
Abstract
Background
Copy number variations (CNVs) are a major type of structural genomic variants that underlie genetic architecture and phenotypic variation of complex traits, not only in humans, but also in livestock animals. We identified CNVs along the chicken genome and analyzed their association with performance traits. Genome-wide CNVs were inferred from Affymetrix® high density SNP-chip data for a broiler population. CNVs were concatenated into segments and association analyses were performed with linear mixed models considering a genomic relationship matrix, for birth weight, body weight at 21, 35, 41 and 42 days, feed intake from 35 to 41 days, feed conversion ratio from 35 to 41 days and, body weight gain from 35 to 41 days of age.
Results
We identified 23,214 autosomal CNVs, merged into 5042 distinct CNV regions (CNVRs), covering 12.84% of the chicken autosomal genome. One significant CNV segment was associated with BWG on GGA3 (q-value = 0.00443); one significant CNV segment was associated with BW35 (q-value = 0.00571), BW41 (q-value = 0.00180) and BW42 (q-value = 0.00130) on GGA3, and one significant CNV segment was associated with BW on GGA5 (q-value = 0.00432). All significant CNV segments were verified by qPCR, and a validation rate of 92.59% was observed. These CNV segments are located nearby genes, such as KCNJ11, MyoD1 and SOX6, known to underlie growth and development. Moreover, gene-set analyses revealed terms linked with muscle physiology, cellular processes regulation and potassium channels.
Conclusions
Overall, this CNV-based GWAS study unravels potential candidate genes that may regulate performance traits in chickens. Our findings provide a foundation for future functional studies on the role of specific genes in regulating performance in chickens.
Similar content being viewed by others
Background
Gallus gallus is an excellent biological model organism for genetic studies [1] and a species of considerable economic relevance worldwide. In 2019, global poultry meat consumption was estimated at 97,000 tons [2], being one of the main sources of protein for humans. Understanding the genetic architecture of performance-related traits may contribute to the development of new genomic strategies to increase production efficiency and sustainability of the chicken industry.
Significant advances have been achieved on chicken genetics [3] since the landmark publication of the first reference genome [4], which has been continuously updated with the most recent genome assembly (GRCg6a) released in 2018. Variations in the genome, especially single nucleotide polymorphisms (SNPs), are known to be associated with phenotypic variation [5]. However, structural variations, such as copy number variations (CNVs) have been increasingly studied and associated with quantitative traits of economic interest in livestock [6,7,8,9].
CNVs associated with phenotypes of economic interest are promising targets for animal breeding programs [10]. They are defined as large DNA fragments (conventionally > 1 kb) that, due to deletion or duplication events, display variable copy number between individuals of a population [11]. When compared to SNPs, CNVs encompass more total bases and seem to have a higher mutation rate and potentially greater effects on gene structure, gene regulation and consequently gene expression [12].
Various techniques are available for CNVs detection in humans and other animal species [13]. Most of them depend on the analysis of signal intensity along the genome, such as the comparative genomic hybridization array (aCGH) [14] and high-density SNP chips [15]. Although sequencing-based CNV analyses pipelines have been developed and seem to be a viable alternative [16], SNP chips have been commonly used for CNV detection [8, 17]. This technology allows CNVs identification due to the abnormal hybridization that occurs for SNPs located in CNV regions (CNVRs) [15]. Simultaneous measurement of both signal intensity variations, measured for each allele of a given SNP, and changes in allelic composition (i.e. B allele frequency) allow the detection of both copy number changes and copy-neutral loss-of-heterozygosity (LOH) events [15].
Several factors, such as detection algorithm, genotyping platform, SNP density and population genetic background may impact CNV scanning performance [18]. Indeed, different algorithms used for CNV detection may demonstrate variable sensitivity, consistency and reproducibility, especially for commercial SNP arrays [19], such as Illumina and Affymetrix SNP chips. One of the most prominent algorithms for CNV detection is the PennCNV software [20], which has been widely applied in several studies on livestock species, including chickens [7], horses [21], pigs [22], cattle [6] and sheep [17]. Moreover, PennCNV has better consistency when compared to other CNV calling algorithms [19]. Nevertheless, CNVs identified through SNP-chip platforms can be associated with a considerable rate of false negative and positive results [18]. Therefore, the quantitative polymerase chain reaction (qPCR) is commonly used for CNV validation, being a molecular method to confirm computationally identified loci [8, 23].
In chickens, several studies have identified quantitative trait loci (QTL) and positional candidate genes flagged by SNPs significantly associated with traits of economic interest such as performance, carcass and abdominal fat [24, 25]. Unsurprisingly, the number of CNV-focused studies is increasing in chicken populations as well [7, 26]. CNVs associated with late feathering [27], pea-comb phenotype [28], dermal hyperpigmentation [29], dark brown plumage color [30] and resistance/susceptibility to Marek’s disease [31] have been reported. None CNV-association study for performance traits in chickens has been described yet.
Herein, we identified CNVs in the genome of a broiler population, performed a CNV-based GWAS for performance traits and validated associated CNV segments by qPCR. In addition, we identified performance-related genes overlapping significant CNV segments to establish relationships between structural genomic variation and such phenotypes.
Results
CNV identification
After applying the initial quality control filters, 223 individuals out of 1461 genotyped chickens from the TT Reference Population presented DishQC< 0.82 and call rate < 97%, and were excluded from further analyses. Therefore, individual-based CNV calls were performed on the remaining 1238 samples. Pedigree information on father-mother-offspring trio was used to update the CNV status for the trios, generating more accurate CNV calls [20]. From the total of 1238 chickens, 709 trios were determined based on complete family information available. Then, the trio-based CNV calling using 779 animals, represented by 709 trios, consisting of 14 sires, 56 dams and 709 offspring, was performed. Several families with incomplete information could not be used as PennCNV is not able to handle trios with missing sire or dam genotypes. After quality control filtering and removal of duplicated CNVs from the dataset, we identified 23,214 unique autosomal CNVs, including 2905 deletions and 20,309 duplications. Finally, a total of 614 chickens had at least one CNV call after the quality control process.
CNVR compilation
CNVRs represent the concatenation of overlapping CNVs into a consensus genomic region. CNVs showing overlap of at least one base pair among samples in this population were summarized across all individuals into CNVRs. After filtering, 23,214 individual CNVs were merged into 5042 distinct CNV regions, which cover 12.84% (136.75 million of base pairs - Mb) of the chicken autosomal genome. The number of regions with copy loss and gain were 424 and 4105, respectively. The presence of both types was observed in 513 regions. The CNVRs had variable sizes ranging from 0.14 kb to 760 kb with an average size of 27.12 kb. The number of chickens with CNVs mapped onto a given CNVR ranged from 1 (0.13%) to 348 (44.67%) from the total of 614 chickens. We identified 656 CNVRs occurring in more than 1% of the population (i.e. ‘polymorphic CNVRs’, as suggested by Itsara et al. [32]). The relative chromosome coverage by CNVRs ranged from 1.55% for GGA24 to 18.38% for GGA2, while the absolute genomic length overlapped by CNVRs varied from 0.10 Mb for GGA24 to 35.98 Mb for GGA1. Detailed information of all CNVRs detected in our population is provided in Additional file 1.
Association of CNV segments with performance traits
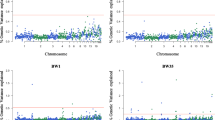
Genome-wide association studies were performed to investigate significant associations of CNV segments, named as CNV-based GWAS, with eight performance-related traits measured in our population: BW, BW21, BW35, BW41, BW42, FI, FCR and BWG. Manhattan plots for CNV segments across the 33 autosomal chromosomes associated with performance traits are presented in Fig. 1. Note that the FDR method for multiple testing correction shrunk the -log10(q-value) for non-significant CNV segments towards zero, while it magnified the -log10(q-value) for significant associated CNV segments. The Manhattan plots of the raw p-values for CNV segments across the 33 autosomal chromosomes associated with performance traits and the QQplots for BW, BW35, BW41, BW42 and BWG are in Additional files 2 and 3, respectively.

