xten-PE150 platform.
Chloroplast genome assembly and annotation
Raw sequencing read (raw read) data were stored in FASTQ format. The FASTQ data were filtered to obtain clean reads. For data filtering, reads were removed if: (1) they had an adapter; (2) the N content exceeded 10%; (3) they had a base value of less than 10 with a quality value exceeding 50%. The filtered reads were assembled using SOAPdenovo software [46] (http://soap.genomics.org.cn/soap denovo.html), and the assembly was then optimized according to the paired-end and overlap of reads result. For some gaps in the sequence, the assembly result was filled and corrected by SOAPdenovo software to obtain a complete cp genome. The OrganellarGenomeDRAW (OGDraw) online annotation software [47] (http://phylocluster.biosci. Ttexas.edu/dogma/) was used to annotate the complete cp genomes of the nine collected subg. Amygdalus species, and the remaining three species of subg. Amygdalus plants genomes were downloaded from the National Center for Biotechnology Information (NCBI) database (Table 1). The complete cp genome of P. pseudocerasus Lindl. (NC030599) [48] was used as the reference and was manually revised and annotated with Geneious R8 software [49]. Finally, OGDraw visualization software [50] (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html) was used to draw a physical map of complete cp genomes of subg. Amygdalus species.
Chloroplast genome sequence comparative analysis
The complete cp genome sequences of the 12 subg. Amygdalus species were compared. The IR, LSC, and SSC areas, and their boundary information were compared; the online IRscope tool (https://irscope.shinyapps.io/irapp/) [51] was used to map the IR boundary contrast figure. The differences of complete cp genome sequences of the 12 subg. Amygdalus species were studied, and the P. dulcis genome was used as the reference sequence. Each complete cp genome sequence annotation file format of “bed” was converted, which were uploaded to the online analysis program mVISTA (http://genome.lbl.gov/vista/mvista/submit.shtml) [52], and the Shuffle-Lagan mode was selected for genome-wide comparison. The complete cp genome sequence was imported into Geneious R8 software, and the mauve plug-in was used to alignment of global [53]. The gene rearrangement was detected by collinearity analysis with P. dulcis (MT019559) as a reference sequence.
Chloroplast genome repeat sequence identification and SSR analysis
Three types of repeated sequences (palindromic repeats, dispersed repeats, and tandem repeats) of all 12 subg. Amygdalus species were searched and identified by the online REPuter software (https://bibiserv.cebitec.uni-bielefeld.de/reputer/manual.html) [54]. In this study, the parameters had a minimal repeat size of 20 bp and the Hamming distance was 3. Tandem repeat sequences were identified by the online software Tandem Repeats Finder (http://tandem.bu.edu/trf/trf.html) [55]. The alignment parameters for match, mismatch, and indels were 2, 7, and 7, respectively. The minimum alignment scores of reported repeats, maximum period size, and maximum TR array size were 80 bp, 500 bp, and 2 bp, respectively. SSR locations were identified by the online MISA software (https://webblast.ipk-gatersleben.de/misa/) [56]. Moreover, the minimum numbers of repeats of mononucleotides, dinucleotides, trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides were 10, 6, 5, 3, 3, and 3, respectively.
Chloroplast phylogenetic relationships and divergence time estimate
Chloroplast phylogenetic relationships
The complete cp genomes of 33 species from the NCBI database (https://www.ncbi.nlm.nih.gov/) and the 12 subg. Amygdalus species were selected to construct a phylogenetic tree; the complete cp genomes were obtained for nine species of Rosoideae, 19 species of Maleae, 1 species of Sporaeeae, 13 species of Amygdaleae, P. discoidea, P. maximowiczii, and P. pseudocerasus (for the NCBI accession numbers, see Table S4). First, the complete cp genomes of the 45 species were examined by multiple sequence alignment in MAFFT using the PhyloSuite software [57, 58]. Subsequently, the comparison results were imported into the ModelFinder program [59], and Akaike information criterion was selected for nucleotide substitution model testing. Then, the “.nex” files in the result were imported into the MrBayes program [60] to build a phylogenetic tree based on the BI method. Prunus discoidea, P. maximowiczii, and P. pseudocerasus were outgroups in reconstruction of the BI phylogenetic tree. We chose the GTR model and GAMMA distribution, and a Markov chain Monte Carlo with one cold and three heated chains [61, 62]. Analyses were run for 2,000,000 generations total, sampled once every 1000 generations, running end when the value of the average standard deviation of split frequencies was less than 0.01. We discarded the less than 25% of aging samples and constructed a consistent tree according to the remaining samples.
