
Abstract
Background
Integrons are genomic elements that mediate horizontal gene transfer by inserting and removing genetic material using site-specific recombination. Integrons are commonly found in bacterial genomes, where they maintain a large and diverse set of genes that plays an important role in adaptation and evolution. Previous studies have started to characterize the wide range of biological functions present in integrons. However, the efforts have so far mainly been limited to genomes from cultivable bacteria and amplicons generated by PCR, thus targeting only a small part of the total integron diversity. Metagenomic data, generated by direct sequencing of environmental and clinical samples, provides a more holistic and unbiased analysis of integron-associated genes. However, the fragmented nature of metagenomic data has previously made such analysis highly challenging.
Results
Here, we present a systematic survey of integron-associated genes in metagenomic data. The analysis was based on a newly developed computational method where integron-associated genes were identified by detecting their associated recombination sites. By processing contiguous sequences assembled from more than 10 terabases of metagenomic data, we were able to identify 13,397 unique integron-associated genes. Metagenomes from marine microbial communities had the highest occurrence of integron-associated genes with levels more than 100-fold higher than in the human microbiome. The identified genes had a large functional diversity spanning over several functional classes. Genes associated with defense mechanisms and mobility facilitators were most overrepresented and more than five times as common in integrons compared to other bacterial genes. As many as two thirds of the genes were found to encode proteins of unknown function. Less than 1% of the genes were associated with antibiotic resistance, of which several were novel, previously undescribed, resistance gene variants.
Conclusions
Our results highlight the large functional diversity maintained by integrons present in unculturable bacteria and significantly expands the number of described integron-associated genes.
Similar content being viewed by others


Background
Integrons are machineries that enables transfer of genetic material between DNA molecules [1, 2]. Through site-specific recombination, integrons have the ability to incise, excise and re-organize genes into, out of, and within a host genome [3–5]. Integrons are estimated to be present in at least 6% of the bacterial genomes [6] and can be located either on chromosomes, as in e.g. Vibrio ssp. and Xanthomonas ssp., or on conjugative elements, as is common for in pathogens such as Escherichia coli and Salmonella enterica [7, 8]. Since integrons enable incorporation of a wide range of genes, they have been suggested to play a major role in the adaptation and evolution of many forms of bacteria [9–11]. Integrons present in pathogenic bacteria often carry antibiotic resistance genes, which enable the bacteria to survive antibiotic treatment. Similarly, chromosomal integrons present on Vibrio ssp. maintain virulence factors, such as genes encoding for toxins, which enable bacteria to gain advantages when colonizing different environments and hosts [7, 12, 13]. However, despite their central role in adaptation, the functional repertoire of integron-associated genes is far from fully characterized.
All integrons are organized according to a common structure. First, they carry an intI gene which encodes an integrase, the enzyme that facilitates the gene transfer by sequential incorporation of genes at the attI recombination site. Furthermore, there is an integron-associated promoter (Pc) that regulates the expression of the incorporated genes. Genes mediated by the integron are organized in gene cassettes. Each cassette consists of an open reading frame (ORF) together with an attC recombination site [9, 14]. AttC sites are imperfect palindromic sequences that are 55 to 141 nucleotides long and exhibit a very low degree of conservation between gene cassettes [4, 15]. During the gene transfer, the bottom strand of the attC site folds into a hairpin secondary structure through alignment of two pairs of complementary motifs, R”/R’ and L”/L’ that are separated by short spacers, which are up to 10 nucleotides long. The L-sites are separated by a region that is 14 to 102 nucleotides long and forms the central loop of the hairpin. R’ and R” are the most conserved parts of the attC site and have the general motifs RYYYAAC and GTTRRRY, respectively (where R is a purine and Y is a pyrimidine). Integrons located on conjugative elements usually consist of up to 8 gene cassettes, many of them with antibiotic resistance genes, while chromosomal integrons can carry hundreds of gene cassettes, which can be spread over the chromosome in multiple arrays [7].
Multiple efforts have been made to study integron-associated genes and their biological functions. The integron database INTEGRALL contains, for example, roughly 1500 integrase and 8000 gene cassettes extracted from public sequence repositories [16]. Also, in a recent study, 2,484 genomes from bacterial isolates were analyzed for the presence of integrons which resulted in 4,597 predicted attC sites [6]. Most bacteria are, however, hard to cultivate under standard lab conditions and their genome is therefore not yet sequenced [17, 18]. Analysis based on genomes from bacterial isolates will thus reflect only a small proportion of the integron-associated genes. To this end, metagenomics offers a cultivation-independent way to analyze the genetic basis of bacterial communities. Indeed, studies using targeted amplicon sequencing have shown that integrons are common in bacterial communities in the environment and the human microbiome [19–24]. However, amplicon-based studies have so far mainly targeted specific types of integron classes or structures (often integrases of class I) and they are therefore unable to capture the full diversity of integron-associated genes. Shotgun metagenomics is, in contrast, free from many of the biases associated with amplicon sequencing and can thus describe the functional potential of a bacterial community in a more holistic way, including the genes located in integrons. However, metagenomic sequence data is fragmented and needs to be assembled prior analysis - a process that is often especially hard for integrons due to their repetitive nature [23, 25]. Consequently, complete fully reconstructed integrons are rare in metagenomic data, which makes their identification and the study of their incorporated gene cassettes challenging.
In this study, we present a comprehensive survey of integron-associated genes present in metagenomes. We used a novel computational approach optimized for highly fragmented sequence data, where the individual attC sites were first detected and then, in a second step, their associated upstream ORFs were identified. This circumvented the need for assembled full-length integrons. We analyzed 375 million contigs assembled from approximately 10 terabases of raw metagenomic data and found 13,397 non-redundant integron-associated genes. The highest abundance of integron-associated genes was found in marine environments, where they were approximately a 100-fold more common than in the human microbiome. The identified genes encoded proteins with a large functional diversity. The most abundant functional classes included defense mechanisms and gene mobility which were also highly overrepresented among the integron-associated genes. We noted furthermore, that genes associated with toxin-antitoxin systems as well as glutathione s-transferases (GST) were especially common. Interestingly, as many as two-thirds of the integron-associated genes had an unknown function and could not be matched to any database. Moreover, less than 1% of the integron-associated genes were antibiotics and biocide/metal resistance genes of which several were novel variants that had not been previously described. In addition, our results describe the extensive functional repertoire associated with bacterial integrons and significantly expand the number of known integron-associated genes.