Manhattan plots for CNV segments across the 33 autosomal chromosomes associated with a birth weight, b body weight at 35 days, c body weight at 41 days and d body weight at 42 days and e body weight gain. The X-axis represents the somatic chromosomes, and Y-axis shows the corresponding -log10 q-value. Red and blue lines indicate FDR-corrected p-values of 0.05 and 0.1, respectively
There were three distinct CNV segments classified as losses and significantly associated (q-value< 0.05) with BWG, BW35, BW41, BW42 and BW (Table 1). One CNV segment was significantly associated with BWG (q-value = 0.00443); one CNV segment was significantly associated with BW35 (q-value = 0.00571), BW41 (q-value = 0.00180) and BW42 (q-value = 0.00130), and one CNV segment was significantly associated with BW (q-value = 0.00432). It is interesting to highlight that the significant CNV segment associated with BW35, BW41 and BW42 was the same (GGA3:97801202–97809208). Note that none significant CNV segments associated with BW21, FI and FCR were detected.
In Fig. 2, each dot represents an animal in the corresponding copy number state (0-3n) on the X-axis and the observed phenotypic value on the Y-axis. For the significant CNV segment associated with BW (GGA5: 12059966–12062666), a decrease in copy number is associated with heavier birth weight. The same trend was observed for the significant CNV segment associated with BW35, BW41 and BW42 (GGA3: 97801202–97809208), i.e. higher copy number was observed in animals with lower body weight. Conversely, the significant CNV segment associated with BWG (GGA3: 64169030–64171297) displayed an opposite behavior.

a Birth weight, b body weight at 35 days, c body weight at 41 days and d body weight at 42 days and e body weight gain distribution in each CN state for the significant CNV segment. Each dot represents an animal in the corresponding copy number state (0-3n) on the X-axis and the observed phenotypic value on the Y-axis. The legend on the right displays the color code for the CN state. See the main text for a detailed description of each segment
qPCR validation
Since that CNV breakpoints depend on the segmentation algorithm used, some variation on CNV segment detection between PennCNV and qPCR is expected. The qPCR results (Fig. 3) revealed a validation rate of 92.59%, which confirms the existence of CNV segments that have been associated with performance traits. In addition, it revealed that the CNV type was concordant between both methods for most of the samples, except for the first sample with primers 7 and 8. Note that for CNV segments where at least one breakpoint was within the target segment, PennCNV results were confirmed by qPCR. It is important to mention that the third tested animal had a copy number status estimated by PennCNV of 0n for the CNV segment on GGA5. Primer information and validation rates are presented in Additional files 4 and 5, respectively.

Quantitative PCR was carried out for significantly associated CNV segments on a GGA3 at 64 Mb, b GGA3 at 97 Mb and c GGA5 at 12 Mb using two groups (control (2n) and experimental) with three different animal samples per group and three distinct primer pairs per CNV. In each panel, bars in different colors represent distinct experimental animals for each segment. The right-most bars depict the relative copy number estimated for each animal in PennCNV. Each bar was calculated from three technical replicates. The error bars show the minimum and maximum value encountered among the replicates
CNV segments overlapping known QTLs
The significant CNV segments associated with body weight gain (GGA3: 64169030–64171297) overlapped with previously mapped QTLs for body weight at 49 days of age (QTL #30854, [33]), comb weight (QTL #127114, [34]), residual feed intake (QTL #64556, [35]), and testis weight (QTL #213559, [36]). The significant CNV segment associated with BW35, BW41 and BW42 (GGA3: 97801202–97809208) also overlapped with two QTLs described above (QTL #30854 and #127114). Moreover, both significant CNV segments overlapped with 18 out of 27 previously published QTLs for growth-related traits mapped in the Embrapa F2 Chicken Resource Population ([37], Table 2). None previously reported QTLs overlapped with the CNV segment significantly associated with birth weight (GGA5: 12059966–12062666).
Identification of regulatory elements
We investigated the presence of CpG islands within the significant CNV segments. However, no CpGs were identified on such regions (Additional file 6). Moreover, we found that the significant CNV segment on chromosome 5 (GGA5:12059966–12062666), previously associated with birth weight, is located nearby the KCNJ11 gene, approximately 3.4 kb downstream of the gene start site. Our analysis of ChIP-seq data for H3K27ac of chicken skeletal muscle, an indicator of cis-regulatory elements, like active enhancers [38], showed overlapping of a H3K27ac enriched region with the aforementioned CNV segment (Additional file 6).
Candidate genes and gene-set analysis
A total of 32 genes, including KCNJ11, MyoD1 and SOX6, were annotated within a 1-Mb window in genomic regions defined by significant CNV segments associated with BWG, BW35, BW41, BW42 and BW (Table 3). A list with detailed information about the 32 genes is provided in the Additional file 7.
Gene enrichment analysis was performed using WebGestalt to search for biological processes, cellular components and molecular functions. WebGestalt top 10 most relevant enriched categories for Biological Process, Cellular Component and Molecular Function, based upon genes annotated to each category, can be observed in Table 4. Noticeably, the most relevant enriched categories for biological processes, such as regulation of striated muscle cell differentiation, regulation of muscle cell differentiation and regulation of muscle tissue development, are directly implicated in muscle growth and development.
Complementary, STRING databases were used to search for enriched pathways and protein domains on genes annotated within 1-Mb window of significant CNV segments (Fig. 4 and Table 5). Interestingly, the three networks identified are related to cell differentiation and muscle functioning (Fig. 4).

Confidence view of the network created by the STRING software. Nodes represent proteins produced by a single protein-coding gene locus. Edges represent protein-protein associations. Line colors indicate types of interaction evidence: known interactions from curated databases (cyan) or experimentally determined (pink); predicted interactions from gene neighborhood (green); and other sorts of interactions such as co-expression (black). The large network, in the middle, and smaller networks, on the right and left extremes, both relate to cell differentiation and muscle functioning
In addition, terms associated with potassium channels and regulation of insulin secretion were enriched for CNV candidate genes related to performance traits (Table 5). Moreover, regarding to protein domains, the calcium homeostasis modulator family, consisting of three members of the FAM26 gene family, was enriched. Furthermore, 78 publications significantly enriched in STRING are presented in Additional file 8.
Discussion
To investigate the effect of CNVs on production-related traits in broilers, we analyzed a Brazilian broiler population, selected for body weight, carcass and cuts yield, feed conversion, fertility, chick viability and reduced abdominal fat. In addition, the availability of information about the family structure of this population allowed the identification of family-based CNVs.
CNVs are significant sources of genetic variation [39] and have been associated with disease, abnormal development, physical appearance as well as many other economic traits in livestock animals [6, 8, 31]. It is noteworthy that CNVs are generally in low LD with SNPs [40], and its taggability is lower than SNP taggability [41]. Therefore, the genetic variation explained by CNVs might not be fully captured in the traditional SNP-based analysis. Thus, CNV-based GWAS studies can provide valuable insights on the genetic control of economically important traits for livestock breeding programs. CNV mapping can be based on different reference genome assemblies, populations and platforms. Hence, variability of CNV breakpoints (i.e., genomic coordinates) can happen due to different biological and technical influences [11]. Therefore, CNV comparison among studies is not prosaic, even in the same species, and, as a consequence, different approaches may be complementary to each other [26, 42].
In our population, copy number gains were more abundant than losses. Likewise, Yi et al. [42], Gorla et al. [7] and Sohrabi et al. [43] reported more gains than losses and mixed regions in chicken populations. One reason is that duplications are more likely to be conserved than deletions because deletion regions are relatively gene-poor and therefore these regions are prone to purifying selection [44]. Nonetheless, deletion polymorphisms might have a significant role in the genetics of complex traits, even though not directly observed in several gene mapping studies [44].