Then, based on the Akaike information criterion, the ModelFinder program was run again to select the optimal nucleotide substitution model from the nucleotide substitution models suitable for BEAST 2 analysis. The results were imported into the IQ-TREE program [63]; the GTR + I + G + F4 model and an ultrafast bootstrap approximation algorithm were selected [64]. The number of re-sampling was 10,000, and the SH-aLRT test was enabled to re-sample 1000 times [65]. The consistent tree file “.contree” was imported into Figtree v1.4.4 software to view and edit the tree, and the output tree diagram was saved to the file [66].
Divergence time estimate
The BEAST v2.6.0 program [62] was used to estimate the divergence time of subg. Amygdalus. First, the MAFFT comparison results were imported to BEAUTi v2, and GTR + Gamma was selected as the nucleotide substitution model (shape = 0.241) [61]. “Empirical” was set of the basic frequency, and the strict molecular clock model was selected. Second, the “Yule Model” was used as the system tree model, and we set the number of iterations and sampling in the Markov chain Monte Carlo algorithm to 3,000,000 and 1000, respectively. Finally, the “.xml” file was obtained from BEAST v2.6.0 [62]. The result was imported into Tracer v1.7.1 software (http://tree.bio.ed.ac.uk /software/tracer/) to check the effective sample size; the effective sample size was greater than 200, which means the results were robust. We used the TreeAnnotator program (https://beast.community/index.html) [67] to perform optimal tree merging. Burn-in was set to 10%, the optimal tree was saved as the final result, and Figtree v1.4.4 software [66] was used to view phylogenetic trees with different time estimates.
We estimated the divergence time of the 12 subg. Amygdalus species with the TimeTree tool (http://www.timetree.org) [68] using the BI tree. We selected four nodes to determine the divergence time: (1) Pentactina rupicola and Vauquelinia californica diverged 62 Mya (range, 30–70 Mya) [69,70,71]; (2) Duchesnea chrysantha and Fragaria pentaphylla diverged 45 Mya (range, 22–52 Mya) [69, 70, 72,73,74,75,76]; (3) M. prattii and Dichotomanthes tristaniicarpa diverged 32.0 Mya (range, 19.4–46.4 Mya) [70, 77]; (5) Photinia prunifolia and H. arbutifolia diverged 32.9 Mya (range, 21.0–46.4 Mya) [70, 77]. The standard deviation of four nodes was 1.0 Mya. Finally, we analyzed the dated phylogeny in Figtree, and the value of node representing the tMRCA and 95%HPD were displayed in Figtree to make the results more intuitive.
Availability of data and materials
The data-sets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Shi WT, Wen J, Lutz SS. Pollen morphology of the Maddenia clade of Prunus and its taxonomic and phylogenetic implications. J Syte Evol. 2013;51(2):164–83. https://doi.org/10.1111/j.1759-6831.2012.00233.x.
Wang L, Guo Z, Shang Q, Sa W, Wang L. The complete chloroplast genome of Prunus triloba var. plena and comparative analysis of Prunus species: genome structure, sequence divergence, and phylogenetic analysis. Braz J Botany. 2021;44(1):85–95. https://doi.org/10.1007/s40415-020-00685-6.
Wen J, Berggren ST, Lee CH, Ickert-Bond S, Yi T, Yoo K-O, et al. Phylogenetic inferences in Prunus (Rosaceae) using chloroplast ndhF and ribosomal ITS sequences. J Sys Evol. 2008;46(3):322–32.
Browicz K. Conspect and chorology of the genera Amygdalus L. and Louiseania Carrière. Arbor Kornickie. 1989;34:31–54.