Results
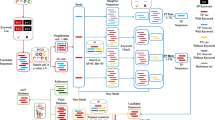
Assembled metagenomic data was analyzed for integron-associated genes using a newly developed computational pipeline (Fig. 1). First, putative attC sites were identified based on their evolutionarily conserved patters using HattCI [15], which implements a generalized Hidden Markov model (gHMM) that individually describes each motif present in the attC site (R’, R”, L’, L”, spacers and loop). Next, the secondary structures of the identified attC sites were validated using a covariance model implemented using Infernal [26]. The model was trained on a structure-based multiple alignment of previously identified and manually annotated attC sites. Afterwards, the results were filtered to remove potential false positives, for that we excluded predicted attC sites that were isolated on the sequence and thus not located in close vicinity to any other attC site (maximum distance between attC sites was set to be 4,000 nucleotides, which was chosen as a conservative upper limit for the gene length in the cassettes). Finally, Prodigal [27] was used to predict open reading frames (ORFs) upstream of the attC sites for the top strand. Evaluation based on 291 gene cassettes demonstrated that the pipeline had a sensitivity of 91% for detecting attC sites. The false positive rate was low with not a single incorrect match in 400 gigabases of sequence data generated by reshuffling eight bacterial genomes. See Methods for full details about the computational pipeline implementation and the evaluation.

Description of the computational pipeline used to detect attC sites in metagenomic data. Assembled metagenomic DNA sequences are used as input. Next, the gHMM-based HattCI is used to detect the attC sites present in the input sequences. Subsequently, the secondary structure of the detected attC sites is evaluated by a covariance model implemented in Infernal, which runs the search in its most sensitive mode. Identified attC sites on the same strand are considered to be part of the same integron when they are at maximum 4,000 nucleotides (nt) apart. Note that integrons with only one attC site are removed from the analysis in order to ensure a high true positive rate. Finally, the ORFs are predicted upstream of the attC sites
The pipeline was used to analyze more than 10 terabases of metagenomic data assembled into 370 million contigs comprising 267 gigabases. The sequence data, which was collected from four major databases and ten metagenomic studies, reflected a wide range of different microbial communities (Table 1). Applying the pipeline to the full dataset resulted in 16,148 predicted gene cassettes, comprising 11,585 unique attC sites and 13,397 unique ORFs (Additional file 1: Table S1).
The relative abundance of attC sites varied between 0.0002 and 0.5 copies per million bases. The highest abundance was found in marine biofilm communities while the level was lowest in the human microbiome. A catalog of the predicted integron-associated genes was formed based on the set of unique ORFs. The length of the genes in the catalog was short, with a median of 402 nucleotides and a standard deviation of 308 nucleotides (Fig. 2a). This was close to the length of the previously identified integron-associated genes reported in the INTEGRALL database [16] (median 474, sd 290) but considerably shorter than the lengths of chromosomal bacterial genes (median 831, sd 735) (Fig. 2a). The G/C-content of the genes in the catalog varied substantially and was between 0.20 and 0.74 with a median of 0.50 and a standard deviation of 0.09. Similar to the gene length, the G/C-content corresponded well with the one found in the genes in INTEGRALL (median 0.51 and standard deviation 0.08). The G/C-distribution was however much wider than what is typically encountered within a single bacterial genome where the G/C-content standard deviation was between 0.04 and 0.05 (Fig. 2b).

Boxplots for a ORF length and b G/C-content for the integron-associated genes identified in this study. For comparisons, the corresponding data for three reference bacterial species have been included, Escherichia coli K-12, Staphylococcus aureus NCTC8325 and Bifidobacterium longum NCC2705. c Cluster analysis of the integron-associated genes. The x-axis shows the cluster threshold in sequence identity (higher value corresponds to a more homogeneous clusters) and the y-axis the number of produced clusters
Next, the diversity of the catalog was assessed using cluster analysis. At a 97% amino acid sequence similarity cut-off, the 13,397 genes formed 12,833 clusters (Fig. 2c), which decreased to 11,946 clusters at a 70% cut-off. At a 50% cut-off, there were still 11,007 clusters formed of which the largest contained 30 genes while 9,517 clusters were singletons. Thus, the number of clusters reduced slowly with a decreasing sequence similarity cut-off, indicating a high diversity with many distinct genes.
The gene catalog was functionally annotated by comparing the genes against three different databases containing functional profiles: Cluster of Orthologous Groups (COG) [28], TIGRFAM 15.0 [29] and PFAM 29.0 [30]. In total, 4817 (36%) of the genes had a match (E-value<10−5) against at least one of the three databases, where 3,497 (26%), 1,727 (13%) and 4,373 (33%) of the ORFs matched functions in the COG, TIGRFAM and PFAM databases, respectively. Among those were 2,277 (17%), 1,203 (9%) and 3,488 (26%) matched to profiles with a known biological function. The most highly abundant functions included toxin-antitoxin systems (e.g. TIGR02607, TIGR02385, PF05016, PF02604, COG2026), GST, in particular, glutathione-dependent formaldehyde-activating genes (PF04828, TIGR02820, COG3791) as well as acetyltransferases (TIGR01575, PF13302, COG0454), endonucleases (PF01844, PF14279), receptor-associated transport activity (TIGR01352) and methylases (COG0863) (Additional file 1: Table S1). The matches to the COG database were assigned to 24 major functional classes (‘COG categories’). The most common functional classes were defense mechanisms (23%) followed by transcription (15%) and mobility (12%). For the TIGRFAM database, the most common functional classes (‘TIGRroles’) were extra-chromosomal functions (29%), protein synthesis (11%) and DNA metabolism (10%) (Additional file 2: Fig. S1b). Gene ontology analysis, based on the matches to the PFAM databases showed that the most common molecular function found is associated with catalytic activities (1.3%), while the most common biological process is related to metabolism (1.1%) and the most common cellular component is part of the membrane (0.42%) (Fig. 3 and Additional file 3: Table S2)).

Functional annotation of the integron-associated genes (solid bars) and other genes found in metagenomes using COG functional categories (striped bars). Of the 13,397 integron-associated genes in our catalog, 2,277 genes matched a COG with a known function. 116,259,264 ORFs were not associated with integrons in metagenomes, out of which 50,201,496 matched a COG with a known function. Percentages on the plot are given in relation to those numbers
Next, we assessed which functional categories were most overrepresented among the integron-associated genes compared to other genes present in the metagenomic data (Fig. 4 and Additional file 4: Table S3)). Using Prodigal, we predicted 116,259,264 unique ORFs that were not associated with any attC site, of which 50,201,496 (43%) matched a COG with a known function. The difference in functional assignments between the two groups of genes was assessed for each COG category using Fisher’s exact test. The three COG categories that were most overrepresented among the integron-associated genes were defense mechanisms (odds ratio 6.46,p<10−15), mobilome (odds ratio 5.06,p<10−15) and function unknown (odds ratio 3.66,p<10−15). Categories that instead were most underrepresented among the integron-associated genes included carbohydrate metabolism and transport (odds ratio 0.158,p<10−15), amino acid transport and metabolism (odds ratio 0.180,p<10−15) and lipid transport and metabolism (odds ratio 0.197,p<10−15).