In the present study, significant CNV segments associated with performance traits on chromosome 3, for body weight at 35, 41 and 42 days and body weight gain from 35 to 41 days, and on chromosome 5 for birth weight were identified. Given that these traits are not independent, and genetic correlations between performance traits have been widely reported in chickens [45], it is expected that certain CNV regions may be concomitantly associated with more than one trait, especially body weight measured in different ages (Fig. 1).
In the qPCR validation, we systematically assessed the overall agreement rate of the significant CNV segments detected in silico with qPCR results. The validation results indicated that all CNV segments were confirmed in at least one qPCR assay, consequently all CNVs may be real. Our results indicated that there is a small discrepancy (7.41%) between qPCR and PennCNV callings, which may be due to variations on the exact genomic coordinates of the CNVs that influenced the hybridization of the qPCR primers and the amplification efficiency.
We identified overlaps of significant CNV segments associated with body weight at 35, 41, 42 days and body weight gain with four previously mapped QTLs for weight traits and residual feed intake (RFI). RFI is defined as the difference between actual feed intake and predicted feed intake based on energy requirements for body weight gain and maintenance [46]. Moreover, we found genomic windows defined by significant CNV segments overlapping published QTLs for growth-related traits in the Embrapa F2 Chicken Resource Population [24]. Many studies, conducted with different chicken lines, have successfully identified QTLs and genes associated with economically important traits [47]. Given that QTLs and genes underlie functional regions of the genome, they may not be prone to structural rearrangements and thus not expected to be subject to CNVs [23]. Therefore, QTLs and genes located inside or nearby CNVs are of special interest.
Noticeably, SNP-based studies [37, 46, 48] have identified many more QTLs associated with the traits analyzed in our study than the CNV-based approach applied here. Indeed, Pértille et al. [37] identified 88 QTLs associated with feed conversion, feed intake, birth weight, and body weight at 35 and 41 days of age in the Embrapa F2 Chicken Resource Population. Mebratie et al. [46] and Moreira et al. [48] identified, respectively, 11 and 19 QTLs associated with body weight traits in a commercial broiler chicken population and in the Embrapa F2 Chicken Resource Population. This difference in QTL mapping is expected since CNVs are more frequently associated with deleterious effects than favorable ones, which is not the case of SNPs, at least those included in the SNP arrays [49]. In addition, since known QTLs were (mostly) mapped using microsatellite markers and SNPs, they will not necessarily capture the same effect as CNVs. If associated CNVs do not overlap with QTLs previously found in other studies, that could occur because specific CNV probes can be excluded from a SNP-GWAS due to Hardy-Weinberg equilibrium deviation or rigorous multiple testing corrections [23].
CNVs that comprise functional genes may induce phenotypic variation by altering gene structure, dosage and regulation, as a consequence of natural evolutionary processes [50], such as genetic drift or artificial selection. We identified 32 genes annotated within a 1-Mb window of significant CNV segments associated with birth weight, body weight at 35, 41 and 42 days and body weight gain from 35 to 41 days.
Note that animals presenting deletions (0n/1n) in significant CNV segments were less frequent in our population, while their average body weights at birth and at 35, 41 and 42 days of age were higher compared to animals with normal copy number (2n) in the same CNV segments (Fig. 2). Two reasons may explain the low frequency of favorable genotypes for body weights at different ages: 1) this meat-type population has been under multiple trait selection, not exclusively focused on improving body weight, and 2) the TT line that gave rise to the TT Reference Population was selected for only 17 generations [51]. In 2010, Johansson et al. [52] conducted a study with two chicken lines (high and low body weight lines) from a single trait selection experiment, where even after 50 generations of selection, the high line is still responding to selection. Conversely, for body weight gain, the increase in the copy number of the respective significant CNV segment was positively associated with the phenotype (Fig. 2). Since these CNV segments are located in proximity of several genes (Table 3) and, as it has been shown that the expression of a gene may be affected by their presence [12], CNVs may act as important modulators of gene expression.
CNVs inserted in regulatory regions like enhancers, promoters or in 3’UTR regions, may modify availability of binding sites to transcription factors or miRNAs, respectively, resulting in the modulation of their associated genes. In addition, a wide variety of cis-regulatory elements have been investigated for the presence of CpG islands and methylation. Despite being frequently found in promoter regions and TSSs, low levels of methylation can be found in enhancers and insulators, modulating gene transcription [53]. However, as expected due to its low frequency distribution in the genome, no overlap between CpG islands and the significant CNV segments was identified.
Our analysis of ChIP-seq data suggests that the significant CNV segment located on chromosome 5 may be affecting expression of its cognate genes due to the presence of regulatory elements (Additional file 6). Once enhancer activity is tissue-specific, our analysis does not reject the possibility that these elements may be present in other tissues related to metabolism and weight gain, like hepatic and adipose tissues [54, 55]. In addition, although the other significant segments have not shown peaks for enhancers, it does not mean that other regulatory elements, such as insulators and silencers, cannot be associated and affect gene expression. In this sense, CNVs may have potential to modulate core regulators located in their proximity, propagating such effects to genome-wide gene expression [12] and accounting for differences manifested at the phenotype level (Fig. 2). Near to the CNV segment on chromosome 5, we identified KCNJ11, MyoD1, PIK3C2A and SOX6 genes, which might have important effects on chicken growth and development regulation.
The KCNJ11 gene, found to be 2217 base pairs upstream of the aforementioned significant CNV segment, is known to regulate insulin secretion [56]. A glucose metabolism disorder is usually linked as a cause of reduced development of chicken muscle tissue under stress, especially in broilers [57]. This gene was enriched for the ATP-sensitive potassium channel (KATP) pathway. KATP subunits are, among other genes, encoded by KCNJ11. KATP channels have a high activity in rat fast-twitch fibers, distinguished by raised muscle strength, and a low activity in slow-twitch fibers, characterized by weakness, fragility and lowered muscle strength [58]. KCNJ11 gene knockout mice present reduced glycogen, slender phenotype and weakness [58]. It has been reported that the effect of the KCNJ11 gene on muscle may occur due to alterations in the KATP channel activity, which, in turn, affects the potassium flow inside the cell, settling the membrane potential needed for muscle activity [59]. For this reason, this gene may promote early growth and development in chickens. In fact, it was found to be highly expressed in the muscle tissue of one-week-old chicks [56], which supports our findings that this gene, closely located to a significantly associated CNV segment, can play a role in the regulation of birth weight. Our analysis of regulatory elements suggests that changes in the copy number in the KCNJ11 gene might be affecting gene transcription and could explain the effect on birth weight associated with these changes. This hypothesis, however, needs to be addressed by RNA data on different tissues. Interestingly, a novel 163-bp indel in the downstream region of this gene was significantly associated with growth traits in chickens [56]. In the same study, synteny analyses found that KCNJ11 maintains a close connection with its neighboring genes. It is interesting to note that one of these genes is the Myogenic differentiation 1 (MyoD1).
The myogenic regulatory factors are a family of vertebrate proteins (MyoD, Myf5, Mrf4 and Myog) that are robust transcription factors for muscle genes [60]. The MyoD1 gene can promote myoblast differentiation and have relevant effects on muscle development [61]. Previous study in quail lines revealed that a delay in MyoD1 expression is associated with increased body weight and muscle mass [62]. A very high degree of synteny is maintained between MyoD1 containing regions of human chromosome 11 and chicken chromosome 5, comprising ABCC8, KCNJ11, PIK3C2A, RPS13, SERGEF, NUCB2 and PLEKHA7 genes [63]. Mutations in PIK3C2A gene were discovered to cause a growth-related genetic syndrome in humans, consisting of dysmorphic features, short stature and skeletal abnormalities [64]. This gene has been attributed to biological functions such as glucose transport, Akt pathway activation, endosomal trafficking, phagosome maturation, mitotic spindle organization, exocytosis and autophagy [65].
