
Abstract
Background
Heat stress (HS) is a major stress event in the life of an animal, with detrimental upshots in production and health. Long-non-coding RNAs (lncRNAs) play an important role in many biological processes by transcriptional regulation. However, no research has been reported on the characterization and functionality of lncRNAs in heat-stressed rats.
Results
We studied expression levels of lncRNAs in rats during HS, using strand-specific RNA sequencing. Six rats, three in each of Control (22 ± 1 °C) and H120 (42 °C for 120 min) experimental groups, were used to screen for lncRNAs in their liver and adrenal glands. Totally, 4498 and 7627 putative lncRNAs were identified in liver and adrenal glands of the Control and H120 groups, respectively. The majority of lncRNAs were relatively shorter and contained fewer exons than protein-coding transcripts. In total, 482 (174 up-regulated and 308 down-regulated) and 271 (126 up-regulated and 145 down-regulated) differentially-expressed lncRNAs (DElncRNAs, P < 0.05) were identified in the liver and adrenal glands of the Control and H120 groups, respectively. Furthermore, 1274, 121, and 73 target differentially-expressed genes (DEGs) in the liver were predicted to interact with DElncRNAs based on trans−/cis- and sequence similarity regulatory modes. Functional annotation analyses indicated that these DEGs were mostly significantly enriched in insulin signalling, myeloid leukaemia, and glucagon signalling pathways. Similarly, 437, 73 and 41 target DEGs in the adrenal glands were mostly significantly enriched in the cell cycle (trans-prediction) and lysosome pathways (cis-prediction). The DElncRNAs interacting with DEGs that encode heat shock proteins (HSPs) may play an important role in HS response, which include Hsf4, Dnaja1, Dnajb4, Hsph1 and Hspb1 in the liver, and Dnajb13 and Hspb8 in the adrenal glands. The strand-specific RNA sequencing findings were also further verified through RT-qPCR.
Conclusions
This study is the first to provide a detailed characterization and functional analysis of expression levels of lncRNAs in liver and adrenal glands of heat-stressed rats, which provides basis for further studies on the biological functions of lncRNAs under heat stress in rats and other mammalian species.
Similar content being viewed by others

Background
Heat stress (HS) is one of the main abiotic stressors that influence human and animal survival, welfare, and development [1,2,3]. Escalating global warming, combined with the global increase in the number of production animals and the intensification of agriculture [4, 5], has resulted in HS becoming a difficult challenge for livestock and poultry production. Heat stress leads to enormous economic losses to the livestock production industry [6], stemming from reduced production of meat, egg, and milk, decreased fertility, and increased morbidity and mortality [4, 7, 8]. The current trends in the increase of global temperature [9, 10] indicate that it is necessary and urgent to comprehensively investigate the genetic and biological mechanisms of HS, as well as develop long-lasting, cumulative, and significant strategies for preventing HS.
Over the past decades, HS research has been carried out in many species, such as humans [11], cattle [12], pigs [13, 14], corals [15], and rats [16, 17]. However, the regulatory mechanisms of HS are still unclear. Transcriptome sequencing technology for animals [18, 19] and cells [20] is becoming a suitable method for exploring HS-related genes and biological pathways. Studies have reported thousands of differentially-expressed genes (DEGs) under certain HS conditions [21,22,23,24]. There are many processes that affect the expression of genes, such as the regulation of long non-coding RNA (lncRNA) [25]. LncRNA is a non-coding RNA longer than 200 nucleotides in length and with more than two exons. LncRNA can regulate gene expression at the transcription and post-transcription levels [26]. Previous studies have reported several lncRNAs playing crucial role in HS response through interaction with transcription factors [27] or feedback regulation of key stress response proteins [28, 29]. Heat shock response is a major and crucial defence mechanism during HS, which contributes to cell recovery from heat shock damages, e.g., protein misfolding and aggregation [30, 31]. Furthermore, several lncRNAs have been identified in animals under HS conditions [32,33,34]. However, the understanding of the contributions of lncRNAs to the cellular HS response is still unclear.
The liver and adrenal glands play a key role in maintaining animal homeostasis during HS [19, 35, 36], but the role of lncRNA during this process still requires in-depth investigation. Therefore, the main aim of this study was to perform a transcriptomic analysis of rat liver and adrenal glands, following exposure to HS, to identify related DEGs, differentially-expressed lncRNAs (DElncRNAs), and key biological pathways related to HS response in rats. Our findings will contribute to a better understanding of the regulatory mechanisms of HS response in rats and other mammals.
Results
Comprehensive identification of lncRNAs in liver and adrenal glands
A total of ~ 29.9 and 28.3 million raw reads in the liver and adrenal glands were obtained (Additional file 2: Table S2), in which 29.8 and 28.1 million clean reads were aligned to the reference genome (Ensemble release version Rnor 6.0.91). The average mapping rate of clean reads in the liver and adrenal glands was 95.71 and 92.99%, respectively. Subsequently, 484,530 and 613,791 unique transcripts, both in liver and adrenal glands, were assembled from H120 and Control rats, respectively.
Five filtering steps were performed for identifying candidate lncRNA (Fig. 1). Firstly, the assembled transcripts were filtered with rat coding gene sequences. Almost 72.72% (352,401) and 72.79% (446,801) of transcripts in liver and adrenal glands are coding genes, and the remaining 27.27% (132,129) and 27.21% (166,990) of transcripts are considered to be non-coding. Secondly, the transcripts that might encode conserved protein domains were further filtered out by comparing them to two protein databases including (National Center for Biotechnology Information) NCBI non-redundant (NR) protein database and Universal Protein Resource (UniProt) database and, as a result, 12,840 and 20,850 transcripts in the liver and adrenal glands were retained. LncRNAs are usually defined as non-coding RNAs longer than 200 nucleotides and having more than two exons. Based on these features, a third filter was applied, and 4840 (37.52%) transcripts in the liver and 8258 (39.61%) transcripts in the adrenal glands were removed. Finally, the coding-non-coding index (CNCI), the coding potential assessment tool (CPAT), and the predictor of lncRNAs and mRNAs based on the k-mer scheme (PLEK) were used to evaluate the protein-coding potential, and 4498 and 7627 transcripts in the liver and adrenal gland tissues were retained (Fig. 2). After employing the four above mentioned stringent filters, transcripts expressed only in one sample were also removed. Finally, 4498 and 7627 transcripts in the liver and adrenal gland tissues were considered as putative lncRNAs (Fig. 2).

The detailed schematic pipeline of long-non-coding RNA (lncRNA) transcripts identification. Control was kept at room temperature (22 ± 1 °C, relative humidity [RH] (%): 50%); H120 were subjected to 42 °C and RH 50% for 120 min. NR: (National Center for Biotechnology Information) NCBI non-redundant (NR) protein database; UniProt: Universal Protein Resource; CNCI: coding-non-coding index; CPAT: the coding potential assessment tool; PLEK: predictor of lncRNAs and mRNAs based on the k-mer scheme

The Venn diagram for prediction of coding potential of non-coding transcripts in liver and adrenal glands. The > 2 SAMPLES means that only transcripts identified in at least two samples were retained for further analyses
Classification and characterization of lncRNAs in liver and adrenal glands
According to the location relative to the nearest protein-coding gene (PCG), lncRNAs in the liver and adrenal glands were further classified into four types, including intergenic, intronic, sense, and antisense (Fig. 3a). About 45.75% of lncRNAs in the liver (Fig. 3a_left panel) and 57.31% of lncRNAs in the adrenal glands (Fig. 3a_right panel) were located in intergenic regions, whereas 23.08 and 30.35% lncRNAs were transcripts most from introns. In addition, 19.45% of lncRNAs in the liver were antisense of PCGs, which were more frequent than those lncRNAs that overlapped with genes (11.72%). The same feature was also found in adrenal glands, i.e. the number of antisense lncRNAs was 2.44 times greater than that of sense lncRNAs (Fig. 3a_right panel).