Gene ontology analysis of the integron-associated genes using PFAM families. Out of the 13,397 integron-associated genes in our catalog, 3,488 matched a PFAM family with a known function, which were in turn mapped to the metagenomics GO slim. Not all PFAM families mapped to a GO term; as a result, 1534 genes had a corresponding GO term. Level 1 terms were removed and those with at least 5 counts were kept (For the whole list GO terms and their counts please see Additional file 3: Table S2)
Next, the catalog was compared to functionally specialized databases containing integron-associated genes (INTEGRALL), antibiotic resistance genes (ResFinder) [31] and biocide and metal resistance genes (BacMet) [32] (Table 2). Interestingly, only 51 (0.38%) of the genes in the catalog had a close match (sequence similarity>97%) to genes previously reported in INTEGRALL. The majority of these genes were either previously known integron-associated resistance genes, hypothetical proteins or genes with unknown function. At a more relaxed sequence similarity cut-off (>70%), the overlap with INTEGRALL increased, but only to 201 (1.5%). The low number of matches to INTEGRALL suggests that the large fraction of the ORFs in the catalog is previously undescribed. The catalog also contained few known antibiotic, metal and biocide resistance genes. Only 25 (0.19%) and 4 (0.030%) of the genes had a close match to genes in the ResFinder and BacMet databases respectively. These matches included several previously reported integron-associated resistance genes, such as the β-lactamases VIM, OXA-2 and OXA-10, the sulfonamide resistance gene sul1, the aminoglycoside resistance genes aadA and the quaternary ammonium compound-resistance protein qacF (Additional file 1: Table S1). Interestingly, when the matching criterion was set to 70% sequence similarity, the number of matches increased to 31 (0.23%) and 7 (0.052%) for ResFinder and BacMet respectively, suggesting the presence of integron-associated resistance genes previously uncharacterized in the literature. Novel putative resistance gene variants included a class D β-lactamase with 93% similarity to OXA-9, several trimethoprim resistance genes ranging between 77% to 96% similarity to known dfr-genes and chloramphenicol resistance gene with 88% similarity to catB (Additional file 1: Table S1).
Finally, structure-based clustering was done to investigate the association between biological function and structure of the attC sites. Based on GraphClust [33], 4102 attC sites were clustered into five distinct groups containing 319 to 1928 attC sites each (Additional file 5: Fig. S2). The remaining 7483 attC sites were removed since GraphClust either 1) assigned them to a cluster with an invalid structural consensus or 2) could not assign them unambiguously to a specific cluster. Tests for overrepresentation showed that several groups were significantly associated with specific COG categories (Additional file 6: Table S4) and GO terms (Additional file 7: Table S5). In particular for the COG categories, clusters (a) and (c) were associated with defense mechanisms (p-values 0.019 and 0.00034, respectively), cluster (b) with inorganic ion transport and metabolism (p-value 0.0272), cluster (d) with cell wall/membrane/envelope biogenesis (p-value 0.0030) and cluster (e) with secondary metabolites biosynthesis, transport and catabolism (p-value 8.6x10-5).
Discussion
In this study we applied a computational pipeline to metagenomic data and identified 13,397 integron-associated genes present in the environment. The analysis was based on 370 million contigs assembled from approximately 10 terabases of sequence data representing microbial communities from a wide range of environments, including the human microbiome. This is, to the best of our knowledge, the most comprehensive characterization of integron-associated genes in uncultured bacteria to date. Indeed, only a small proportion of the identified genes (51 out of 13,397) has previously been reported in the extensive INTEGRALL database, which suggests that most of our findings are not represented in public repositories. Analysis of the identified genes showed a high functional diversity, where only 36% of the genes could be assigned to a known biological function. The functional role of as many as 64% remained unknown. In addition, structured-based clustering of attC sites resulted five groups which showed a weak, but significant, association with specific biological functions.
The relative abundance of gene cassettes differed substantially between the analyzed metagenomes; the levels were found to be especially high in the epipelagic and mesopelagic communities and biofilms. Here, the number of attC sites ranged between 0.05 and 0.50 copies per million bases, which, assuming an average genome size of 2 megabases [34], corresponds to up to approximately 1 gene cassette per cell. High levels of horizontal mobile elements and, in particular, integrons, have previously been reported in marine microbial communities. For example, a large diversity of integrases as well as gene cassettes has been described in marine sediments [20, 35] and deep-sea hydrothermal vent fluid [19]. Also, integrase genes have previously been reported to be common in marine periphyton biofilms [36]. Many forms of bacterial species commonly occurring in marine ecosystems, such as Vibrio spp. [13] and Pseudomonas spp. [37], are known to maintain chromosomal integrons, which may contribute to the high level of gene cassettes observed in these environments [7, 38, 39]. In contrast, low levels of integron-associated genes were found in the human gut metagenomes. Indeed, we found less than 0.01 gene cassettes per cell, which is a 100-fold lower abundance than in the marine metagenomes. This suggests that integron-associated genes are relatively rare in the human microbiome. These findings are in line with previous studies where the abundance of integron-associated integrases has been shown to be substantially lower in the human microbiome compared to many other microbial communities [40]. It should, however, be pointed out that these results will, most likely, not reflect the true diversity of integron-associated genes in any of these environments. Microbial communities are highly diverse and, due to limited sequencing depth, metagenomic studies will only describe integron-associated genes with highest abundance. Nevertheless, our results underline that there are substantial differences in the abundance of integron-associated genes between environmental compartments.