MyoD1 was enriched in biological processes associated with the SOX6, a gene related with muscle physiology, such as regulation of striated muscle cell differentiation and development, regulation of muscle cell differentiation and regulation of muscle organ development. The expression level of the SOX6 gene was positively associated with CNV and increased during skeletal muscle cell differentiation, by upregulating expression levels of muscle-growth-related genes in chickens as well as in other animal species [66].
We found genes nearby significant CNV segments associated with body weight at 35, 41 and 42 days (LPIN1 and TRIB2) and body weight gain (GPRC6A and NT5DC1) that may be of special importance and have potential effects on chicken growth. A significant association was found between a variant in the 3′ UTR of chicken LPIN1 gene and breast muscle fiber diameter [67], suggesting that this gene has a potential effect on muscle fiber development. The TRIB2 gene, a novel regulator of thymocyte cellular proliferation, was found to be involved in reproduction and growth in White Leghorn chickens, and consequently might represent footprints of the selection process [68]. The GPRC6A gene was found to have functions related to testis growth and development in broilers [69]. In addition, another interesting gene is the NT5DC1, previously related to muscle tissue, angiogenesis and amino acid metabolism [70]. We found an enriched cluster for calcium homeostasis modulator (CALHM) gene family, which included three members: FAM26D, FAM26E and FAM26F. Even though CALHMs have been classified as pore-forming subunits of plasma membrane ion channels, questions about their function remain unanswered, hence their role needs to be ascertained on further investigations [71].
In summary, from SNP-chip data of a broiler population, we identified novel structural variation regions in the genome that, based on gene enrichment and literature information, harbor potential candidate genes, with important roles in a wide range of biological, cellular, and molecular processes, linked with muscle differentiation, growth, and development. Our findings reveal that alterations in copy number within or nearby these genes could result in phenotypic variation, thus contributing to a better understanding of performance regulation in chickens.
Conclusions
This study identified structural variations associated with five complex traits of interest in a broiler population using a probe-level based CNV association approach. Our results provide substantial information about the potential CNV impacts on animal production, growth, development, and performance-related traits, laying a foundation for incorporating CNVs into the future poultry breeding programs and contributing to expand scientific research on genetics, particularly on structural variations involved in animal biology and physiology.
Methods
Population description
A paternal broiler line (TT) belonging to the Chicken Breeding Program of Embrapa Swine and Poultry National Research Center, in Concórdia, Santa Catarina State, South of Brazil, was developed in 1992. This line originated from White Plymouth Rock and White Cornish breeds and has been under multiple trait selection to improve body weight, feed conversion, carcass and breast yields, viability, fertility, reduction of abdominal fat and metabolic syndromes [51]. The experimental broiler population evaluated in this study, called TT Reference Population, was generated by an expansion of the paternal broiler line TT and consisted of approximately 1500 chickens, which were all slaughtered in 2008, generated in five hatches from 20 males and 92 females (1:5). More details can be found in Marchesi et al. [51].
Phenotype measurement
Body weight was recorded at 1 (birth weight), 21, 35, 41 and 42 (after fasting) days of age. Over the period between 35 and 41 days of age, chickens were transferred to individual cages for measuring feed intake and body weight gain, to evaluate feed conversion. At 42 days of age, all chickens (~ 1500 individuals) were weighted and euthanized by cervical dislocation followed by exsanguination. By then, a blood sample from each animal was collected for subsequent DNA extraction. In this study, we analyzed eight performance traits: (i) birth weight (BW), (ii) body weight at 21 days of age (BW21), (iii) body weight at 35 days of age (BW35), (iv) body weight at 41 days of age (BW41), (v) body weight at 42 days of age (BW42), (vi) feed intake measured from 35 to 41 days of age (FI), (vii) feed conversion ratio measured from 35 to 41 days of age (FCR) and, (viii) body weight gain measured from 35 to 41 days of age (BWG). More detailed descriptions on this population, rearing conditions and phenotype measurements are available in Marchesi et al. [51]. The descriptive statistics for the analyzed phenotypes are shown in Table 6.
DNA extraction, genotyping and quality control
Genomic DNA from 1461 blood samples was extracted using the PureLink® Genomic DNA (Invitrogen, Carlsbad, CA, USA) kit and then quantified using Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). After extraction, DNA integrity was evaluated on agarose gel (1%) and diluted to 10 ng/μL. Diluted genomic DNA was prepared following recommended Affymetrix protocols in order to perform the genotyping analysis using the 600 K Affymetrix Axiom Genotyping Array (Affymetrix®, Inc. Santa Clara, CA, USA, [49]), that contains segregating SNPs for different populations, including commercial broiler lines.
Initially, Axiom™ Analysis Power Tools (Affymetrix®) software v.2.10.2.2 was used to filter genotypes based on DishQC and call rate parameters. A minimum default quality control of 0.82 and a minimum sample call rate of 97% were used. Therefore, only samples with DishQC ≥0.82 and call rate ≥ 97% were kept for following analyses. SNPs in sex chromosomes, and those not mapped in the chicken genome assembly (GRCg6a) were excluded. Only SNPs annotated to autosomal chromosomes from GGA1 to GGA33 were included in the analysis.
From a total of 1461 genotyped chickens, 223 samples were removed from the analysis after applying the DishQC criteria, and a filter on sample call rate ≥ 97% loci. From the total of 580,961 SNPs available on the SNP array, 476,254 informative polymorphic SNPs on the autosomal chromosomes (GGA1–33) were kept after filtering.
Input construction and CNV calling
CNV calling was performed using PennCNV v.1.0.5 [20], an integrated hidden Markov model (HMM) that merges various sources of information, including relative signal intensities (log R Ratio, LRR) and relative allele frequencies (B allele frequency, BAF) at each SNP, the distance between adjacent SNPs, and the population frequency of the B allele (PFB).
The files denominated ‘summary’, ‘calls’ and ‘confidences’ that are built during SNP genotyping and initial data filtering, and are required for signal intensities estimation, were used to extract the LRR and BAF values. First, these files were used to generate canonical clusters [72] by the PennCNV-Affy ‘generate_affy_ geno_cluster.pl’ function, which allows the estimation of the LRR and BAF values by the PennCNV-Affy ‘normalize_affy_geno_cluster.pl’ function. Then, the PFB file was estimated from marker’s individual BAF values, using the PennCNV ‘compile_pfb.pl’ function. Next, the individual-based CNV calling was performed using the -test option with default parameters for the HMM model. Given that the GC ratio content around each SNP marker is known to influence signal strength, creating the so-called genomic waves [73], the LRR of each sample was corrected using the chicken GC content file (i.e., GC content of 1-Mb genomic regions surrounding each SNP) by the -gcmodel option. As long as family structure can be used for generating more accurate CNV calls [20], and pedigree information for a father-mother-offspring trio was available, a family-based CNV detection algorithm was used to jointly update CNV status previously obtained in the individual-based calling step.
For CNV filtering, the default PennCNV standard deviation (SD) criteria for LRR ≤ 0.35, BAF drift< 0.01, and waviness factor ≤ 0.05 were used. Note that the waviness factor represents the dispersion in signal intensity over the genome. Following, CNVs with minor allele frequencies (MAF) less than 0.05 were removed to avoid calling artefacts. Moreover, CNVs smaller than 1 kb were also excluded and only CNVs consisting of at least three consecutive SNPs were retained in the analysis [7]. Lastly, all duplicated CNVs (i.e., same event in the same parental) were removed. Duplicated CNV entries occurred due to half sib families, as some sires and dams were included more than once in PennCNV analysis. The CNV calling was focused only on autosomal chromosomes GGA1 to GGA33 as PennCNV results for sex chromosomes are unreliable and difficult to interpret [20].