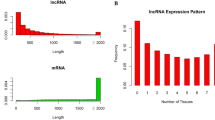
The classification and characterization of lncRNAs identified in liver and adrenal glands. a Number of lncRNAs in different categories. b Transcript lengths of protein-coding transcripts and lncRNAs. c Number of exons per transcript for protein-coding transcripts and lncRNAs. Left panel depicts results for liver and right panel for adrenal glands
Figure 3b and c show the transcript length and number of exons of lncRNAs compared to protein-coding transcripts. Figure 3b shows that almost 70.8% of lncRNAs in the liver ranged in size from 200 to 1000 nucleotides, with only 29.20% > 1000 nucleotides. In contrast, about 86.15% of protein-coding transcripts were > 1000 nucleotides (Fig. 3b_left panel). In the adrenal glands, similar characteristics of lncRNAs and protein-coding transcripts were observed with 55.79% of lncRNAs having > 1000 nucleotides and 90.09% of protein-coding transcripts having> 1000 nucleotides (Fig. 3b_right panel). Interestingly, most of the lncRNAs of the liver and adrenal glands (86.11 and 86.89%, respectively) contained two to three exons, while the number of exons of protein-coding transcripts ranged from two to over ten (Fig. 3c). These statistics indicated that the majority of lncRNAs were relatively shorter and contained fewer exons than protein-coding transcripts.
Identification of temperature-dependent differentially-expressed lncRNAs (DElncRNAs)
A total of 482 and 271 DElncRNAs (P < 0.05) in the liver and adrenal glands were obtained and further divided into six categories according to fold change (FC) values (Table 1 and Additional file 3: Table S3). The top 20 DElncRNAs in the liver (12 up-regulated and 8 down-regulated) and adrenal glands (11 up-regulated and 9 down-regulated) were used for clustering analyses, which indicated clearly-clustered results (Fig. 4a). Three samples (rats) for each treatment group were clustered together. Additionally, 13 DElncRNAs were shared between the liver and adrenal glands (Fig. 4b), 469 and 258 DElncRNAs were identified in the liver and adrenal glands, respectively, as having tissue-specific expression. Among which, most lncRNAs (63.54%) were down-regulated in the liver, and over half (54.6%) of lncRNAs were down regulated in the adrenal glands. The log-transformed relative expression FC of ten lncRNAs in H120 and Control groups generated from real-time quantitative PCR (RT-qPCR) were in line with the results of RNA-seq data (Fig. 4c). The Pearson correlation coefficient (PCC) between RT-qPCR and RNA-seq was as high as 0.88, which confirmed the reliability of the RNA-seq analysis.

Hierarchical clustering and validation analysis of the specific differentially-expressed lncRNAs (DElncRNAs). a The Pheatmap of the top20 DElncRNAs in liver and adrenal glands. b The Pheatmap of commonly identified DElncRNAs in liver and adrenal glands. c The comparative analysis of the expression level of randomly selected lncRNAs in liver and adrenal glands using RNA-seq and RT-qPCR. The log (10 + 1)-transformed FPKM values of DElncRNAs (rows) are clustered using hierarchical clustering, and the samples are grouped according to the similarity of expression profiles of DElncRNAs
Functional prediction of DElncRNAs
Construction of co-expression network between DElncRNAs and target DEGs
A total of 3909 and 4953 DEGs (q < 0.05) were identified in rat liver and adrenal glands in a previous study [22]. The co-expression network between DElncRNAs and DEGs in the liver and adrenal gland tissues was created (Fig. 5). 1,935,712 connections between DElncRNAs and DEGs in the liver were identified, in which 44.46% were positive connections, and 55.54% were negative connections (Fig. 5a_left panel). Furthermore, among all relationships, the PCCs of 14.79% were between − 0.8 and − 0.6 and followed by 13.16% connections between 0.8 and 1.0. In the adrenal glands, 1,492,397 links were identified between DElncRNAs and DEGs; the positive and negative associations were 47.00 and 53.00%, respectively. Moreover, most PCCs between DElncRNAs and DEGs in the adrenal glands (15.41%) ranged from − 0.8 to − 0.6, followed by 12.14% PCCs between − 0.6 and − 0.4 (Fig. 5a_right panel). In order to better indicate the relationship between the DElncRNAs and DEGs, the connections with high correlation |PCC| > 0.99 were selected for further analyses (Fig. 5b). Three thousand seven hundred twenty-five connections including 317 DElncRNAs and 1274 DEGs, and 1969 connections including 139 DElncRNAs and 437 DEGs in the liver and adrenal glands were retained (Additional file 4: Table S4). All connections between DElncRNAs and DEGs were then divided into 6 or 7 categories in the liver and adrenal glands, respectively (Fig. 5b). The largest number of connections between DElncRNAs and DEGs in the liver was identified in the cluster of one DElncRNAs interacting with 11 ~ 20 DEGs, which includes 81 unique DElncRNAs and 648 DEGs. Only one DElncRNA (TCONS_00000716) was found to interact with 57 DEGs when HS occurred. Four hundred eighty-two connections in the adrenal glands were clustered in the classification of one DElncRNA interacting with 21 ~ 30 DEGs, which includes 20 unique DElncRNAs and 207 unique DEGs. Therefore, Fig. 5 shows that multiple lncRNAs might regulate one DEG and, on the contrary, multiple DEGs may be regulated by a single lncRNA.

Statistical analysis of co-expression and the classification of the correlation frequency between DElncRNAs and differentially-expressed genes (DEGs). a Pearson correlation coefficient (PCC) analyses between DElncRNAs and DEGs in liver and adrenal glands. b Statistical analysis of the number of DEGs related to the same DElncRNAs
The functions of 1274 and 437 DEGs interacting with 317 and 139 DElncRNAs in the liver and adrenal glands were annotated (Additional file 5: Table S5 and Additional file 6: Figure S1A). In the liver, 1274 DEGs were significantly enriched (P < 0.05) in 124 biological process (BP) terms, such as response to heat (GO: 0009408), response to hypoxia (GO: 0001666), response to unfolded protein (GO: 0006986), and biosynthesis and metabolism of glucose and fat acid (e.g., GO: 0042593, GO: 0006633 and GO: 0071397). The Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed 20 significantly enriched pathways (P < 0.05) in liver (Additional file 6: Figure S1A_left panel), some of which were associated with glucose and fat acid metabolism (e.g., adipocytokine signaling pathway), hormone regulation (e.g., estrogen signaling pathway), and cancer pathways (e.g., PPAR signaling pathway), suggesting that HS response may be a complex process comprising of neurohormonal regulation, energy metabolism, and immune response. Twenty-six BPs were significantly enriched (P < 0.05) by 437 DEGs in the adrenal glands (Additional file 5: Table S5), with three of them shared with liver, i.e. glycosaminoglycan biosynthetic process (GO: 0006024), protein phosphorylation (GO: 0006468) and cellular response to amino acid starvation (GO: 0034198). Furthermore, five significant pathways (P < 0.05) were detected (Additional file 6: Figure S1A_right panel), but none of them were shared in the liver.