Functional analysis of the 13,397 integron-associated genes demonstrated a large functional diversity and a wide range of biochemical roles. Commonly occurring functional classes included defense mechanisms, gene mobility, transcription, protein synthesis, DNA metabolism and gene expression regulation. Genes associated with defense mechanisms and mobility were highly overrepresented and more than five times more common among genes in integrons than among other genes in the communities. Moreover, toxin-antitoxin systems (TA-systems) were found to be especially common in the gene catalog. TA-systems typically contains two types of genes, one that encodes a toxin that can destroy the bacterial cell and one that encodes an antitoxin that inhibits the toxin. The eventual loss of the antitoxin gene(s), caused by illegitimate recombination events that impairs genes in the integrons, would allow the toxin to kill the host cell. Therefore, TA-systems are hypothesized to stabilize mobile elements and to ensure that they are properly inherited after cell division [13, 41–44]. The stability of chromosomal integrons, which can contain more than 200 gene cassettes and often more than one TA-system [45], may thus be improved by these systems. In our gene catalog, we identified as many as 14 different classes of toxins and 15 classes of antitoxins of which 9 were part of the same system. This included, for example, BrnT/BrnA, RelE/RelB, ParE/ParD, HigB/HigA, YoeB/YefM and HicA/HicB. Several of these TA-systems have been previously found in integrons, where e.g. HigB/HigA have been detected in chromosomal integrons of Vibrio spp. [41] and HicA/HicB and HigA/HigB have been found in gene cassettes in humanassociated bacterial communities [46]. Another common gene found in the catalog was GST, which are detoxification enzymes that can catalyze glutathione to a wide range of xenobiotic substances [47]. Most common were glutathione-dependent formaldehyde-activating enzymes (Gfa), which condenses formaldehyde, a toxin commonly found in natural environments that binds and inactivates proteins [48, 49]. Previous reports have found GSTs and Gfa enzymes among integron-associated genes from marine sediments [35], and its prevalence suggests that this gene confers a selective advantage to the host cell.
Approximately two thirds (64%) of the genes in the catalog were ORFans, i.e. genes that did not match any known gene, function or domain in any entry in the reference databases. Genes with unknown function were also found to be highly overrepresented among the integron-associated genes. This suggests that a large fraction of the genes maintained in the integrons of the analyzed bacterial communities has uncharacterized biological functions or are too evolutionary distant to be annotated using sequence homology. These findings are in line with previous amplicon-based studies. For example, analysis of gene cassettes from deep-sea hydrothermal vents showed that up to 82% of their genes did not have any significant match in sequence databases [19]. Similarly, analyses of integrons in the human microbiome have demonstrated that up to 85% of the gene cassettes had ORFs of unknown function [23, 46]. The number of unknown genes present in environmental microbial communities has, regardless of their genomic contexts, shown to be substantial and can reach up to 80% [9, 35, 36, 50]. The exact role and biological functions of the large number of ORFans maintained by environmental bacteria is currently not clear. A recent study applied sensitive probabilistic alignment algorithms and showed that a proportion of the ORFans are likely to be distant homologs, which share too low similarity to known genes to be properly annotated using standard approaches [51]. Studies of the Escherichia coli pan-genome have, furthermore, shown that ORFans often have evolutionary conserved protein-coding capacity and that many are properly transcribed and translated [52, 53]. The large proportion of unknown genes in integrons found in our analysis further supports the hypothesis that many ORFans have important biological roles. Indeed, integrons are highly plastic and genes that would not provide any form of evolutionary advantage would be expected to be excised and not further maintained by the community. Thus, our results suggest that analysis of integrons using culture-independent techniques, such as shotgun metagenomics, may be used to guide the identification of novel mobile genes that encode previously uncharacterized biochemical functions that enables bacteria to adapt to different selection pressures [1, 54, 55].
Analysis of the gene catalog revealed that integron-associated genes were in general short with a length distribution centered around 400 nt. In contrast, chromosomal ORFs are often substantially longer, e.g. with a median of 849 bp in Escherichia coli. This difference was most likely not a result of the de novo identification of the ORFs, even if this sometimes estimates the length of the predicted gene incorrectly [27]. In fact, when we de novo predicted ORFs in the Escherichia coli genome using the exact same setting as in the analysis of the gene cassettes, the median length changed only slightly (from 849 to 831 bp). In addition, the lengths of the integron-associated genes predicted in this study were also relatively similar to the length of the genes reported in the integron database INTEGRALL (median length 474 bp). The short length of the ORFs is thus likely a biological effect and not a technical artifact, suggesting that the length of the genes present in integrons are under a strong evolutionary selection pressure. Indeed, it is well-known that incorporation of genes located on extrachromosomal DNA is typically associated with reduced fitness due to increasing the cost of cell replication, DNA maintenance and gene expression [56]. It can, however, not be excluded that there are other mechanisms associated with how genes are incised, excised and expressed in integrons that results in additional selection pressures on the gene length. Moreover, the G/C-content of the genes in the catalog varied substantially. Centered at an average G/C-content of 0.5, the distribution was more than twice as wide as the distribution encountered for chromosomal genes. The G/C-content of non-mobile chromosomal genes is known to differ significantly between bacterial genomes and has been shown to be correlated with genome size, habitat and living conditions, such as temperature [57–59]. Thus, the wide distribution of G/C-content among the integron-associated genes suggest that the ORFs have been mobilized from a diverse set of hosts with a wide range of different G/C-content. It also suggests that there is no, or little, selection pressures towards a more narrow G/C-content distribution.
Previous studies have demonstrated that integrons, in particular those of class 1, have gene cassettes that are dominated by antibiotic resistance genes [60–62]. This is especially true for clinical variants that are commonly occurring in pathogenic bacteria, such as Enterobacteriacea. In contrast, a recent systematic study of integron-associated genes in bacterial genomes demonstrated that only 4% of the genes could be associated with antibiotic resistance [6]. Our results, based on the analysis of DNA sequenced directly from bacterial communities, showed that only 0.19% of the genes were known antibiotic resistance genes. When relaxing the similarity threshold to also include genes that were homologous to known resistance genes, this number was still less than 0.5%. These findings show that integron-associated genes are, in general, not coding for antibiotic resistance determinants but typically contain genes with a much more diverse functional repertoire. It also suggests that integron-associated genes reported into existing repositories, such as INTEGRALL and GenBank, are likely to be heavily biased towards cultivable pathogenic bacteria and does only reflect a small proportion of the diversity present many environmental communities. Nevertheless, 38 resistance genes were present in our data. Interestingly, nine of these were novel and have previously not been reported in resistance gene databases. This suggests that integrons in environmental bacterial communities maintain resistance genes that have not yet been incorporated into human pathogens [24]. This is in line with previous studies that have reported a surprisingly large diversity of novel resistance genes present in metagenomes [63–66]. In fact, several forms of clinically relevant resistance genes have also been hypothesized to originate from environmental bacteria. In particular, Shewanella spp., which are naturally occurring in marine ecosystems, have been suggested to be the source of resistance genes such as the beta-lactamase OXA-48 and the fluoroquinolone resistance gene qnrA [67, 68]. Another example are the beta-lactamases PER and MOX, which were recently shown to be mobilized from the bacterial genera Pararheinheimera and Aeromonas, respectively, which both are ubiquitously present in the environment [69, 70]. Thus, our results further underline that environmental bacterial communities are sources of antibiotic resistance genes. They also show that analysis of metagenomes can be used to identify novel integron-associated resistance gene variants that have not yet been encountered in clinical settings. It should, however, be pointed out that further experimental work is needed to confirm the phenotypes of the novel resistance gene variants.