CNVR compilation
Individual CNV calls filtered by PennCNV overlapping at least one base pair were concatenated into CNV regions (CNVRs) using the populationRanges (grl, density = 0.1) function from the CNVRanger R/Bioconductor package [74]. Genomic areas with density < 10% were deleted to avoid false positive predictions. The CNVRs were classified as gain or loss. The overlapping CNVRs of ‘gain’ and ‘loss’ were merged into single regions to account for genomic regions in which both events can occur (i.e., ‘both’ CNVRs). The frequency of each CNVR was estimated based on the number of samples mapped at the genomic interval covered by the CNVR.
Genome-wide association analyses
Genome-wide association analyses between performance traits and CNV segments were carried out using the CNVRanger R/Bioconductor package [74]. This procedure was originally proposed by da Silva et al. [23] . First, the CNV segments to be used in the association analyses were established. For that, a state was assigned for each of the SNP probes overlapping a CNV call. Then, we estimated the CNV frequency in each probe and selected only those with frequency above 5% [74]. Finally, selected probes were used to construct the CNV segments based on a CNV-genotype similarity, in which subsequent probes with identical genotype in ≥95% of our population were concatenated to CNV segments. A raw p-value was independently generated for each probe. Following, the raw p-values were corrected using genomic inflation and, the probe with the lowest p-value was selected to represent the CNV segment. Multiple testing correction was carried out using the FDR method [75] generating the q-values for each CNV segment. The following statistical models were used for association analyses:
-
a)
yijkl = μ + Si + Hj + CNVk + al + eijkl
-
b)
\( {y}_{ijklm}=\mu +{S}_i+{H}_j+{CNV}_k+b\left( BW{35}_l-\overline{BW35}\right)+{a}_m+{e}_{ijklm} \)
where yijkl and yijklm are the phenotypic record on the lth or mth animal, respectively, μ is the overall intercept, Si is the fixed effect of the ith sex (i = 1, 2), Hj is the fixed effect of the jth hatch (j = 1, 2, 3, 4, 5), CNVk is the number of copies of a given allele in the genotype of the kth CNV segment (k = 1, …, 191, represented as gain, loss and normal (2n), and coded as 1, − 1 and 0, respectively), b is the linear regression coefficient related to the BW35 effect considered as deviation from the mean (\( \overline{BW35} \)), al and am are the random direct additive genetic effect for the lth or mth animal, respectively, and eijkl and eijklm are the random residual effect for the lth or mth animal, respectively. Note that sex and hatch were included in the models as class effects for all phenotypes, and BW35 was fit as continuous effect only for FI, FCR and BWG (model b). The random components of the models were distributed as \( \mathbf{a}\sim \mathrm{N}\left(0,\mathbf{G}{\upsigma}_{\mathrm{a}}^2\right) \) and \( \mathbf{e}\sim \mathrm{N}\left(0,\mathbf{I}{\upsigma}_{\mathrm{e}}^2\right) \), where \( {\upsigma}_{\mathrm{a}}^2 \) and \( {\upsigma}_{\mathrm{e}}^2 \) are the genetic and residual variances, respectively, G is the CNV-based genomic relationship matrix, and I is an identity matrix.
Lastly, we established two different thresholds. The first corresponded to a suggestive association (FDR-corrected p-value< 0.1) and should be used to identify CNVs for consideration in future studies. The second one corresponded to a significant association (FDR-corrected p-value< 0.05), consequently, highlighting regions more likely to be truly associated with the investigated phenotypes [76].
Validation by qPCR
Quantitative PCR (qPCR) was carried out to validate the significant CNV segments associated with performance traits. Copy number was determined in the 3 significant CNV segments using primer pairs designed to target each segment. It is noteworthy that 3 different primer pairs were designed for each CNV segment to account for possible breakpoints variations. A total of 18 samples, consisting of 3 reference animals (2n) and 3 different testing animals per CNV segment, were selected for the validation process based on the amount of double-stranded DNA (dsDNA) measured with Qubit® 2.0 Fluorometer. Primers designed using Primer3plus [77] were quality tested through NetPrimer (http://www.premierbiosoft.com/netprimer). Additionally, we used the SNPdb [78] against the Ensembl-Biomart tool (http://www.ensembl.org/biomart/martview, [79]) to check the presence of SNPs in the genomic region targeted by the primers.
All primers were previously PCR-tested to verify non-specific amplicons and to optimize qPCR conditions. A qPCR solution of a final 10 μl was used consisting of 5.0 μl PowerUp™ SYBR™ Green Master Mix 2x (Applied Biosystems®, catalog number: A25742), 0.5 μl forward primer (10 mM), 0.5 μl reverse primer (10 mM) and 4.0 μl of genomic DNA (2.5 ng/μl). The reference and testing samples were amplified with the designed primers sets in technical triplicates carried out in QuantStudio™ 12 k Flex machine coupled to QuantStudio 12 K Flex Software v.1.2.2 (Applied Biosystems®). The qPCR thermocycling steps were as follows: 50 °C for 2 min, 95 °C for 2 min and 40 cycles of amplification (95 °C for 15 s, 55–60 °C (primer-dependent) for 15 s and 72 °C for 1 min). The reference samples were randomly chosen from a set predicted by PennCNV to have normal copy number status on all the tested regions.
Cycle thresholds (Ct) were corrected by primer mean efficiency calculated by LinReg [80] and copy number was estimated from normalized ratio method (NR): 2 × 2-(∆∆Ct) [81]. The primers for the propionylcoenzyme A carboxylase gene (PCCA, GGA1) were used as references [82]. Moreover, the control value was estimated based on the average value of ΔCt from reference diploid animals, and copy number states were categorized based on the geometric mean between copy number 1, 2 and 3 [83], where lack of amplification was considered as 0n (complete deletion).
CNV segments overlapping known QTLs
Overlaps of the significant associated CNV segments with previously mapped QTLs for performance traits were determined using information from the Chicken QTLdb - release 43 (https://www.animalgenome.org/cgi-bin/QTLdb/GG/index, [5]. We used the available .bed files with the QTL coordinates based on the GRCg6a genome assembly to check for overlaps using the subsetByOverlaps function from the GenomicRanges R/Bioconductor package [84]. All previously mapped QTLs were reported by QTL ID numbers, available at the Chicken QTLdb [5].
Additionally, we checked the overlapping between the genomic windows covered by the significant CNV segments and the QTLs for growth-related traits reported for the Embrapa F2 Chicken Resource Population, which was originated by crossing sires of the paternal broiler line TT (same line used to obtain the TT Reference Population) and dams of a maternal layer line [37]. The genomic coordinates of the reported QTLs were converted from Gallus_gallus-5.0 to GRCg6a using the LiftOver tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver).
CNV segments overlapping regulatory elements
The overlaps of the significant associated CNV segments with CpG islands were determined using information from the GRCg6a genome assembly, available on UCSC Genome Browser (http://hgdownload.soe.ucsc.edu/goldenPath/galGal6/bigZips/). In addition, NCBI Refseq data, equally obtained from the UCSC Genome Browser (http://hgdownload.soe.ucsc.edu/goldenPath/galGal6/bigZips/genes/galGal6.ncbiRefSeq.gtf.gz), were used to locate TSSs (Transcription Start Sites) in the significant CNV segments and infer promoter regions based on their genomic location (2 kb upstream of a TSS).