Yazbek M, Oh SH. Peaches and almonds: phylogeny of Prunus subg. Amygdalus (Rosaceae) based on DNA sequences and morphology. Plant Sys and Evol. 2013;299(8):1403–18. https://doi.org/10.1007/s00606-013-0802-1.
Maatallah S, Dabbou S, Castagna A, Guizani M, Hajlaoui H, Ranieri AM, et al. Prunus persica by-products: a source of minerals, phenols and volatile compounds. Sci Hortic. 2020;261(1):109016. https://doi.org/10.1016/j.scienta.2019.109016.
Socias I, Company R. Fruit tree genetics at a turning point: the almond example. Theor Appl Genet. 1998;96(5):588–601. https://doi.org/10.1007/s001220050777.
Ahmad R, Potter D, Southwick SM. Genotyping of peach and nectarine cultivars with SSR and SRAP molecular markers. J Am Soc Horticultural Sci Am Soc Horticultural Sci. 2004;129(2):204–10. https://doi.org/10.21273/JASHS.129.2.0204.
Du ZY, Duan YZ. The complete chloroplast genome of the Amygdalus ferganensis (Rosaceae) in Xinjiang, China. Mitochondrial DNA Part B. 2019;4(2):3575–6. https://doi.org/10.1080/23802359.2019.1676670.
Pavelková P, Kiss T, Nečas T. Evaluation of presence and concentration of PPV in rootstocks derived from Prunus davidiana (Carr.) Franch. Acta Univ Agric Silviculturae Mendelianae Brunensis. 2019;67(1):121–31. https://doi.org/10.11118/actaun201967010121.
Suelves M, Puigdomènech P. Different lipid transfer protein mRNA accumulate in distinct parts of Prunus amygdalus flower. Plant Sci. 1997;129(1):49–56. https://doi.org/10.1016/S0168-9452(97)00103-9.
Zheng QN, Wang J, Zhou HB, Niu SF, Liu QL, Yang ZJ, et al. Effectiveness of Amygdalus mongolica oil in hyperlipidemic rats and underlying antioxidant processes. J Toxicol Environ Health A. 2017;80(22):1193–8. https://doi.org/10.1080/15287394.2017.1367124.
Rezaei A, Nasirpour A, Tavanai H. Fractionation and some physicochemical properties of almond gum (Amygdalus communis L.) exudates. Food Hydrocoll. 2016;60(1):461–9. https://doi.org/10.1016/j.foodhyd.2016.04.027.
Amar M, Elmosallamy M, Wang L, Zhou H, Zheng B, Jiang X, et al. Peach chloroplast genome variation architecture and phylogenomic signatures of cpDNA introgression in Prunus. Can J of Plant Sci. 2019;99(6):885–96. https://doi.org/10.1139/cjps-2019-0129.
Fang JP, Zhong ZC, Zhong GH. The Age Structure of Tibetan Prunus mira Koehne Kov et Kpsl Population in Tibet Linzhi Region China. Forestry Sci & Tec. 2008;22(1):53–56.
Hao HP, Jiang CD, Shi L, Tang YD, Yao J. Effects of root temperature on thermostability of photosynthetic apparatus in Prunus mira seeding. Chin J Plant Ecol. 2009;33:984–92.
Zhu JZ, Li CX, Zhang CJ, Wang Y, Li XG, Zhong J. Fusarium solani causing fruit rot of peach (Prunus persica) in Hunan, China. Crop Prot. 2019;122(1):171–4. https://doi.org/10.1016/j.cropro.2019.05.009.
Eilers EJ, Klein A-M. Landscape context and management effects on an important insect pest and its natural enemies in almond. Biol Control. 2009;51(3):388–94. https://doi.org/10.1016/j.biocontrol.2009.07.010.
Vafadar M, Attar F, Maroofi H, Mirtadzadini M. Pollen morphology of Amygdalus L. (Rosaceae) in Iran. Acta Soc Bot Pol. 2010;79(1):63–71.
Wen J, Berggren ST, Lee CH, Ickert-Bond S, Yi TS, Yoo KO, et al. Phylogenetic inferences in Prunus (Rosaceae) using chloroplast ndhF and nuclear ribosomal ITS sequences. J Syst Evol. 2008;46(3):322–32.