The gene catalog presented in this papers was identified using a novel computational pipeline developed to search metagenomic data for integron-associated genes. The implementation can accurately identify attC sites directly, without requiring a complete integron to be present, which makes it applicable to the often short contigs produced by shotgun metagenomics. This is especially important when analyzing integrons, since they are often hard to assemble due to the presence of repetitive sequences [23, 25]. The computational pipeline combines two different steps to ensure a high performance. First, HattCI, a probabilistic model in the form of a generalized hidden Markov model was used to identify potential attC sites. The HattCI model uses pattern matching for the specific regions of the attC sites and therefore has both a high sensitivity and a high computational performance, which enables efficient processing of large data volumes. In the second step, HattCI predictions were evaluated by analyzing their secondary structure using a covariance model created using Infernal. Evaluation based on a 291 attC sites large testing set confirmed that this combination resulted in the ability to identify more than 90% of the attC sites. Furthermore, to keep the number of false positives to a minimum, we only considered attC sites that are found sufficiently close to each other. Since most integrons are expected to contain more than one gene, and thus more than one attC site, this efficiently removed many of the false positives caused by spurious isolated hits. Consequently, when analyzing 100,000 reshuffled Escherichia coli genomes, corresponding to ∼500 gigabases of sequence data, not a single false positive was found. However, the strict filtering suggest that there are likely additional integron-associated genes present in the analyzed datasets, e.g. singletons present on short contigs, that are not considered by our method. Furthermore, HattCI and the covariance model were trained using a gold standard dataset containing manually verified annotation of attC sites. The dataset is however based on data from public sequence repositories and therefore biased towards gene cassettes encountered in class 1 integrons [62]. Different classes of integrases are known to have different affinities for certain attC sites [71] and it is therefore likely that our approach misses many forms of attC sites. Consequently, the number of gene cassettes identified in this study is likely to be a conservative estimate. Reanalyzing the dataset when more comprehensive and unbiased training data becomes available is therefore supposed to further expand the presented gene catalog.
Conclusions
In this study we present a systematic survey of gene cassettes present in metagenomic data. This is, to the best of our knowledge, the most comprehensive analysis of integron-associated genes in bacterial communities to date. We observed that the relative abundance of gene cassettes varies between communities, being more abundant in marine environments, and less common in the human gut. Also, our results show that integrons in bacterial communities maintain genes with a diverse set of biological functions. This is further emphasized by the large number of unknown genes for which no close homologue could be found in the sequence databases. Furthermore, the presence of previously undescribed antibiotic resistance gene variants supports the hypothesis that bacterial communities are a reservoir for novel resistance determinants that can be transferred into human pathogens. Our study also shows that metagenomic data can, regardless of its fragmented nature, be an important source of information for the characterization of integron-associated genes, and potentially other genes and genomic structures. Thus, further studies, including even larger volumes of metagenomic data, are warranted.
Methods
Description of the computational method
To accurately identify gene cassettes located on metagenomic contigs, a computational method was developed. The result, MIG-finder (Metagenomic Integron-associated Gene finder), operates by identifying the two main components of the gene cassettes: the recombination site (attC) and the associated open reading frame (ORF). First, the method predicts attC sites based on their sequence similarity using HattCI 1.0b which implements a seven-state generalized hidden Markov model (gHMM) to describe each conserved motif or variable region of the attC site (i.e. R’, R”, L’, L”, two spacers and loop). The gHMM was trained using 231 manually curated attC sites [15]. HattCI was run in the mode where both strands are analyzed, with a batch of 1000 sequences and 6 threads (‘-b -s 1000 -t 6’). All matches with a score above 0 was kept. Next, the secondary structure of the predicted attC sites was validated by a covariance model (CM) implemented using Infernal v1.1.1 [26]. The CM was constructed using the command cbuild in Infernal (using default parameters) from a structure-based multiple alignments produced by LocaRNA v1.8.9 [72] using 109 attC sites, which were chosen out of the initial 231 in order to produce a valid consensus for the attC site secondary structure. The model construction was guided by anchoring complementary positions in the alignment. Then, Infernal, in its maximum sensitive mode (‘—max’), was used to analyze all potential attC sites. AttC sites predicted with an Infernal score less than 20 were removed from the analysis. Due to the palindromic nature of the attC sites, overlapping matches could appear on both strands. In these cases, only the match with the highest score was selected. Next, the identified attC sites were filtered to remove false positives. This was done by only keeping attC sites located at a maximum distance of 4000 nucleotides from another attC site. This distance was assumed as the maximum distance of two attC sites located on the same integron. Consequently, attC sites found in isolation from any other attC site, i.e. more than 4000 nucleotides up or downstream were thus considered as potential false positives and discarded. Finally, ORFs were predicted using Prodigal v2.6.2 running in metagenomic mode [27], not allowing for genes to run off edges and saving the predictions in a fasta file with nucleotide and translated versions (‘-p meta -f gff -q –c’). Predicted ORFs were kept if they i) had a score larger than 0, ii) had maximum overlap of 50 nucleotides with any attC site and iii) for the first gene cassette in the array, the ORF was no further than 500 nucleotides away from the attC site. On the top strand, the predicted ORF was the associated with its closest upstream attC site, and reversely for the bottom strand. Note that, attC sites were required to be on the same strand to be part of the same integron.
The sensitivity of the method was evaluated using 291 manually curated attC sites [6] that represents the diversity of the attC sites present in INTEGRALL [16]. The pipeline was capable of detecting 90.4% of these sequences. The specificity was evaluated using data generated by reshuffling eight genomes, representing different part of the bacterial phylogeny, 10,000 times each (Escherichia coli (NC_000913.3), Staphylococcus aureus (NC_007795.1), Bifidobacterium longum (NC_004307.2), Rhizobium marinum (GCF_000705355.1), Burkholderia cepacia (GCF_001411495.1), Acidobacterium capsulatum (NC_012483.1), Desulfobacter vibrioformis (GCA_000745975.1), Streptomyces griseus (GCF_000010605.1)). For this data, the pipeline did not generate a single false positive.