Cis-prediction of DElncRNAs
A total of 512 and 545 genes were predicted in the liver and adrenal glands, with 121 and 191 DEGs (Additional file 7: Table S6). Functional annotation of all the predicted genes (Additional file 8: Table S7) and only the DEGs (Additional file 9: Table S8 and Additional file 6: Figure S1B) were performed. In the liver, 121 DEGs were significantly enriched (P < 0.05) in 13 BPs, including the radial glial cell differentiation (GO: 0060019) with the highest fold enrichment score of 67.44, followed by CDP-choline pathway (GO: 0006657) and JAK-STAT cascade involved in growth hormone signaling pathway (GO: 0060397). All DEGs in the liver were enriched in five pathways, and only one pathway, acute myeloid leukemia (rno05221), was significantly enriched (P < 0.05) under the H120 treatment. In the adrenal glands, 33 BPs (Additional file 9: Table S8), as well as four pathways [e.g., lysosome (rno04142), peroxisome (rno04146), gap junction (rno04540) and NF-kappa B signaling pathway (rno04064)], were significantly enriched (P < 0.05). Furthermore, the NF-kappa B signaling pathway has been shown to play a crucial and major role during heat stress response through activating autophagy [37].
Identification of DElncRNAs & DEGs interaction based on similarity search method
In order to perform the functional prediction for the DElncRNAs more comprehensively, the potential lncRNA-mRNA interactions based on the similarity-search method was investigated. Overall, 17,251 potential RNA-mRNA interactions in the liver were detected between 1180 DElncRNAs and 364 genes, and 9917 potential RNA-mRNA interactions between 1985 DElncRNAs and 171 genes were identified in the adrenal glands (Additional file 10: Table S9). In the liver, functional enrichment analysis of the 364 genes revealed 28 significantly enriched BPs (P < 0.05), which were mainly engaged in cell proliferation, positive regulation of GTPase activity and vesicle-mediated transport (Additional file 11: Table S10). For the KEGG analysis, eight pathways were detected, two of which are related to cellular growth and development (P < 0.05; Additional file 6: Figure S1C_left panel). Furthermore, 26 BPs were identified in the adrenal glands (P < 0.05), with some pathways positively regulating the transcription from the RNA polymerase II promoter, positively regulating transcription, DNA-template, and stimulating cell and neuron development (Additional file 11: Table S10). Twenty significantly regulated pathways were detected in the adrenal glands (Additional file 6: Figure S1C_right panel), 15 of them are directly engaged in different types of cancer, suggesting that innate immunity and inflammation-related pathways in the adrenal glands could be mobilized to respond to HS. In addition, pathways such as the MAPK signaling pathway [38] and the GnRH signaling pathway [39] have been reported in previous HS studies.
By combining the DEGs identified in the current study, 73 (48 up-regulated and 25 down-regulated) and 41 (12 up-regulated and 29 down-regulated) target genes in the liver and adrenal glands were differentially expressed. Functional annotation of these DEGs is shown in Additional file 12: Table S11. Seventy-three DEGs in the liver were significantly enriched (P < 0.05) in the process of muscle development, such as striated muscle contraction (GO: 0006941), myosin filament assembly (GO: 0031034), muscle contraction (GO: 0006936), and forebrain development (GO: 0030900). Whilst, these DEGs were also significantly involved (P < 0.05) in the pathways of glucagon signaling and tight junction. Functional annotation for the 41 DEGs obtained in the adrenal glands revealed that the positive regulation of axon regeneration (GO: 0048680), which showed the highest fold enrichment score of 106.27, was significantly enriched (P < 0.05) and the DEGs Ndel1 and Braf were both up-regulated. Furthermore, the other significantly-enriched BP term was phospholipid translocation (GO: 0045332) and the enriched genes ATP10a and ATP8b5p were all down-regulated. A remarkable finding was the significantly-enriched (P = 0.026) cellular component (CC) term of extracellular exosome (GO: 0070062) with 11 genes in the adrenal glands being part of the extracellular exosome, of which four genes were up-regulated and seven genes were down-regulated (Additional file 12: Table S11). No pathway was significantly enriched in the adrenal glands.
The interaction between DElncRNAs and DEGs encoding the heat shock proteins (HSPs)
Heat stress may induce misfolding and aggregation of proteins, halt the whole proteins translation, and further cause the apoptosis of cells [31]. The HSP/chaperone network is a major component of multiple stress responses, which is recruited under HS and manage protein folding [30]. From a previous study, 30 and 33 genes of the HSP family genes in the liver and adrenal glands were differentially expressed, with fold change ranging from 0.19 to 28.03 and from 0.11 to 183.84 [22]. All of these 30 HSP encoding genes in the liver were significantly enriched (P < 0.05) in 5 BP terms, such as chaperone-mediated protein folding (GO: 0061077), response to heat (GO: 0009408), ATPase regulator activity (GO: 0060590), protein refolding (GO: 0042026), and regulation of transcription from RNA polymerase II promoter in response to stress (GO: 0043618) (Fig. 6a). The 33 genes in the adrenal glands were significantly enriched (P < 0.05) in 6 BP terms, and 2 of them (GO: 0061077 and GO: 0009408) were shared with liver tissue (Fig. 6b). The predictive analysis of target genes for the lncRNAs also displayed 19 and 6 HSP encoding genes in the liver and adrenal glands, respectively (Fig. 6a). Among which, 15 and 5 DEGs in the liver and adrenal gland tissues were regulated by DElncRNAs (Fig. 6c and d). The interactions among different HSP encoding DEGs and DElncRNAs indicate that lncRNAs play a crucial role in post-transcriptional regulation of HS-related genes.

Functional annotation of DEGs encoding proteins of the heat shock protein (HSP) family and their potential interactions with DElncRNAs. a and b Functional interaction networks among various DEGs that encode heat shock proteins (HSPs) identified in liver (a) and adrenal gland (b) tissues. c and d Interaction networks among different HSP-encoding DEGs and DElncRNAs. Interactions between DEGs and DElncRNAs were predicted by trans-, cis- and sequencing similarity regulatory module. The interaction network was visualized by Cytoscape software. For the nodes, the red ovals mean DElncRNAs, blue rectangles mean DEGs; for the lines, the dark orange lines mean positive correlation between DEGs and DElncRNAs (trans prediction), the blue lines mean negative correlation between DEGs and DElncRNAs (trans prediction), the black lines mean DElncRNAs regulated DEGs by cis-action and the green lines mean DElncRNAs regulated DEGs based on the sequence similarity
Discussion
Over the past decades, with the discovery of few new classes of regulatory non-coding RNAs, an increasing amount of evidences reveals that lncRNAs are engaged in many BPs, such as transcriptional [40, 41] and epigenetic regulation [42, 43], regulation of cell growth [44, 45] and those contributing towards disease etiology [46]. The HS response process is no exception, as lncRNAs are being identified as mediators of general transcription repression of genes [47, 48]. However, a comprehensive analysis of the profiles of lncRNAs showing differential expression under low and mild HS conditions has not yet been reported. In the present study, a well-established HS-rat model was used to explore the expression profile and potential functions of lncRNAs involved in the HS response process in rat liver and adrenal glands by strand-specific RNA-seq. Notably, this study investigated for the first time the expression pattern of lncRNAs in adrenal glands under HS treatment. A total of 4498 and 7627 transcripts were identified in the liver and adrenal glands, which were considered as putative lncRNAs (Figs. 1 and 2). The majority of lncRNAs in the liver and adrenal glands is located in intergenic regions, which is consistent with previous studies performed in other mammalian [49, 50], plant [51, 52] and fungal [53, 54] species. In addition, previous research showed that animal lncRNAs are shorter and have fewer exons than PCGs [55, 56], which is in line with our findings (Fig. 3b and c).