Liu Y, Xu L, Cheng Z. Phylogenetic analysis of stone fruits such as peach, plum, apricot, Mume and cherry based on ITS sequences. Acta Horticulturae Sin. 2007;34(1):23–8.
Lee S, Wen J. A phylogenetic analysis of Prunus and the Amygdaloideae (Rosaceae) using ITS sequences of nuclear ribosomal DNA. Am J Bot. 2001;88(1):150–60. https://doi.org/10.2307/2657135.
Yang Q, Liu S, Yu X. Research progress on DNA barcoding analysis methods. Chin J Appl Ecol. 2018;29(3):1006–14.
Ren BQ, Chen ZR. DNA barcoding plant life. Chin Bull Botany. 2010;45(1):1–12.
Nock CJ, Waters DL, Edwards MA, Bowen SG, Rice N, Cordeiro GM, et al. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol J. 2011;9(3):328–33. https://doi.org/10.1111/j.1467-7652.2010.00558.x.
Neuhaus HE, Emes MJ. Nonphotosynthetic metabolism in plastids. Ann Rev Plant Phy Plant Mol Bio. 2000;51(1):111–40. https://doi.org/10.1146/annurev.arplant.51.1.111.
Jansen RK, Raubeson LA, Boore JL, Depamphilis CW, Chumley TW, Haberle RC, et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol Acad Press. 2005;395:348–84.
Wicke S, Schneeweiss GM, Depamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Bio. 2011;76(3):273–97. https://doi.org/10.1007/s11103-011-9762-4.
Xue S, Shi T, Luo W, Ni X, Iqbal S, Ni Z, et al. Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Horticulture Res. 2019;6(1):89.
Khan AL, Asaf S, Lubna A-RA, Al-Harrasi A. Decoding first complete chloroplast genome of toothbrush tree (Salvadora persica L.): insight into genome evolution, sequence divergence and phylogenetic relationship within Brassicales. BMC Genomics. 2021;22(1):312.
Frailey DC, Chaluvadi SR, Vaughn JN, Coatney CG, Bennetzen JL. Gene loss and genome rearrangement in the plastids of five Hemiparasites in the family Orobanchaceae. BMC Plant Bio. 2018;18(1):30. https://doi.org/10.1186/s12870-018-1249-x.
Zhou T, Zhu H, Wang J, Xu Y, Xu F, Wang X. Complete chloroplast genome sequence determination of Rheum species and comparative chloroplast genomics for the members of Rumiceae. Plant Cell Rep. 2020;39(6):811-24.
Dong W, Xu C, Li D, Jin X, Li R. Lu Q, Suo Z. Comparative analysis of the complete chloroplast genome sequences in psammophytic Haloxylon species (Amaranthaceae). 2016;4(2):e2699.
He L, Qian J, Li X, Sun Z, Xu X, Chen S. Complete chloroplast genome of medicinal plant Lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Mol. 2017;22(2):249. https://doi.org/10.3390/molecules22020249.
Li P, Lu RS, Xu WQ, Ohi-Toma T, Cai MQ, Qiu YX, et al. Comparative genomics and phylogenomics of East Asian tulips (Amana, Liliaceae). Front Plant Sci. 2017;8:451.
Lu RS, Li P, Qiu YX. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front Plant Sci. 2017;7:2054.
Naydenov K, Naydenov M, Alexandrov A, Vasilevski K, Gyuleva V, Matevski V, et al. Ancient split of major genetic lineages of European black pine: evidence from chloroplast DNA. Tree Genet Genomes. 2016;12(4):68. https://doi.org/10.1007/s11295-016-1022-y.
Cavalier S. T. Chloroplast evolution: secondary Symbiogenesis and multiple losses. Curr Bio. 2002;12(2):R62–4. https://doi.org/10.1016/S0960-9822(01)00675-3.
Raman G, Choi KS, Park S. Phylogenetic relationships of the Fern Cyrtomium falcatum (Dryopteridaceae) from Dokdo Island based on chloroplast genome sequencing. Gene. 2016;7(12):115. https://doi.org/10.3390/genes7120115.