Processing of metagenomic data
The computational method was applied to 370 million metagenomic DNA sequences consisting either of assembled contigs or sufficiently long reads (average length of 712 nt). This data corresponded to approximately 10 Tbases of metagenomic sequenced reads. The data was compiled from four metagenomic databases, CAMERA [73], MG-RAST [74] [downloaded August 2016], NTenv [75] [downloaded March 2016] and EBI Metagenomics [76] [downloaded June 2016]. From the MG-RAST repository, only datasets that had at least 10,000 sequences and an average fragment length above 1,000 nucleotides were selected (i.e. containing contigs or long sequence reads). Redundant datasets that were present in several databases (e.g. CAMERA and MG-RAST) were removed. Ten additional metagenomic studies containing assembled contigs that were not present in the databases were also included [downloaded between Feb and July 2016]. One of these studies contained samples from marine periphyton biofilms from the coastal waters of the Swedish west coast collected and prepared by the authors. The other studies were identified through literature searchers and the data was downloaded from repositories or directly from the authors. In particular, Tara Oceans is a comprehensive study from epipelagic and mesopelagic communities around the world. For further details on the datasets see Table 1.
Creation and analysis of the gene catalog
A catalog of integron-associated genes was created based on the ORFs and their corresponding attC sites predicted from metagenomic data. The catalog was made non-redundant by removing identical pairs of attC sites and ORFs (100% sequence similarity for both, in their nucleotide and aminoacid sequences, respectively). Each ORF in the catalog was functionally annotated using three independent general databases containing gene functions: Cluster of Orthologous Groups (2003-2014 COG) [28], TIGRFAM 15.0 [29] and PFAM 29.0 [30]. Each ORF was compared against each database using hmmscan in HMMER 3.1b [77] with default parameters and saving a table of hits per-sequence. Only the best match per sequence, with a maximum E-value of 0.001 was kept. Gene Ontology analysis was done by mapping the PFAM hits to the metagenomics GO-slim annotation [78]. Next, the gene catalog was compared with three specialized databases: the INTEGRALL [16], ResFinder [31] and BacMet [32]. The comparison was performed using BLAST v2.2.31+ [79] in ‘tblastn’ mode for INTEGRALL and Resfinder and in ‘blastp’ mode for BacMet. The comparison was done using two separate cut-offs, one strict (97% sequence similarity) and one relaxed (70%). In both cases, only matches that covered at least 50% of the subject were kept. If there were multiple matches, the match with the highest sequence similarity in each database was kept.
All unique attC sites were clustered based on sequence-structure similarity using GraphClust [33] with default parameters modified to “input_win_shift = 100, input_win_size =200, OPTS_fasta2shrep_gspan = ‘-t “3=0,5=80” -M 5 -c 20 --cue -u --stack --seq-graph-t –seq-graph-alph’, GLOBAL_iterations = 4 and GLOBAL_num_clusters = 2”, which pre-defined the number of clusters to be 8. The resulting cluster were manually evaluated to contain the key attC site motifs, i.e. R”/R’ and matching L”/L’, with an extra-helix nucleotide formed in L” due to difference in length with L’, and in addition the presence of second extra-helix nucleotide found in the loop [71]. Then clusters with a structural consensus that did not contained these features were discarded, resulting in 5 structural clusters. Tests for functional enrichment of COG Categories and Gene Ontology were done for each generated cluster. The test was done using Fisher’s exact test [80] comparing the relative number of occurrences of a specific function within the cluster compared to all other ORFs in the catalog. For the test, ORF’s that did not have a functional annotation were discarded together with their attC site. Similarly, attC sites that did not belong to one of the five valid structural clusters were discarded together with their ORFs.
Availability of data and materials
The data analyzed in this study consisted of pre-existing datasets which are specified in Table 1.
Abbreviations
- CM:
-
Covariance model
- COG:
-
Cluster of orthologous groups
- gHMM:
-
Generalized hidden Markov model
- ORF:
-
Open reading frame
- PCR:
-
Polymerase chain reaction
References
Gillings MR. Integrons: past, present, and future. Microbiol Mol Biol Rev. 2014; 78(2):257–77. https://doi.org/10.1128/mmbr.00056-13.
Stokes HW, Hall RM. A novel family of potentially mobile DNA elements encoding site specific gene-integration functions: integrons. Mol Microbiol. 1989; 3(12):1669–83. https://doi.org/10.1111/j.1365-2958.1989.tb00153.x.
Collis CM, Hall RM. Gene cassettes from the insert region of integrons are excised as covalently closed circles. Mol Microbiol. 1992; 6(19):2875–85. https://doi.org/10.1111/j.1365-2958.1992.tb01467.x.
Hall RM, Brookes DE, Stokes HW. Site-specific insertion of genes into integrons: role of the 59-base element and determination of the recombination cross-over point. Mol Microbiol. 1991; 5(8):1941–19. https://doi.org/10.1111/j.1365-2958.1991.tb00817.x.
MacDonald D, Demarre G, Bouvier M, Mazel D, Gopaul DN. Structural basis for broad DNA-specificity in integron recombination. Nature. 2006; 440(7088):1157–62. https://doi.org/10.1038/nature04643.
Cury J, Jove T, Touchon M, Neron B, Rocha EPC. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res. 2016; 44(10):4539–50. https://doi.org/10.1093/nar/gkw319.
Mazel D. Integrons: agents of bacterial evolution. Nat Rev Microbiol. 2006; 4(8):608–20. https://doi.org/10.1038/nrmicro1462.
Johnning A, Karami N, Hallbäck ET, Müller V, Nyberg L, Pereira MB, Stewart C, Ambjörnsson T, Westerlund F, Adlerberth I, et al. The resistomes of six carbapenem-resistant pathogens–a critical genotype–phenotype analysis. Microb Genomics. 2018; 4(11):e000233.
Boucher Y, Labbate M, Koenig JE, Stokes HW. Integrons: mobilizable platforms that promote genetic diversity in bacteria. Trends Microbio. 2007; 15(7):301–9. https://doi.org/10.1016/j.tim.2007.05.004.
Holmes AJ, Gillings MR, Nield BS, Mabbutt BC, Nevalainen KMH, Stokes HW. The gene cassette metagenome is a basic resource for bacterial genome evolution. Environ Microbiol. 2003; 5(5):383–94. https://doi.org/10.1046/j.1462-2920.2003.00429.x.