LncRNAs have been reported to be involved in regulation of the HS response in some species [19, 34, 57, 58]. In our analysis, 482 (174 up-regulated and 308 down-regulated) and 271 (126 up-regulated and 145 down-regulated) DElncRNAs in rat liver and adrenal glands were identified under H120 condition, respectively (Table 1). Out of them, 13 DElncRNAs were shared in the liver and adrenal gland tissues (Fig. 4b), which might be meaningful for exploring the regulatory mechanism between the hypothalamic-pituitary-adrenal axis (HPA axis) and liver under HS. It has been reported that lncRNA modulate the transcription of genes by several different actions, including trans-regulatory, in which lncRNAs themselves co-regulate their distant genes; cis-regulatory, in which lncRNAs control the expression of their neighbouring genes; or they interact with the pre-mRNA based on the sequence similarity [26, 50, 59]. Therefore, in order to further demonstrate the potential function of lncRNAs in the HS process, the putative tran- and cis-regulatory, as well as sequencing similarity regulatory module, were analysed. Furthermore, all the 3909 and 4953 DEGs in the liver and adrenal glands were used as targets for DElncRNAs and performed functional annotation for the DEGs that met various rules of function prediction (Additional file 6: Figure S1 and Additional file 5: Table S5, Additional file 8: Table S7, Additional file 9: Table S8, Additional file 11: Table S10, Additional file 12: Table S11). The findings are consistent with results of our previous study [60] and other literature reports [54]. Furthermore, the shared pathways produced from target DEGs in the liver and adrenal glands might contribute to the understanding the potential crosstalk mechanisms between two tissues under HS treatment [61].
The importance of HSP family in regulating the process of the HS response has been highlighted in very early studies [62, 63]. The protection provided by HSPs gives ability for organisms to cope with HS at high temperatures. Furthermore, previous studies have discovered the crucial role of several lncRNAs in the regulation of the HS response [64, 65]. For instance, the drosophila hsr-omega (hsrω) non-coding RNA, which might be the most extensively studied lncRNA, is induced by heat shock [66,67,68]. The heat shock RNA-1 (HSR1) is one of the earliest reported lncRNAs participating in the HS response by regulating the activation of HSF1 [69]. In stress-free cells, HSF1 can bind to some chaperones, such as HSPC1/HSP90. When HS occurs, HSF1 is released and binds to heat shock elements in the HSP encoding gene promoter, thereby inducing its transactivation [64]. In summary, it is important to investigate the impact of lncRNAs on HS response from the perspective of heat shock response. In the present study, the DEGs that encode HSPs were functionally annotated (Fig. 6a) and participated in interaction networks with DElncRNAs by trans-, cis- and similarity regulatory analyses (Fig. 6b). Two HSF genes, Hsf4 and Hsf2, in the liver were found to interact with one (TCONS_00002046) and four (TCONS_00002592, TCONS_00001714, TCONS_00003953 and TCONS_00003953) DElncRNAs by cis- and trans-action, respectively. Furthermore, the Hsf4 was down-regulated (q < 0.05) with a FC greater than five after H120. It has been reported that Hsf4 can interact with the canonical heat shock element of the alphaB-crystallin gene (Hspb6 in rat), whilst, Hsf2 is heat-inducible and functions in heat shock-induced autophagic cell death [70, 71]. In addition, among all the DEGs encoding HSPs with a fold change greater than 5, the Dnaja1, Dnajb4, Hsph1, Hspb1 genes in the liver, and Dnajb13 and Hspb8 genes in the adrenal glands are all known to play diverse roles in HS regulation [21, 72,73,74,75]. The DElncRNAs that interacted with DEGs encoding HSPs are worth exploring further to find out the specific role of lncRNAs in mRNA transcription and post-transcriptional regulation during HS.
Conclusions
Many lncRNAs involved in the HS response in rat liver and adrenal gland tissues were discovered and characterized, and their potential cis-, trans-, and sequencing similarity acting functions were predicted based on their corresponding target DEGs. These findings provide a catalogue of rat liver and adrenal glands lncRNAs and will contribute to a better understanding of the regulatory mechanisms of HS responses in rats and other mammalian species. Future studies are required to explore the physiological functions and the mechanisms by which these lncRNAs respond to HS.
Methods
Animals and treatments
In a previous research [22], a total of 99 eight-week-old pathogen-free female Sprague-Dawley rats (Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China), weighing 205 ± 7.16 g, were used to establish HS model under various HS conditions. Eighteen of them were assigned to the HS treatment (H120; 42 °C for 120 min). Ten rats, including 5 rats from Control group and 5 from the H120 group were used to perform the RNA-seq. Based on the PCC of samples, 3 rats in each group were selected for lncRNA-seq. Prior to the beginning of the HS experiment, all experimental rats were housed (3 rats per cage) at 22 ± 1 °C and 50% room humidity with the light/dark cycle (on 06:00 AM, off 18:00 PM) for 1 week. During the whole experiment, rats were given access to feed and water ad libitum. Moreover, the experiment was performed in the floor-standing artificial climate incubator (BIO250, BOXUN Medicine Instrument Co, Shanghai, China). Rats in the Control group were never introduced to the incubator and were placed at room temperature. The experiment was conducted with healthy and conscious rats.
Tissue collection and RNA extraction
The experimental rats were anesthetized by intraperitoneal injection of 1%, 1.2 mL sodium pentobarbital (40 mg/kg of body weight) [76]. After 2 min, all anesthetized animals were immediately euthanized by cardiac blood collection just after loss of consciousness. The protocols of blood samples collection were reported previously [77, 78]. Briefly, the hair on the abdomen of animals was cut with sterile surgical scissors (Shinva Medical Instrument Co.Ltd., Shandong, China) and the skin was disinfected with 75% alcohol. Then, rats were held with left hand and kept in dorsal recumbency position at an approximately 30° angle (head lowermost). A #4 needle was inserted into the rat’s heart by the right hand at the 3rd and 4th intercostal space. About 4 mL blood sample was collected, the rat’s abdomen was quickly dissected by sterile surgical scissors and forceps (Shinva Medical Instrument Co.Ltd., Shandong, China). Then the liver and adrenal glands were collected, washed in ice-cold phosphate buffer solution (PBS) and snap-frozen immediately in liquid nitrogen. Total RNA of the liver and adrenal glands was extracted using RNA regent (HUAYUEYANG Biotechnology (Beijing) Co. Ltd) according to the manufacture’s protocol. NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA) was used to measure the concentrations and purity of RNA. The RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA) was employed to assess the integrity of RNA integrity.
Transcriptome library construction and paired-end strand-specific transcriptome (rRNA-free) sequencing
The protocols of transcriptome library construction and paired-end strand-specific transcriptome sequencing were previously reported [79]. Briefly, 3 μg RNA per sample was used for RNA-seq library construction. Firstly, due to some lncRNAs lacking the poly (A) tail, Epicentre Ribo-zero™ rRNA Removal Kit (Epicentre, USA) was used to remove the ribosomal RNA from the total RNA, and ethanol precipitation was used to clean up the rRNA free residue. Subsequently, the rRNA-depleted RNA by NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA) was used to produce sequencing libraries according to the manufacturer’s recommendations. Then, the random hexamer primer and M-MuLV Reverse Transcriptase (RNaseH-) were employed to synthesize the first-strand cDNA, and the DNA Polymerase I and RNase H was subsequently used to synthesize the second-strand cDNA. The 3′ ends of DNA fragments was adenylated and NEBNext Adaptor with hairpin loop structure was ligated to prepare for hybridization. With the purpose of selecting cDNA fragments of favorably 250 ~ 300 bp in length, the AMPure XP system (Beckman Coulter, Beverly, USA) was selected to purify the library fragments. Before PCR experiment, 3 μL USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min followed by 5 min at 95 °C. In the PCR experiment, the Universal PCR primers, Index (X) Primer and Phusion High-Fidelity DNA polymerase were added in the reaction system. At last, AMPure XP system and the Agilent Bioanalyzer 2100 system was used to purify the products and evaluate the library quality, respectively. A cBot Cluster Generation System using HiSeq 4000 PE Cluster Kit (Illumina, NEB, USA) was used to cluster the index-coded samples following the manufacturer’s instructions. Subsequently, an Illumina Hiseq 4000 platform was used to sequence the library preparations and produced 150 bp paired-end reads.