Vafadar M, Osaloo SK, Attar F. Molecular phylogeny of the genus Amygdalus (Rosaceae) based on nrDNA ITS and cpDNA trnS-trnG sequences. Turk J Botany. 2014;38:439–52. https://doi.org/10.3906/bot-1303-46.
Su T, Wilf P, Huang Y, Zhang S, Zhou Z. Peaches preceded humans: fossil evidence from SW China. Sci Rep. 2015;5(1):16794. https://doi.org/10.1038/srep16794.
Liu XL, Wen J, Nie ZL, Johnson G, Liang ZS, Chang ZY. Polyphyly of the Padus group of Prunus (Rosaceae) and the evolution of biogeographic disjunctions between eastern Asia and eastern North America. J Plant Res. 2013;126(3):351–61. https://doi.org/10.1007/s10265-012-0535-1.
Devore ML, Pigg KB. A brief review of the fossil history of the family Rosaceae with a focus on the Eocene Okanogan highlands of eastern Washington state, USA, and British Columbia, Canada. Plant Syst Evol. 2007;266(1):45–57. https://doi.org/10.1007/s00606-007-0540-3.
Doyle J, Doyle J. A rapid DNA isolation procedure from small quantities of fresh leaf tissues. Phyt Bull. 1986;19:11–5.
Yang JB, Li DZ, Li H-T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol Ecol Res. 2014;14(5):1024–31.
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1(1):18. https://doi.org/10.1186/2047-217X-1-18.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–5. https://doi.org/10.1093/bioinformatics/bth352.
Feng Y, Liu T, Wang XY, Li B-B, Liang C-L, Cai Y-L. Characterization of the complete chloroplast genome of the Chinese cherry Prunus pseudocerasus (Rosaceae). Conserv Genet Res. 2018;10(1):85–8. https://doi.org/10.1007/s12686-017-0770-9.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–9. https://doi.org/10.1093/bioinformatics/bts199.
Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41(W1):W575–81. https://doi.org/10.1093/nar/gkt289.
Amiryousefi A, Hyvönen J, Poczai P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 2018;34(17):3030–1.
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32(suppl_2):W273–9.
Darling A, Mau B, Blattner F, Perna N. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–403. https://doi.org/10.1101/gr.2289704.
Kurtz S, Schleiermacher C. REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics. 1999;15(5):426–7. https://doi.org/10.1093/bioinformatics/15.5.426.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80. https://doi.org/10.1093/nar/27.2.573.
Thiel T, Michalek W, Varshney R, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 2003;106(3):411–22. https://doi.org/10.1007/s00122-002-1031-0.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Bio Evol. 2013;30(4):772–80. https://doi.org/10.1093/molbev/mst010.
Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, et al. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Res. 2020;20(1):348–55. https://doi.org/10.1111/1755-0998.13096.
Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14(6):587–9. https://doi.org/10.1038/nmeth.4285.
Fredrik R. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Bio. 2012;3(61):539–42.
Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–8. https://doi.org/10.1093/bioinformatics/14.9.817.
Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Bio. 2007;7(1):214. https://doi.org/10.1186/1471-2148-7-214.
Nguyen L-T, Schmidt H, Von Haeseler A, Minh B. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Bio Evol. 2015;32(1):268–74. https://doi.org/10.1093/molbev/msu300.
Minh BQ, Nguyen M. A T, Haeseler a V. ultrafast approximation for phylogenetic bootstrap. Mol Bio Evol. 2013;5(30):1188–95.
Guindon S, Dufayard J, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Bio. 2010;59(3):307–21. https://doi.org/10.1093/sysbio/syq010.
Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4(vey016). https://doi.org/10.1093/ve/vey1016.
Rambaut A. FigTree version 1.4.0.Available at http://tree.bio.ed.ac.uk/software/figtree/. 2012.
Kumar S, Stecher G, Suleski M, Hedges SB. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. 2017;34(7):1812–9. https://doi.org/10.1093/molbev/msx116.
Hermant M, Hennion F, Bartish IV, Yguel B, Prinzing A. Disparate relatives: life histories vary more in genera occupying intermediate environments. Perspect Plant Ecol Evol Syst. 2012;14(4):283–301. https://doi.org/10.1016/j.ppees.2012.02.001.