Escudero JA, Loot C, Mazel D. In: Rampelotto PH, (ed).Integrons as Adaptive Devices. Cham: Springer; 2018, pp. 199–239. https://doi.org/10.1007/978-3-319-69078-0.
Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Tettelin Â, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, Mcdonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, Mcdonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000; 406(6795):477–83. https://doi.org/10.1038/35020000.
Rowe-Magnus DA, Guerout A-M, Biskri L, Bouige P, Mazel D. Comparative analysis of superintegrons: engineering extensive genetic diversity in the Vibrionaceae. Genome Res. 2003; 13(3):428–42. https://doi.org/10.1101/gr.617103.
Hall RM, Collis CM, Kim MJ, Partridge SR, Recchia GD, Stokes HW. Mobile gene cassettes and integrons in evolution. Ann N Y Acad Sci. 1999; 870:68–80. https://doi.org/10.1111/j.1749-6632.1999.tb08866.x.
Pereira MB, Wallroth M, Kristiansson E, Axelson-Fisk M. HattCI: fast and accurate attC site identification using hidden Markov models. J Comput Biol. 2016; 23(11):891–902. https://doi.org/10.1089/cmb.2016.0024.
Moura A, Soares M, Pereira C, Leitao N, Henriques I, Correia A. INTEGRALL: A database and search engine for integrons, integrases and gene cassettes. Bioinformatics. 2009; 25(8):1096–8. https://doi.org/10.1093/bioinformatics/btp105.
Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998; 180(18):4765–74.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J-F, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu W-T, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013; 499(7459):431–7. https://doi.org/10.1038/nature12352.
Elsaied H, Stokes HW, Nakamura T, Kitamura K, Fuse H, Maruyama A. Novel and diverse integron integrase genes and integron-like gene cassettes are prevalent in deep-sea hydrothermal vents. Environ Microbiol. 2007; 9(9):2298–312. https://doi.org/10.1111/j.1462-2920.2007.01344.x.
Elsaied H, Stokes HW, Kitamura K, Kurusu Y, Kamagata Y, Maruyama A. Marine integrons containing novel integrase genes, attachment sites, attI, and associated gene cassettes in polluted sediments from Suez and Tokyo Bays. ISME J. 2011; 5(7):1162–77. https://doi.org/10.1038/ismej.2010.208.
Elsaied H, Stokes HW, Yoshioka H, Mitani Y, Maruyama A. Novel integrons and gene cassettes from a Cascadian submarine gas-hydrate-bearing core. FEMS Microbiol Ecol. 2014; 87(2):343–56. https://doi.org/10.1111/1574-6941.12227.
Stokes HW, Holmes AJ, Nield BS, Holley MP, Nevalainen KMH, Mabbutt BC, Gillings MR. Gene cassette PCR: sequence-independent recovery of entire genes from environmental DNA. Appl Environ Microbiol. 2001;67(11): 5240–6. https://doi.org/10.1128/aem.67.11.5240-5246.2001.
Wu YW, Rho M, Doak TG, Ye Y. Oral spirochetes implicated in dental diseases are widespread in normal human subjects and carry extremely diverse integron gene cassettes. Appl Environ Microbiol. 2012; 78(15):5288–96. https://doi.org/10.1128/aem.00564-12.
Razavi M, Marathe NP, Gillings MR, Flach C-F, Kristiansson E, Larsson DJ. Discovery of the fourth mobile sulfonamide resistance gene. Microbiome. 2017; 5(1):160. https://doi.org/10.1186/s40168-017-0379-y.
Bengtsson-Palme J, Boulund F, Fick J, Kristiansson E, Larsson DG. Shotgun metagenomics reveals a wide array of antibiotic resistance genes and mobile elements in a polluted lake in India. Front Microbiol. 2014; 5:648. https://doi.org/10.3389/fmicb.2014.00648.
Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013; 29(22):2933–5. https://doi.org/10.1093/bioinformatics/btt509.
Hyatt D, Locascio PF, Hauser LJ, Uberbacher EC. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics. 2012; 28(17):2223–30. https://doi.org/10.1093/bioinformatics/bts429.
Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded Microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015; 43(D1):261–9. https://doi.org/10.1093/nar/gku1223.
Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003; 31(1):371–3. https://doi.org/10.1093/nar/gkg128.
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2015; 44(D1):279–85. https://doi.org/10.1093/nar/gkv1344.
Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012; 67(11):2640–4. https://doi.org/10.1093/jac/dks261.
Pal C, Bengtsson-Palme J, Rensing C, Kristiansson E, Larsson DGJ. BacMet: antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 2014; 42(D1):737–3. https://doi.org/10.1093/nar/gkt1252.
Heyne S, Costa F, Rose D, Backofen R. Graphclust: Alignment-free structural clustering of local RNA secondary structures. Bioinformatics. 2012; 28(12):224–32. https://doi.org/10.1093/bioinformatics/bts224.
Raes J, Korbel JO, Lercher MJ, von Mering C, Bork P. Prediction of effective genome size in metagenomic samples. Genome Biol. 2007; 8(1):10. https://doi.org/10.1186/gb-2007-8-1-r10.
Koenig JE, Boucher Y, Charlebois RL, Nesbø C, Zhaxybayeva O, Bapteste E, Spencer M, Joss MJ, Stokes HW, Doolittle WF. Integron-associated gene cassettes in Halifax Harbour: assessment of a mobile gene pool in marine sediments. Environ Microbiol. 2008; 10(4):1024–38. https://doi.org/10.1111/j.1462-2920.2007.01524.x.
Sanli K, Bengtsson-Palme J, Henrik Nilsson R, Kristiansson E, Rosenblad MA, Blanck H, Eriksson KM. Metagenomic sequencing of marine periphyton: Taxonomic and functional insights into biofilm communities. Front Microbiol. 2015; 6(10):1192. https://doi.org/10.3389/fmicb.2015.01192.
Vaisvila R, Morgan RD, Posfai J, Raleigh EA. Discovery and distribution of super-integrons among Pseudomonads. Mol Microbiol. 2001; 42(3):587–601. https://doi.org/10.1046/j.1365-2958.2001.02604.x.
Mazel D, Dychinco B, Webb VA, Davies J. A distinctive class of integron in the Vibrio cholerae genome. Science. 1998; 280(5363):605–8. https://doi.org/10.1126/science.280.5363.605.