Transcriptome library construction and paired-end RNA-sequencing
In a previous research [22], the mRNA profiles in the liver (n = 5) and adrenal glands (n = 5), including the same samples of liver and adrenal glands sequenced by strand-specific transcriptome sequencing in the current study, were analyzed using the RNA-seq technique. Briefly, the library was sequenced in a paired-end reads modus of 150 bp per read using Illumina® HiSeq 2000 platform. Furthermore, in this previous study, genome-wise false discovery rate (FDR), termed as q-value was calculated for differential expression analysis, and genes with q < 0.05 were selected as DEGs [22].
Quality control for sequencing data and assembly of RNA transcripts
All sequencing reads were quality checked and trimmed to remove reads containing adapters, reads with more than 10 Ns and low-quality reads (i.e., more than 50% of the reads with a quality score of less than 10 or read length < 30) from raw data. The quality of clean data was assessed via the software of FastQC version 0.11.7 [80]. At the same time, Q20, Q30, and GC content of the clean data were calculated. All the downstream analyses were based on clean data with high quality. The clean reads were aligned with the rat reference genome (Ensemble release version Rnor 6.0.91, ftp://ftp.ensembl.org/pub/release-95/genbank/rattus_norvegicus/) using Hisat2 version 2.1.0 [81]. Reads met with the minimum isoform fraction 0.01 and minimum reads per bp coverage were assembled and quantified using String Tie version 1.3.6 [82]. The fragments per kilobase of exon model per million reads mapped (FPKM [controlling for fragment length and sequencing depth]) values were used to estimate the expression of genes and transcripts.
Bioinformatics analysis for identification of LncRNA
All transcripts were divided into PCGs and non-coding transcripts after the reference genome annotation. A non-coding transcript that overlapped with PCGs, shorter than 200 nucleotides and containing single exon were filtered out. Then filtered transcripts were aligned against to the NCBI NR database (www.ncbi.nlm.nih.gov/protein) and UniProt rat_10116 protein database (www.uniprot.org) [83]; any transcripts which shown sequence similarity with any of these proteins with a cut-off E value of 10− 5 were removed. After these two filtering steps, the software CNCI version 2 [84], PLEK version 1.2 [85], CPAT version 2.0 [86] were used to predict the coding potential of transcripts. The candidate transcripts with no putative coding potential (CNCI score < 0), PLEK score < 0, and coding probability cut-off value of CPAT < 0.44 were considered as final lncRNAs. Furthermore, the final lncRNAs only identified in at least two samples were used for further analyses.
Classification and characteristic analysis of LncRNAs
The identified lncRNAs were further classified as intergenic, intronic, sense, and antisense lncRNAs based on the spatial relationships of their transcripts loci with PCGs using Cuffcompare tool [50]. The transcripts length and exon number for protein-coding transcripts and lncRNA transcripts were assessed [52]. The proportion of different kinds of lncRNAs and protein-coding transcripts was calculated.
DElncRNAs between H120 and control
The identification of DElncRNAs in the liver and adrenal glands between H120 and Control was carried out using Ballgown package of R version 3.5.3 (TUNA Team, Tsinghua University, Beijing, China). The FC of lncRNAs were calculated as log2 (FPKM H120/FPKM Control) and lncRNAs with P < 0.05 were identified as DElncRNAs.
Validation of LncRNAs transcripts by real-time quantitative PCR (RT-qPCR)
A total of 2 mL RNA of the liver were transcribed into cDNA using the cDNA Synthesis SuperMix kit (Trans, Beijing, China). Ten lncRNAs, including nine top 20 DElncRNAs and one non-significantly expressed lncRNA, but with high expression levels, were randomly selected to carry out the RT-qPCR. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal standard to normalize the expression level of target genes. Primers for GAPDH and lncRNAs were designed using Primer-BLAST (www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome) [87] and double-checked by Oligo v7 (Molecular Biology Insights, Inc., Cascade, CO, USA). All the primers were synthesized by Shanghai Sangon Biotech Co., Ltd. (Shanghai, China) and were specified in Additional file 1: Table S1. Each reaction was performed in 20 mL mixtures, including 2 mL diluted cDNA sample as template, 10 mL SYBR Premix Ex Taq (2х) (TaKaRa, Kyoto, Japan), 1 mL forward and 1 mL reverse gene-specific primers and 6 mL ddH2O. The PCR reaction procedure comprised an initial degeneration at 95 °C for 10 min, 45 cycles of degeneration at 95 °C for 10 s, annealing at 58 °C for 15 s, and extension at 72 °C for 15 s, followed by a final extension at 72 °C for 30 s. The comparative threshold cycle (Ct) value method was adopted to analyze the relative gene expression. Triplicate RT-qPCRs were accomplished on each cDNA. RNA expression levels relative to the GAPDH gene were calculated as 2-△△Ct according to previous research [88]. In order to compare the results of the RNA-seq analysis and RT-qPCR, fold change values were log2 transformed.
Investigation of trans/cis/binding nuclei-target genes’ regulation and functional annotation of the target genes
Three methods were used to predict the target genes of the DElncRNAs based on the different action modes of lncRNA. One was trans-prediction [89], in which the principle of prediction was the co-expression relationship between lncRNA and their target protein-coding genes. Pearson correlation coefficient between expression patterns were calculated by R software, using the expression values of DElncRNAs, target genes, and target DEGs identified in our previous research [22]. In order to show a more accurate and intuitive relationship between DElncRNAs and DEGs, only highly correlated (|PCC| > 0.99) expressions were selected to construct the interaction network. The other action mode analyzed was cis-prediction [90], which was based on the adjacent positional relationship between DElncRNAs and target genes. The DElncRNA-mRNA pairs located on the same chromosome within 100 kb were identified as potentially cis-regulated. This analysis was completed using the BioMart tool of Ensemble genome browser 98 online (http://useast.ensembl.org/biomart/martview/8cae5041d3bb22709301ea05f556fc84) [91]. The last prediction was based on the similarity-search method. The detailed analysis process was according to a previously published paper [50]. All the predicted target genes of DElncRNAs were annotated with publicly available databases, including Gene Ontology terms (GO, http://geneontology.org/docs/go-enrichment-analysis/) and KEGG (https://www.genome.jp/kegg/pathway.html) databases. The functional enrichment results of genes with P < 0.05 were considered as significant.
Construction of interaction network for heat shock proteins (HSPs) encoding DEGs and DElncRNAs
All DEGs encoding HSPs were searched in the DEGs list and annotated by the ClueGO software [92]. The interaction networks among DEGs encoding HSPs and their interacting DElncRNAs were constructed and visualized by the Cytoscape version 3.7.2 [93].
Availability of data and materials
The strand-specific RNA sequencing datasets generated during the current study are available in the Sequence Read Archive (SRA) database at the National Center for Biotechnology Information (NCBI) with the BioProject ID PRJNA624751.