Töpel M, Antonelli A, Yesson C, Eriksen B. Past climate change and plant evolution in Western North America: a case study in Rosaceae. PLoS One. 2012;7(12):e50358. https://doi.org/10.1371/journal.pone.0050358.
Khan G, Zhang FQ, Gao QB, Fu PC, Xing R, Wang JL, et al. Phylogenetic analyses of Spiraea (Rosaceae) distributed in the Qinghai-Tibetan plateau and adjacent regions: insights from molecular data. Plant Syst Evol. 2016;302(1):11–21. https://doi.org/10.1007/s00606-015-1238-6.
Hohmann N, Wolf EM, Lysak MA, Koch MA. A time-calibrated road map of Brassicaceae species radiation and evolutionary history. Plant Cell. 2015;27(10):2770–84. https://doi.org/10.1105/tpc.15.00482.
Ndiribe C, Pellissier L, Dubuis A, Vittoz P, Salamin N, Guisan A. Plant functional and phylogenetic turnover correlate with climate and land use in the Western Swiss Alps. J Plant Ecol. 2013;7(5):439–50.
Pouget M, Youssef S, Dumas PJ, Baumberger T, San Roman A, Torre F, et al. Spatial mismatches between plant biodiversity facets and evolutionary legacy in the vicinity of a major Mediterranean city. Ecol Indic. 2016;60:736–45. https://doi.org/10.1016/j.ecolind.2015.07.017.
Burns JH, Strauss SY. More closely related species are more ecologically similar in an experimental test. Proc Natl Acad Sci. 2011;108(13):5302–7. https://doi.org/10.1073/pnas.1013003108.
Gehrke B, Kandziora M, Pirie MD. The evolution of dwarf shrubs in alpine environments: a case study of Alchemilla in Africa. Ann Bot. 2015;117(1):121–31. https://doi.org/10.1093/aob/mcv159.
Lo EYY, Donoghue MJ. Expanded phylogenetic and dating analyses of the apples and their relatives (Pyreae, Rosaceae). Mol Phy Evol. 2012;63(2):230–43. https://doi.org/10.1016/j.ympev.2011.10.005.
Acknowledgements
Thanks to the supports by National Natural Science Foundation of China, the Cooperation Project of Industry-University-Research of Yulin, the Doctoral Research Start-up Foundation Project of Yulin University, the Project of Science and Technology Bureau of Yulin High-tech Area.
Funding
This work was supported by the National Natural Science Foundation of China (32060095), the Project of Science and Technology Bureau of Yulin High-tech Area (CXY-2020-18), the Doctoral Research Start-up Foundation Project of Yulin University (18GK15) and the Cooperation Project of Industry-University-Research of Yulin (2016CXY-13, CXY-2019-107-2, HL2020–1).
Author information
Authors and Affiliations
Contributions
Yizhong Duan conceived and designed the study. Zhongyu Du, Ke Lu, Kai Zhang, Haitao Wang and Yiming He performed the experiments. Yizhong Duan, Yiming He, Kai Zhang, Haitao Wang, Ke Lu, Guaiqiang Chai and Jianguo Shi contributed materials/analysis tools. Zhongyu Du, Ke Lu drafted the manuscript. Zhongyu Du, Ke Lu, Haitao Wang and Kai Zhang revised the paper. All authors approved and helped shape the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The plant leaves material of 9 subg. Amygdalus we used were kept in our lab in College of Life Science, Yulin University. Our experimental methods conducted in the study complied with current Chinese laws and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The repeats distribution in the chloroplast genomes of 12 subg. Amygdalus species. Table S2. The tandem repeats distribution in the chloroplast genomes of 12 subg. Amygdalus species. Table S3. Simple sequence repeats in the chloroplast genomes of 12 subg. Amygdalus species. Table S4. List of species accessions numbers were used in phylogenetic analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Du, Z., Lu, K., Zhang, K. et al. The chloroplast genome of Amygdalus L. (Rosaceae) reveals the phylogenetic relationship and divergence time. BMC Genomics 22, 645 (2021). https://doi.org/10.1186/s12864-021-07968-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07968-6