Moreover, to check for overlaps of the significant segments with enhancers, we used publicly available H3K27ac ChIP-seq (Chromatin Immunoprecipitation and sequencing) data from two replicates of chicken skeletal muscle, obtained from the Functional Annotation of Animal Genomes Consortium (FAANG, https://www.ncbi.nlm.nih.gov/geo, GSM4799754 and GSM4799755). First, reads were trimmed with Trim Galore! 0.6.5 (https://github.com/FelixKrueger/TrimGalore) using the default arguments, and then aligned with BWA mem 0.7.17 [85] to the GRCg6a genome. The alignments were sorted and indexed with Samtools 1.11 [86] using the arguments -F 1804 and -q 30. We marked the duplicates with Picard toolkit 2.25.1 (http://broadinstitute.github.io/picard/) and removed them with Samtools. Then, aligned reads in .bam files were converted to .bedgraph. The .bed files with significant peaks (−q 0.01) were obtained from GEO (https://www.ncbi.nlm.nih.gov/geo, GSE158430) and then, the final data (.bedgraph and .bed files) were uploaded in IGV (Integrative Genome Viewer) [87] for visualization.
Identification of candidate genes and gene-set analysis
The gene content of significant CNV segments was assessed using Ensembl Release 101 BioMart tool (https://www.ensembl.org/biomart/martview, [79]), based on the GRCg6a genome assembly. We investigated genes flanking genomic intervals for the significant associated CNV segments, corresponding to 1 Mb windows (500 kb up and downstream).
Enrichment analyses were performed with WebGestalt (http://www.webgestalt.org/), a “WEB-based GEne SeT AnaLysis Toolkit” designed for functional genomics, proteomics and large-scale genetic studies [88]. GO-terms for biological process (BP), cellular component (CC) and molecular function (MF) were investigated. Multiple testing correction was carried out using the default option (i.e., Benjamini-Hochberg method [75]). In addition, STRING v.11 (http://string-db.org/, [89]) was used as a complementary approach to search for enriched pathways and protein domains. Conversely to WebGestalt, the STRING database intends to integrate all publicly available sources of protein–protein direct and indirect interaction information to obtain a comprehensive global network.
Availability of data and materials
All data generated and/or analyzed during this study are public and included in this published article. The chicken genome assembly (GRCg6a) was retrieved from the UCSC Genome Browser website (http://hgdownload.soe.ucsc.edu/goldenPath/galGal6/bigZips/). All previously mapped QTLs were reported by QTL ID numbers, available at the Chicken QTLdb (https://www.animalgenome.org/cgi-bin/QTLdb/GG/index). NCBI Refseq data was obtained from the UCSC Genome Browser (http://hgdownload.soe.ucsc.edu/goldenPath/galGal6/bigZips/genes/galGal6.ncbiRefSeq.gtf.gz). The H3K27ac ChIP-seq data are publicly available at the Functional Annotation of Animal Genomes Consortium repository (FAANG, https://www.ncbi.nlm.nih.gov/geo, GEO accession GSM4799754 and GSM4799755). The datasets used and/or analyzed during the current study (genotypes and phenotypes) are available from the corresponding author on reasonable request.
Abbreviations
- aCGH:
-
Comparative Genomic Hybridization array
- BAF:
-
B Alelle Frequency
- BW:
-
Birth Weight
- BW21:
-
Body Weight at 21 days of age
- BW35:
-
Body Weight at 35 days of age
- BW41:
-
Body Weight at 41 days of age
- BW42:
-
Body Weight at 42 days of age
- BWA:
-
Burrows-Wheeler Aligner
- BWG:
-
Body Weight Gain
- ChIP-seq:
-
Chromatin Immunoprecipitation and sequencing
- CNV:
-
Copy Number Variation
- CNVR:
-
Copy Number Variation Region
- Ct:
-
Cycle threshold
- dsDNA:
-
double-stranded DNA
- FAANG:
-
Functional Annotation of Animal Genomes
- FCR:
-
Feed Conversion Ratio
- FDR:
-
False Discovery Rate
- FI:
-
Feed Intake
- GO:
-
Gene Ontology
- GWAS:
-
Genome-Wide Association Study
- H3K27ac:
-
Histone H3 acetylated at lysine 27
- IGV:
-
Integrative Genome Viewer
- KATP:
-
ATP-sensitive potassium channel
- LD:
-
Linkage Disequilibrium
- LOH:
-
Loss of Heterozygosity
- LRR:
-
Log R Ratio
- MAF:
-
Minor Allele Frequency
- NCBI:
-
National Center for Biotechnology Information
- PCCA:
-
propionylcoenzyme A carboxylase
- PFB:
-
Population Frequency of the B Allele
- qPCR:
-
quantitative Polymerase Chain Reaction
- QTL:
-
Quantitative Trait Locus
- RFI:
-
Residual Feed Intake
- SNP:
-
Single Nucleotide Polymorphism
- STRING:
-
Search Tool for the Retrieval of Interacting Genes/Proteins
- TSS:
-
Transcription Start Site
- USCS:
-
University of California Santa Cruz
- WebGestalt:
-
WEB-based GEne SeT AnaLysis Toolkit
References
Wolpert L. Much more from the chicken's egg than breakfast--a wonderful model system. Mech Dev. 2004;121(9):1015–7.
USDA. Livestock and Poultry: World Markets and Trade. 2020.
Dodgson J, Delany M, Cheng H. Poultry genome sequences: progress and outstanding challenges. Cytogenet Genome Res. 2011;134(1):19–26.
Hillier L, Miller W, Birney E, Warren W, Hardison R, Ponting C, et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432(7018):695–716.
Hu Z, Park C, Wu X, Reecy J. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013;41(Database issue):D871–9.
Santana M, Junior G, Cesar A, Freua M, da Costa Gomes R, da Luz E Silva S, et al. Copy number variations and genome-wide associations reveal putative genes and metabolic pathways involved with the feed conversion ratio in beef cattle. J Appl Genet. 2016;57(4):495–504.
Gorla E, Cozzi M, Román-Ponce S, Ruiz López F, Vega-Murillo V, Cerolini S, et al. Genomic variability in Mexican chicken population using copy number variants. BMC Genet. 2017;18(1):61.
Bai H, Sun Y, Liu N, Liu Y, Xue F, Li Y, et al. Genome-wide detection of CNVs associated with beak deformity in chickens using high-density 600K SNP arrays. Anim Genet. 2018;49(3):226–36.
Zheng X, Zhao P, Yang K, Ning C, Wang H, Zhou L, et al. CNV analysis of Meishan pig by next-generation sequencing and effects of AHR gene CNV on pig reproductive traits. J Anim Sci Biotechnol. 2020;11(1):42.
Clop A, Vidal O, Amills M. Copy number variation in the genomes of domestic animals. Anim Genet. 2012;43(5):503–17.
Redon R, Ishikawa S, Fitch K, Feuk L, Perry G, Andrews T, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–54.
Geistlinger L, da Silva V, Cesar A, Tizioto P, Waldron L, Zimmer R, et al. Widespread modulation of gene expression by copy number variation in skeletal muscle. Sci Rep. 2018;8(1):1399.
Carter N. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat Genet. 2007;39(7 Suppl):S16–21.
Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20(2):207–11.
Yau C, Holmes C. CNV discovery using SNP genotyping arrays. Cytogenet Genome Res. 2008;123(1–4):307–12.
Zhou B, Ho S, Zhang X, Pattni R, Haraksingh R, Urban A. Whole-genome sequencing analysis of CNV using low-coverage and paired-end strategies is efficient and outperforms array-based CNV analysis. J Med Genet. 2018;55(11):735–43.
Wang Z, Guo J, Guo Y, Yang Y, Teng T, Yu Q, et al. Genome-wide detection of CNVs and association with body weight in sheep based on 600K SNP arrays. Front Genet. 2020;11:558.
Zhang D, Qian Y, Akula N, Alliey-Rodriguez N, Tang J, Gershon E, et al. Accuracy of CNV Detection from GWAS Data. PLoS One. 2011;6(1):e14511.
Zhang X, Du R, Li S, Zhang F, Jin L, Wang H. Evaluation of copy number variation detection for a SNP array platform. BMC Bioinformatics. 2014;15(1):50.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant S, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17(11):1665–74.