Abbreviations
- HS:
-
Heat stress
- lncRNAs:
-
Long-non-coding RNAs
- H120:
-
42 °C for 120 min
- DElncRNAs:
-
Differentially expressed lncRNAs
- DEGs:
-
Differentially expressed genes
- HSPs:
-
Heat shock proteins
- PBS:
-
Phosphate buffer solution
- FPKM:
-
Fragments per kilobase of exon model per million reads mapped values
- PCG:
-
Protein-coding gene
- NCBI NR protein database:
-
National Center for Biotechnology Information non-redundant protein database
- UniProt:
-
Universal Protein Resource
- CNCI:
-
Coding-non-coding index
- PLEK:
-
K-mer scheme
- CPAT:
-
Coding potential assessment tool
- RT-qPCR:
-
Real-Time Quantitative PCR
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- Ct:
-
Threshold cycle
- PCC:
-
Pearson correlation coefficient
- GO:
-
Gene Ontology terms
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- BP:
-
Biological process
- CC:
-
Cellular component
- MF:
-
Molecular function
- hsrω:
-
Hsr-omega
- HSR1:
-
Heat shock RNA-1
References
Belhadj Slimen I, Najar T, Ghram A, Abdrrabba M. Heat stress effects on livestock: molecular, cellular and metabolic aspects, a review. J Anim Physiol Anim Nutr. 2016;100(3):401–12.
Saeed M, Abbas G, Alagawany M, Kamboh AA, Abd El-Hack ME, Khafaga AF, Chao S. Heat stress management in poultry farms: a comprehensive overview. J Therm Biol. 2019;84:414–25.
Michelozzi P, Accetta G, De Sario M, D'Ippoliti D, Marino C, Baccini M, Biggeri A, Anderson HR, Katsouyanni K, Ballester F, et al. High temperature and hospitalizations for cardiovascular and respiratory causes in 12 European cities. Am J Respir Crit Care Med. 2009;179(5):383–9.
Renaudeau D, Collin A, Yahav S, de Basilio V, Gourdine JL, Collier RJ. Adaptation to hot climate and strategies to alleviate heat stress in livestock production. Animal. 2012;6(5):707–28.
He XF, Lu Z, Ma B, Zhang L, Li JL, Jiang Y, Zhou GH, Gao F. Effects of chronic heat exposure on growth performance, intestinal epithelial histology, appetite-related hormones and genes expression in broilers. J Sci Food Agr. 2018;98(12):4471–8.
St-Pierre NR, Cobanov B, Schnitkey G. Economic losses from heat stress by US livestock industries. J Dairy Sci. 2003;86:E52–77.
Hu RZ, He YJ, Arowolo MA, Wu SS, He J. Polyphenols as potential attenuators of heat stress in poultry production. Antioxidants-Basel. 2019;8(3):67.
Zaboli G, Huang X, Feng X, Ahn DU. How can heat stress affect chicken meat quality? - a review. Poult Sci. 2019;98(3):1551–6.
Williams JW, Jackson ST, Kutzbach JE. Projected distributions of novel and disappearing climates by 2100 AD. Proc Natl Acad Sci U S A. 2007;104(14):5738–42.
Ebi KL. Healthy people 2100: modeling population health impacts of climate change. Clim Change. 2008;88(1):5–19.
Shvartz E. Heat stress - a strategic military factor. Mil Med. 1970;135(11):1045.
Collier RJ, Renquist BJ, Xiao Y. A 100-year review: stress physiology including heat stress. J Dairy Sci. 2017;100(12):10367–80.
Seibert JT, Abuajamieh M, Sanz Fernandez MV, Johnson JS, Kvidera SK, Horst EA, Mayorga EJ, Lei S, Patience JF, Ross JW, et al. Effects of heat stress and insulin sensitizers on pig adipose tissue. J Anim Sci. 2018;96(2):510–20.
Cui Y, Hao Y, Li J, Bao W, Li G, Gao Y, Gu X. Chronic heat stress induces immune response, oxidative stress response, and apoptosis of finishing pig liver: a proteomic approach. Int J Mol Sci. 2016;17(5):393
Cziesielski MJ, Schmidt-Roach S, Aranda M. The past, present, and future of coral heat stress studies. Ecol Evol. 2019;9(17):10055–66.
Kopalli SR, Cha KM, Hwang SY, Jeong MS, Kim SK. Korean Red Ginseng (Panax ginseng Meyer) with enriched Rg3 ameliorates chronic intermittent heat stress-induced testicular damage in rats via multifunctional approach. J Ginseng Res. 2019;43(1):135–42.
Song JH, Kim KJ, Choi SY, Koh EJ, Park J, Lee BY. Korean ginseng extract ameliorates abnormal immune response through the regulation of inflammatory constituents in Sprague Dawley rat subjected to environmental heat stress. J Ginseng Res. 2019;43(2):252–60.
Lees AM, Lees JC, Lisle AT, Sullivan ML, Gaughan JB. Effect of heat stress on rumen temperature of three breeds of cattle. Int J Biometeorol. 2018;62(2):207–15.
Li Y, Kong L, Deng M, Lian Z, Han Y, Sun B, Guo Y, Liu G, Liu D. Heat stress-responsive transcriptome analysis in the liver tissue of Hu sheep. Genes. 2019;10(5):395.
Chen HS, Tong HS, Zhao Y, Hong CY, Bin JP, Su L. Differential expression pattern of exosome long non-coding RNAs (lncRNAs) and MicroRNAs (miRNAs) in vascular endothelial cells under heat stroke. Med Sci Monit. 2018;24:7965–74.
Islam A, Deuster PA, Devaney JM, Ghimbovschi S, Chen Y. An exploration of heat tolerance in mice utilizing mRNA and microRNA expression analysis. PLoS One. 2013;8(8):e72258.
Dou J, Khan A, Khan MZ, Mi S, Wang Y, Yu Y, Wang Y. Heat stress impairs the physiological responses and regulates genes coding for extracellular exosomal proteins in rat. Genes. 2020;11(3):306.
Srikanth K, Lee E, Kwan A, Lim Y, Lee J, Jang G, Chung H. Transcriptome analysis and identification of significantly differentially expressed genes in Holstein calves subjected to severe thermal stress. Int J Biometeorol. 2017;61(11):1993–2008.
Sun L, Lamont SJ, Cooksey AM, McCarthy F, Tudor CO, Vijay-Shanker K, DeRita RM, Rothschild M, Ashwell C, Persia ME, et al. Transcriptome response to heat stress in a chicken hepatocellular carcinoma cell line. Cell Stress Chaperones. 2015;20(6):939–50.
Sun Q, Hao Q, Prasanth KV. Nuclear long noncoding RNAs: key regulators of gene expression. Trends Genet. 2018;34(2):142–57.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62.
Lellahi SM, Rosenlund IA, Hedberg A, Kiaer LT, Mikkola I, Knutsen E, Perander M. The long noncoding RNA NEAT1 and nuclear paraspeckles are up-regulated by the transcription factor HSF1 in the heat shock response. J Biol Chem. 2018;293(49):18965–76.
Bernabò PVG, Lencioni V. A long noncoding RNA acts as a post-transcriptional regulator of heat shock protein (HSP70) synthesis in the cold hardy Diamesa tonsa under heat shock. PLoS One. 2020;15(14):e0227172.
Xu J, Zheng Y, Pu S, Zhang X, Li Z, Chen J. Third-generation sequencing found LncRNA associated with heat shock protein response to heat stress in Populus qiongdaoensis seedlings. BMC Genomics. 2020;21(1):572.
Jacob P, Hirt H, Bendahmane A. The heat-shock protein/chaperone network and multiple stress resistance. Plant Biotechnol J. 2017;15(4):405–14.
Leon LR, Bouchama A. Heat stroke. Compr Physiol. 2015;5(2):611–47.
Deng Q, Chen S, Fu C, Jiang J, Zou M, Tan Y, Wang X, Xia F, Feng K, Ma K, et al. Long noncoding RNA expression profiles in sub-lethal heat-treated hepatoma carcinoma cells. World J Surg Oncol. 2017;15(1):136.
Han DX, Sun XL, Fu Y, Wang CJ, Liu JB, Jiang H, Gao Y, Chen CZ, Yuan B, Zhang JB. Identification of long non-coding RNAs in the immature and mature rat anterior pituitary. Sci Rep. 2017;7(1):17780.
Li Q, Qiao J, Zhang Z, Shang X, Chu Z, Fu Y, Chu M. Identification and analysis of differentially expressed long non-coding RNAs of Chinese Holstein cattle responses to heat stress. Anim Biotechnol. 2020 Feb;31(1):9–16.
Li JY, Yong YH, Gong DL, Shi L, Wang XM, Gooneratne R, Yadnyavalkya P, Ju XH. Proteomic analysis of the response of porcine adrenal gland to heat stress. Res Vet Sci. 2019;122:102–10.
Kang D, Park J, Shim K. Heat treatment at an early age has effects on the resistance to chronic heat stress on broilers. Animals-Basel. 2019;9(12):1022.
Nivon M, Richet E, Codogno P, Arrigo AP, Kretz-Remy C. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy. 2009;5(6):766–83.
Li H, Liu Y, Wen M, Zhao F, Zhao Z, Liu Y, Lin X, Wang L. Hydroxysafflor yellow A (HSYA) alleviates apoptosis and autophagy of neural stem cells induced by heat stress via p38 MAPK/MK2/Hsp27–78 signaling pathway. Biomed Pharmacother. 2019;114:108815.
Xiong Y, Dong S, Zhao X, Guo KJ, Gasco L, Zoccarato I. Gene expressions and metabolomic research on the effects of polyphenols from the involucres of Castanea mollissima Blume on heat-stressed broilers chicks. Poult Sci. 2016;95(8):1869–80.
Kwek KY, Murphy S, Furger A, Thomas B, O'Gorman W, Kimura H, Proudfoot NJ, Akoulitchev A. U1 snRNA associates with TFIIH and regulates transcriptional initiation. Nat Struct Biol. 2002;9(11):800–5.
Goodrich JA, Kugel JF. Non-coding-RNA regulators of RNA polymerase II transcription. Nat Rev Mol Cell Biol. 2006;7(8):612–6.
Sanchez-Elsner T, Gou D, Kremmer E, Sauer F. Noncoding RNAs of trithorax response elements recruit Drosophila Ash1 to Ultrabithorax. Science. 2006;311(5764):1118–23.
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–60.
Lukiw WJ, Handley P, Wong L, Crapper McLachlan DR. BC200 RNA in normal human neocortex, non-Alzheimer dementia (NAD), and senile dementia of the Alzheimer type (AD). Neurochem Res. 1992;17(6):591–7.
Ma Q, Qi X, Lin X, Li L, Chen L, Hu W. LncRNA SNHG3 promotes cell proliferation and invasion through the miR-384/hepatoma-derived growth factor axis in breast cancer. Hum Cell. 2020 Jan;33(1):232–42.
Ma L, Cao J, Liu L, Du Q, Li Z, Zou D, Bajic VB, Zhang Z. LncBook: a curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019;47(D1):D128–34.
Liu WM, Chu WM, Choudary PV, Schmid CW. Cell stress and translational inhibitors transiently increase the abundance of mammalian SINE transcripts. Nucleic Acids Res. 1995;23(10):1758–65.
Li TH, Spearow J, Rubin CM, Schmid CW. Physiological stresses increase mouse short interspersed element (SINE) RNA expression in vivo. Gene. 1999;239(2):367–72.
Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309(5740):1559–63.
Zhao P, Zheng X, Feng W, Wang H, Kang H, Ning C, Du H, Yu Y, Li B, Zhao Y, et al. Profiling long noncoding RNA of multi-tissue transcriptome enhances porcine noncoding genome annotation. Epigenomics. 2018;10(3):301–20.
X-h J, Xu H-j, Yong Y-h, An L-l, Xu Y-m, Jiao P-r, Liao M. Heat stress upregulates the expression of TLR4 and its alternative splicing variant in bama miniature pigs. J Integr Agric. 2014;13(11):2479–87.
Zhu B, Yang Y, Li R, Fu D, Wen L, Luo Y, Zhu H. RNA sequencing and functional analysis implicate the regulatory role of long non-coding RNAs in tomato fruit ripening. J Exp Bot. 2015;66(15):4483–95.
Till P, Mach RL, Mach-Aigner AR. A current view on long noncoding RNAs in yeast and filamentous fungi. Appl Microbiol Biotechnol. 2018;102(17):7319–31.
Wang ZX, Jiang YY, Wu H, Xie XY, Huang B. Genome-wide identification and functional prediction of long non-coding RNAs involved in the heat stress response in Metarhizium robertsii. Front Microbiol. 2019;10:2336.
Wang F, Li L, Xu H, Liu Y, Yang C, Cowley AW Jr, Wang N, Liu P, Liang M. Characteristics of long non-coding RNAs in the Brown Norway rat and alterations in the Dahl salt-sensitive rat. Sci Rep. 2014;4:7146.
Lu C, Xing Y, Cai H, Shi Y, Liu J, Huang Y. Identification and analysis of long non-coding RNAs in response to H5N1 influenza viruses in duck (Anas platyrhynchos). BMC Genomics. 2019;20(1):36.
Shamovsky I, Ivannikov M, Kandel ES, Gershon D, Nudler E. RNA-mediated response to heat shock in mammalian cells. Nature. 2006;440(7083):556–60.
Yang Z, Li W, Su X, Ge PF, Zhou Y, Hao YY, Shu HY, Gao CL, Cheng SH, Zhu GP, et al. Early response of radish to heat stress by strand-specific transcriptome and miRNA analysis. Int J Mol Sci. 2019;20(13):3321.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66.
Khan A, Jiang X, Dou J, Khan MZ, Usman T, Luo H, Zhu H, Wang Y. Evaluation of heat stress effects on cellular and transcriptional adaptation of bovine granulosa cells. J Anim Sci Biotechnol. 2020;11:25.
Bu DP, Bionaz M, Wang MZ, Nan XM, Ma L, Wang JQ. Transcriptome difference and potential crosstalk between liver and mammary tissue in mid-lactation primiparous dairy cows. PLoS One. 2017;12(3):e0173082.
Moseley PL, Wallen ES, Mccafferty JD, Flanagan S, Kern JA. Heat-stress regulates the human 70-Kda heat-shock gene through the 3′-Untranslated region. Am J Physiol. 1993;264(6 Pt 1):L533–7.
Parsell DA, Lindquist S. The function of heat-shock proteins in stress tolerance - degradation and reactivation of damaged proteins. Annu Rev Genet. 1993;27:437–96.
Place RF, Noonan EJ. Non-coding RNAs turn up the heat: an emerging layer of novel regulators in the mammalian heat shock response. Cell Stress Chaperones. 2014;19(2):159–72.
Audas TE, Lee S. Stressing out over long noncoding RNA. Biochim Biophys Acta. 2016;1859(1):184–91.
Jolly C, Lakhotia SC. Human sat III and Drosophila hsr omega transcripts: a common paradigm for regulation of nuclear RNA processing in stressed cells. Nucleic Acids Res. 2006;34(19):5508–14.
Audas TE, Jacob MD, Lee S. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol Cell. 2012;45(2):147–57.
Lakhotia SC, Mallik M, Singh AK, Ray M. The large noncoding hsromega-n transcripts are essential for thermotolerance and remobilization of hnRNPs, HP1 and RNA polymerase II during recovery from heat shock in Drosophila. Chromosoma. 2012;121(1):49–70.