Dupuis M, Zhang Z, Durkin K, Charlier C, Lekeux P, Georges M. Detection of copy number variants in the horse genome and examination of their association with recurrent laryngeal neuropathy. Anim Genet. 2013;44(2):206–8.
Chen C, Qiao R, Wei R, Guo Y, Ai H, Ma J, et al. A comprehensive survey of copy number variation in 18 diverse pig populations and identification of candidate copy number variable genes associated with complex traits. BMC Genomics. 2012;13(1):733.
da Silva V, Regitano L, Geistlinger L, Pértille F, Giachetto P, Brassaloti R, et al. Genome-Wide Detection of CNVs and Their Association with Meat Tenderness in Nelore Cattle. PLoS One. 2016;11(6):e0157711.
Nones K, Ledur M, Ruy D, Baron EE, Melo C, Moura A, et al. Mapping QTLs on chicken chromosome 1 for performance and carcass traits in a broiler x layer cross. Anim Genet. 2006;37(2):95–100.
Moreira G, Boschiero C, Cesar A, Reecy J, Godoy T, Trevisoli P, et al. A genome-wide association study reveals novel genomic regions and positional candidate genes for fat deposition in broiler chickens. BMC Genomics. 2018;19(1):374.
Yi G, Qu L, Chen S, Xu G, Yang N. Genome-wide copy number profiling using high-density SNP array in chickens. Anim Genet. 2015;46(2):148–57.
Elferink M, Vallée A, Jungerius A, Crooijmans R, Groenen M. Partial duplication of the PRLR and SPEF2 genes at the late feathering locus in chicken. BMC Genomics. 2008;9(1):391.
Wright D, Boije H, Meadows J, Bed'hom B, Gourichon D, Vieaud A, et al. Copy number variation in intron 1 of SOX5 causes the Pea-comb phenotype in chickens. PLoS Genet. 2009;5(6):e1000512.
Dorshorst B, Okimoto R, Ashwell C. Genomic regions associated with dermal hyperpigmentation, polydactyly and other morphological traits in the Silkie chicken. J Hered. 2010;101(3):339–50.
Gunnarsson U, Kerje S, Bed'hom B, Sahlqvist A, Ekwall O, Tixier-Boichard M, et al. The Dark brown plumage color in chickens is caused by an 8.3-kb deletion upstream of SOX10. Pigment Cell Melanoma Res. 2011;24(2):268–74.
Yan Y, Yang N, Cheng H, Song J, Qu L. Genome-wide identification of copy number variations between two chicken lines that differ in genetic resistance to Marek's disease. BMC Genomics. 2015;16:843.
Itsara A, Cooper G, Baker C, Girirajan S, Li J, Absher D, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84(2):148–61.
Demeure O, Duclos M, Bacciu N, Le Mignon G, Filangi O, Pitel F, et al. Genome-wide interval mapping using SNPs identifies new QTL for growth, body composition and several physiological variables in an F2 intercross between fat and lean chicken lines. Genet Sel Evol. 2013;45(1):36.
Sun Y, Liu R, Zhao G, Zheng M, Sun Y, Yu X, et al. Genome-Wide Linkage Analysis Identifies Loci for Physical Appearance Traits in Chickens. G3 (Bethesda, Md). 2015;5(10):2037–41.
Mignon-Grasteau S, Rideau N, Gabriel I, Chantry-Darmon C, Boscher M, Sellier N, et al. Detection of QTL controlling feed efficiency and excretion in chickens fed a wheat-based diet. Genet Sel Evol. 2015;47:74.
Zhang H, Shen L, Xu Z, Kramer L, Yu J, Zhang X, et al. Haplotype-based genome-wide association studies for carcass and growth traits in chicken. Poult Sci. 2020;99(5):2349–61.
Pértille F, Moreira G, Zanella R, Nunes J, Boschiero C, Rovadoscki G, et al. Genome-wide association study for performance traits in chickens using genotype by sequencing approach. Sci Rep. 2017;7(1):41748.
Creyghton M, Cheng A, Welstead G, Kooistra T, Carey B, Steine E, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107(50):21931–6.
Hastings P, Lupski J, Rosenberg S, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10(8):551–64.
Lee Y, Bosse M, Mullaart E, Groenen M, Veerkamp R, Bouwman A. Functional and population genetic features of copy number variations in two dairy cattle populations. BMC Genomics. 2020;21(1):89.
Schrider D, Hahn M. Lower linkage disequilibrium at CNVs is due to both recurrent mutation and transposing duplications. Mol Biol Evol. 2010;27(1):103–11.
Yi G, Qu L, Liu J, Yan Y, Xu G, Yang N. Genome-wide patterns of copy number variation in the diversified chicken genomes using next-generation sequencing. BMC Genomics. 2014;15(1):962.
Sohrabi S, Mohammadabadi M, Wu D, Esmailizadeh A. Detection of breed-specific copy number variations in domestic chicken genome. Genome. 2018;61(1):7–14.
Conrad D, Andrews T, Carter N, Hurles M, Pritchard J. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006;38(1):75–81.
Momen M, Mehrgardi A, Sheikhy A, Esmailizadeh A, Fozi M, Kranis A, et al. A predictive assessment of genetic correlations between traits in chickens using markers. Genet Sel Evol. 2017;49(1):16.
Mebratie W, Reyer H, Wimmers K, Bovenhuis H, Jensen J. Genome wide association study of body weight and feed efficiency traits in a commercial broiler chicken population, a re-visitation. Sci Rep. 2019;9(1):922.
Abasht B, Dekkers J, Lamont S. Review of quantitative trait loci identified in the chicken. Poult Sci. 2006;85(12):2079–96.
Moreira G, Poleti M, Pértille F, Boschiero C, Cesar A, Godoy T, et al. Unraveling genomic associations with feed efficiency and body weight traits in chickens through an integrative approach. BMC Genet. 2019;20(1):83.
Kranis A, Gheyas A, Boschiero C, Turner F, Yu L, Smith S, et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genomics. 2013;14(1):59.
Zhang F, Gu W, Hurles M, Lupski J. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10(1):451–81.
Marchesi J, Buzanskas M, Cantão M, Ibelli A, Peixoto J, Joaquim L, et al. Relationship of runs of homozygosity with adaptive and production traits in a paternal broiler line. Animal. 2018;12(6):1126–34.
Johansson A, Pettersson M, Siegel P, Carlborg O. Genome-wide effects of long-term divergent selection. PLoS Genet. 2010;6(11):e1001188.
Stadler M, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480(7378):490–5.
Willson N, Forder R, Tearle R, Williams J, Hughes R, Nattrass G, et al. Transcriptional analysis of liver from chickens with fast (meat bird), moderate (F1 layer x meat bird cross) and low (layer bird) growth potential. BMC Genomics. 2018;19(1):309.
Lindholm-Perry A, Freetly H, Oliver W, Rempel L, Keel B. Genes associated with body weight gain and feed intake identified by meta-analysis of the mesenteric fat from crossbred beef steers. PLoS One. 2020;15(1):e0227154.
Wang X, Wei C, Zhang Z, Liu D, Guo Y, Sun G, et al. Association of growth traits with a structural variation downstream of the KCNJ11 gene: a large population-based study in chickens. Br Poult Sci. 2020;61(3):320–7.
Liu W, Zhao J. Insights into the molecular mechanism of glucose metabolism regulation under stress in chicken skeletal muscle tissues. Saudi J Biol Sci. 2014;21(3):197–203.
Tricarico D, Selvaggi M, Passantino G, De Palo P, Dario C, Centoducati P, et al. ATP Sensitive Potassium Channels in the Skeletal Muscle Function: Involvement of the KCNJ11(Kir6.2) Gene in the Determination of Mechanical Warner Bratzer Shear Force. Front Physiol. 2016;7:167.
Alekseev A, Reyes S, Yamada S, Hodgson-Zingman D, Sattiraju S, Zhu Z, et al. Sarcolemmal ATP-sensitive K(+) channels control energy expenditure determining body weight. Cell Metab. 2010;11(1):58–69.
Pownall M, Gustafsson M, Emerson C. Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu Rev Cell Dev Biol. 2002;18(1):747–83.
Ling B, Bharathy N, Chung T, Kok W, Li S, Tan Y, et al. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc Natl Acad Sci U S A. 2012;109(3):841–6.
Coutinho L, Morris J, Marks H, Buhr R, Ivarie R. Delayed somite formation in a quail line exhibiting myofiber hyperplasia is accompanied by delayed expression of myogenic regulatory factors and myosin heavy chain. Development (Cambridge, England). 1993;117(2):563–9.
Macqueen D, Johnston I. An update on MyoD evolution in teleosts and a proposed consensus nomenclature to accommodate the tetraploidization of different vertebrate genomes. PLoS One. 2008;3(2):e1567.
Tiosano D, Baris H, Chen A, Hitzert M, Schueler M, Gulluni F, et al. Mutations in PIK3C2A cause syndromic short stature, skeletal abnormalities, and cataracts associated with ciliary dysfunction. PLoS Genet. 2019;15(4):e1008088.
Behrends C, Sowa M, Gygi S, Harper J. Network organization of the human autophagy system. Nature. 2010;466(7302):68–76.
Lin S, Lin X, Zhang Z, Jiang M, Rao Y, Nie Q, et al. Copy Number Variation in SOX6 Contributes to Chicken Muscle Development. Genes. 2018;9(1):42.
Zhang S, Li S, Chen W, Lu W, Huang Y. A single-nucleotide polymorphism in the 3′ untranslated region of the LPIN1 gene and association analysis with performance traits in chicken. Br Poult Sci. 2013;54(3):312–8.
Zhao Q, Liao R, Sun H, Zhang Z, Wang Q, Yang C, et al. Identifying Genetic Differences Between Dongxiang Blue-Shelled and White Leghorn Chickens Using Sequencing Data. G3 (Bethesda, Md). 2018;8(2):469–76.
Zhang H, Na W, Zhang H, Wang N, Du Z, Wang S, et al. TCF21 is related to testis growth and development in broiler chickens. Genet Sel Evol. 2017;49(1):25.
Marchesi J, Ibelli A, Peixoto J, Cantão M, Pandolfi J, Marciano C, et al. Whole transcriptome analysis of the pectoralis major muscle reveals molecular mechanisms involved with white striping in broiler chickens. Poult Sci. 2019;98(2):590–601.
Foskett J. Structures of CALHM channels revealed. Nat Struct Mol Biol. 2020;27(3):227–8.
Peiffer D, Le J, Steemers F, Chang W, Jenniges T, Garcia F, et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16(9):1136–48.
Diskin S, Li M, Hou C, Yang S, Glessner J, Hakonarson H, et al. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008;36(19):e126.
da Silva V, Ramos M, Groenen M, Crooijmans R, Johansson A, Regitano L, et al. CNVRanger: association analysis of CNVs with gene expression and quantitative phenotypes. Bioinformatics (Oxford, England). 2019;36(3):972–3.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300.
Duggal P, Gillanders E, Holmes T, Bailey-Wilson J. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genomics. 2008;9(1):516.
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen J. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35(Web Server issue):W71–4.
Sherry S, Ward M, Kholodov M, Baker J, Phan L, Smigielski E, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Kinsella R, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database. 2011;2011:bar030.
Ramakers C, Ruijter J, Deprez R, Moorman A. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003;339(1):62–6.
Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods (San Diego, Calif). 2001;25(4):402–8.
Wang X, Nahashon S, Feaster T, Bohannon-Stewart A, Adefope N. An initial map of chromosomal segmental copy number variations in the chicken. BMC Genomics. 2010;11(1):351.
D'haene B, Vandesompele J, Hellemans J. Accurate and objective copy number profiling using real-time quantitative PCR. Methods (San Diego, Calif). 2010;50(4):262–70.
Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9(8):e1003118.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 2010;26(5):589–95.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England). 2009;25(16):2078-9.
Robinson J, Thorvaldsdóttir H, Winckler W, Guttman M, Lander E, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6.
Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33(Web Server issue):W741–8.
Szklarczyk D, Gable A, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Acknowledgements
The authors are grateful for support from the CNPq mentioned in the funding statement. The authors would like to acknowledge the collaborative efforts between EMBRAPA and University of São Paulo.
Funding
The TT Reference Population was subsidized by the National Council of Scientific and Technological Development (CNPq) grant number 481755/2007–1 from the Brazilian Government. This study was conducted with support from the CNPq (process number 407970/2018–5), the Brazilian Agricultural Research Corporation – Embrapa (project number 01.11.07.002.04.02) and the São Paulo Research Foundation (FAPESP, process number 2014/08704–0). A.C. Fernandes received fellowship from CNPq (grant 134211/2019–7). L.L. Coutinho and M.C. Ledur are recipients of productivity fellowship from CNPq. FAPESP, CAPES, CNPq and Brazilian Government through Embrapa provided financial support to generate the data, however, they did not participate in the design of the study, sample collection, analysis, data interpretation and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
ACF, GCMM, JOP, MCL and LLC conceived the idea of this research and participated in the experimental design. AMGI carried out the DNA extraction, quantification and sample preparation for genotyping. GCMM and MEC conducted the quality control analysis. ACF, VHS and CPG performed experiments and data analysis. ACF drafted the manuscript. ACF, VHS, CPG, GCMM, TFG, AMGI, JOP, MEC, MCL, FMR and LLC collaborated with interpretation, discussion and writing of the manuscript. JOP, MCL and LLC provided support for the experimental environment, phenotype collection and data analysis. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All protocols related to animal experimentation in this study were conducted in agreement with protocol number 011/2010, approved by the Embrapa Swine and Poultry Ethical Committee for Animal Use (CEUA) in Concórdia, Santa Catarina State – South of Brazil, following international guidelines for animal welfare.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Information of identified CNVRs
Additional file 2
Manhattan plots for CNV segments across the 33 autosomal chromosomes associated with (a) birth weight, (b) body weight at 35 days, (c) body weight at 41 days and (d) body weight at 42 days and (e) body weight gain. The X-axis represents the somatic chromosomes, and Y-axis shows the corresponding -log10 p-value. Red and blue lines indicate p-values of 0.05 and 0.1, respectively
Additional file 3
QQ-plots show the relation of normal theoretical quantiles of the probability distributions between expected (X-axis) and observed (Y-axis) p-values from (a) birth weight, (b) body weight at 35 days, (c) body weight at 41 days, (d) body weight at 42 days and (e) body weight gain
Additional file 4.
Information of the CNV segments validated by qPCR and the primers used for qPCR
Additional file 5.
Validation rates of 9 samples
Additional file 6.
H3K27ac and CpG islands tracks alingment with significant CNV segments. H3K27ac ChIP-seq data of chicken skeletal muscle (blue and red tracks) overlap with the significant CNV segment on chr5:12059966–12062666 (green bar) near the KCNJ11 gene (black bar). The blue rows represent replicate A, while red rows represent replicate B. The overlap with the H3K27ac enriched region is indicated by the dashed box. No overlap was found between the significant CNV segments on chr3:64169030–64171297 or chr3:97801202–97809208 and H3K27ac regions. CpG islands were only located in promoter regions and TSSs (orange track)
Additional file 7.
List with the 32 genes annotated within a 1-Mb window of significant CNV segments
Additional file 8.
78 publications significantly enriched in the STRING network
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fernandes, A.C., da Silva, V.H., Goes, C.P. et al. Genome-wide detection of CNVs and their association with performance traits in broilers. BMC Genomics 22, 354 (2021). https://doi.org/10.1186/s12864-021-07676-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07676-1