Shamovsky I, Nudler E. Isolation and characterization of the heat shock RNA 1. Methods Mol Biol. 2009;540:265–79.
Prasad KV, Taiyab A, Jyothi D, Srinivas UK, Sreedhar AS. Heat shock transcription factors regulate heat induced cell death in a rat histiocytoma. J Biosci. 2007;32(3):585–93.
Gaudet P, Livstone MS, Lewis SE, Thomas PD. Phylogenetic-based propagation of functional annotations within the gene ontology consortium. Brief Bioinform. 2011;12(5):449–62.
Guan J, Yuan L. A heat-shock protein 40, DNAJB13, is an axoneme-associated component in mouse spermatozoa. Mol Reprod Dev. 2008;75(9):1379–86.
Arrigo AP, Gibert B. Protein interactomes of three stress inducible small heat shock proteins: HspB1, HspB5 and HspB8. Int J Hyperthermia. 2013;29(5):409–22.
Wu D, Zhang M, Xu J, Song E, Lv Y, Tang S, Zhang X, Kemper N, Hartung J, Bao E. In vitro evaluation of aspirin-induced HspB1 against heat stress damage in chicken myocardial cells. Cell Stress Chaperones. 2016;21(3):405–13.
Deane CAS, Brown IR. Differential targeting of Hsp70 heat shock proteins HSPA6 and HSPA1A with components of a protein disaggregation/refolding machine in differentiated human neuronal cells following thermal stress. Front Neurosci. 2017;11:227.
Jeon HJ, Han SR, Lim KH, Won KA, Bae YC, Ahn DK. Intracisternal administration of NR2 subunit antagonists attenuates the nociceptive behavior and p-p38 MAPK expression produced by compression of the trigeminal nerve root. Mol Pain. 2011;7:46.
Paulose CS, Dakshinamurti K. Chronic catheterization using vascular-access-port in rats: blood sampling with minimal stress for plasma catecholamine determination. J Neurosci Methods. 1987;22(2):141–6.
Crandall CG, Gonzalez-Alonso J. Cardiovascular function in the heat-stressed human. Acta Physiol. 2010;199(4):407–23.
Hua X, Wang YY, Jia P, Xiong Q, Hu Y, Chang Y, Lai S, Xu Y, Zhao Z, Song J. Multi-level transcriptome sequencing identifies COL1A1 as a candidate marker in human heart failure progression. BMC Med. 2020;18(1):2.
Davis H, Bardsley EN, Paterson DJ. Transcriptional profiling of stellate ganglia from normotensive and spontaneously hypertensive rat strains. Scientific data. 2018;5:180123.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5.
Soudy M, Anwar AM, Ahmed EA, Osama A, Ezzeldin S, Mahgoub S, Magdeldin S. UniprotR: retrieving and visualizing protein sequence and functional information from universal protein resource (UniProt knowledgebase). J Proteomics. 2020;213:103613.
Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C, Liu Y, Chen R, Zhao Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013;41(17):e166.
Li A, Zhang J, Zhou Z. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014;15:311.
Wang L, Park HJ, Dasari S, Wang SQ, Kocher JP, Li W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41(6):e74.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134.
Kanchiswamy CN, Mohanta TK, Capuzzo A, Occhipinti A, Verrillo F, Maffei ME, Malnoy M. Differential expression of CPKs and cytosolic Ca2+ variation in resistant and susceptible apple cultivars (Malus x domestica) in response to the pathogen Erwinia amylovora and mechanical wounding. BMC Genomics. 2013;14:760.
Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grutzner F, Kaessmann H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014;505(7485):635–40.
Wang F, Tian X, Zhou J, Wang GM, Yu WL, Li Z, Fan ZY, Zhang WJ, Liang AB. A three-lncRNA signature for prognosis prediction of acute myeloid leukemia in patients. Mol Med Rep. 2018;18(2):1473–84.
Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S, et al. Ensembl 2019. Nucleic Acids Res. 2019;47(D1):D745–51.
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–3.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Acknowledgements
We thank Pengju Zhao for the help on target genes prediction based on the sequence similarity.
Funding
This work was supported by the fund of Modern Agro-industry Technology Research System of China (No.CARS-36), the program for Changjiang Scholar and Innovation Research Team in University of China (No.IRT_15R62). The first author acknowledges the scholarship provided by the China Scholarship Council (CSC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
YW2 and JD conceived and designed the experiments; YW2 managed the project; JD collected samples, performed the experiments, analyzed the data and drafted the manuscript; FS discussed the results, provided comments and revised the manuscript; LH conducted the validation experiment; AK and MK revised the manuscript and edited the discussion section; YW1 and YY guided the experimental implementation, provided comments and revised the manuscript. All the authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures and protocols for the experimental rat were approved by the Institutional Animal Care and Use Committee at China Agricultural University, China under Permission number: DK996. The in vivo rat experiments were performed at the College of Animal Science and Technology, China Agricultural University. The Institutional Animal Care and Use Committee approved all the experimental procedures, which complied with the China Physiological Society’s guiding principles for research involving animals and adhered to the high standard (best practice) of veterinary care as stipulated in the Guide for Care and Use of Laboratory Animals.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1 : Table S1.
Primers used for real-time quantitative PCR (RT-qPCR) experiment of lncRNAs.
Additional file 2 : Table S2.
Statistics summary of reads generated from 12 RNA-seq libraries in Control and H120 groups.
Additional file 3 : Table S3.
Summary of the DElncRNAs and DEGs identified in liver and adrenal gland tissues.
Additional file 4 Table S4.
Pearson Correlation Coefficient (PCC) analysis of DElncRNAs and DEGs identified in liver and adrenal gland tissues.
Additional file 5 : Table S5.
GO analysis of target DEGs of DElncRNAs predicted based on trans-regulation. The coexpression level between DEGs and DElncRNAs met the criterion of |PCC| > 0.99 for performing the functional annotation.
Additional file 6 : Figure S1.
The top 20 pathways from the enrichment analysis of the target DEGs in liver and adrenal glands under heat stress (H120). (A, B and C) The top 20 pathways from the enrichment analysis of the target DEGs, which were functionally predicted based on trans- and cis-regulatory action, as well as on sequence similarity method, respectively. The pathway analysis was performed by the Kyoto Encyclopedia of Genes and Genomes; Rich factor means enrichment factor.
Additional file 7 : Table S6.
The list of target DEGs of DElncRNAs predicted by cis-regulatory action in liver and adrenal gland tissues.
Additional file 8 : Table S7.
GO analysis of target genes of DElncRNAs predicted by cis-regulatory action in liver and adrenal gland tissues. The DElncRNA-mRNA pairs located on the same chromosome within 100 kb were assumed as potentially cis-regulated.
Additional file 9 : Table S8.
GO analysis of target DEGs of DElncRNAs predicted by cis-regulatory action in liver and adrenal gland tissues. The DElncRNA-mRNA pairs located on the same chromosome within 100 kb were assumed as potentially cis-regulated.
Additional file 10 : Table S9.
The list of target genes of DElncRNAs in liver and adrenal glands predicted based on the sequence similarity search method.
Additional file 11 : Table S10.
GO analysis of target genes of DElncRNAs predicted based on the sequence similarity.
Additional file 12 : Table S11.
GO analysis of target DEGs of DElncRNAs predicted based on the sequence similarity.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dou, J., Schenkel, F., Hu, L. et al. Genome-wide identification and functional prediction of long non-coding RNAs in Sprague-Dawley rats during heat stress. BMC Genomics 22, 122 (2021). https://doi.org/10.1186/s12864-021-07421-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07421-8