Rowe-Magnus DA, Guérout AM, Mazel D. Super-integrons. Res Microbiol. 1999; 150(9-10):641–51. https://doi.org/10.1016/s0923-2508(99)00127-8.
Pal C, Bengtsson-Palme J, Kristiansson E, Larsson DJ. The structure and diversity of human, animal and environmental resistomes. Microbiome. 2016; 4(1):54. https://doi.org/10.1186/s40168-016-0199-5.
Christensen-Dalsgaard M, Gerdes K. Two higBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol Microbiol. 2006; 62(2):397–411. https://doi.org/10.1111/j.1365-2958.2006.05385.x.
Iqbal N, Guérout AM, Krin E, Le Roux F, Mazel D. Comprehensive functional analysis of the 18 Vibrio cholerae N16961 toxin-antitoxin systems substantiates their role in stabilizing the superintegron. J Bacteriol. 2015; 197(13):2150–9. https://doi.org/10.1128/jb.00108-15.
Szekeres S, Dauti M, Wilde C, Mazel D, Rowe-Magnus DA. Chromosomal toxin-antitoxin loci can diminish large-scale genome reductions in the absence of selection. Mol Microbiol. 2007; 63(6):1588–605. https://doi.org/10.1111/j.1365-2958.2007.05613.x.
Van Melderen L, De Bast MS. Bacterial toxin–antitoxin systems: more than selfish entities?PLoS Genet. 2009; 5(3):1000437. https://doi.org/10.1371/journal.pgen.1000437.
Cambray G, Guerout A-M, Mazel D. Integrons. Annu Rev Genet. 2010; 44(1):141–66. https://doi.org/10.1146/annurev-genet-102209-163504.
Tansirichaiya S, Rahman MA, Antepowicz A, Mullany P, Roberts AP. Detection of novel integrons in the metagenome of human saliva. PLoS ONE. 2016; 11(6):1–20. https://doi.org/10.1371/journal.pone.0157605.
Vuilleumier S, Pagni M. The elusive roles of bacterial glutathione S-transferases: new lessons from genomes. Appl Microbiol Biotechnol. 2002; 58(2):138–46.
Chen NH, Djoko KY, Veyrier FJ, McEwan AG. Formaldehyde stress responses in bacterial pathogens. Front Microbiol. 2016; 7(3):1–17. https://doi.org/10.3389/fmicb.2016.00257.
Goenrich M, Bartoschek S, Hagemeier CH, Griesinger C, Vorholt JA. A glutathione-dependent formaldehyde-activating enzyme (Gfa) from Paracoccus denitrificans detected and purified via two-dimensional proton exchange NMR spectroscopy. J Biol Chem. 2002; 277(5):3069–72. https://doi.org/10.1074/jbc.C100579200.
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, Djahanschiri B, Zeller G, Mende DR, Alberti A, Cornejo-Castillo FM, Costea PI, Cruaud C, D’Ovidio F, Engelen S, Ferrera I, Gasol JM, Guidi L, Hildebrand F, Kokoszka F, Lepoivre C, Lima-Mendez G, Poulain J, Poulos BT, Royo-Llonch M, Sarmento H, Vieira-Silva S, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Bowler C, de Vargas C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Jaillon O, Not F, Ogata H, Pesant S, Speich S, Stemmann L, Sullivan MB, Weissenbach J, Wincker P, Karsenti E, Raes J, Acinas SG, Bork P, Boss E, Bowler C, Follows M, Karp-Boss L, Krzic U, Reynaud EG, Sardet C, Sieracki M, Velayoudon D. Structure and function of the global ocean microbiome. Science. 2015; 348(6237):1261359. https://doi.org/10.1126/science.1261359.
Lobb B, Kurtz DA, Moreno-Hagelsieb G, Doxey AC. Remote homology and the functions of metagenomic dark matter. Front Genet. 2015; 6(7):1–12. https://doi.org/10.3389/fgene.2015.00234.
Neuhaus K, Landstorfer R, Fellner L, Simon S, Schafferhans A, Goldberg T, Marx H, Ozoline ON, Rost B, Kuster B, Keim DA, Scherer S. Translatomics combined with transcriptomics and proteomics reveals novel functional, recently evolved orphan genes in Escherichia coli O157: H7 (EHEC). BMC Genomics. 2016; 17(1):133. https://doi.org/10.1186/s12864-016-2456-1.
Yu G, Stoltzfus A. Population diversity of ORFan genes in Escherichia coli. Genome Biol Evol. 2012; 4(11):1176–87. https://doi.org/10.1093/gbe/evs081.
Culligan EP, Sleator RD, Marchesi JR, Hill C. Metagenomics and novel gene discovery: promise and potential for novel therapeutics. Virulence. 2014; 5(3):399–412. https://doi.org/10.4161/viru.27208.
Ufarté L, Potocki-Veronese G, Laville É. Discovery of new protein families and functions: New challenges in functional metagenomics for biotechnologies and microbial ecology. Front Microbiol. 2015; 6(6):1–10. https://doi.org/10.3389/fmicb.2015.00563.
Lawrence JG, Hendrickson H. Genome evolution in bacteria: order beneath chaos. Curr Opin Microbiol. 2005; 8(5):572–8. https://doi.org/10.1016/j.mib.2005.08.005.
Bentley SD, Parkhill J. Comparative genomic structure of prokaryotes. Annu Rev Genet. 2004; 38(38):771–91. https://doi.org/10.1146/annurev.genet.38.072902.094318.
Foerstner KU, von Mering C, Hooper SD, Bork P. Environments shape the nucleotide composition of genomes. EMBO reports. 2005; 6(12):1208–13. https://doi.org/10.1038/sj.embor.7400538.
Musto H, Naya H, Zavala A, Romero H, Alvarez-Valín F, Bernardi G. Correlations between genomic GC levels and optimal growth temperatures in prokaryotes. FEBS Lett. 2004; 573(1-3):73–7. https://doi.org/10.1016/j.febslet.2004.07.056.
Lee JJ, Kim M-N, Park KS, Lee JH, Karim AM, Park M, Kim JH, Lee SH. Complex class 1 integron carrying qnrB62 and blaVIM-2 in a Citrobacter freundii clinical isolate. Antimicrob Agents Chemother. 2016; 60(11):6937–40. https://doi.org/10.1128/aac.00614-16.
Lee K, Yum JH, Yong D, Lee HM, Kim HD, Docquier J-D, Rossolini GM, Chong Y. Novel acquired metallo- β-lactamase gene, blaSIM-1, in a class 1 integron from Acinetobacter baumannii clinical isolates from Korea. Antimicrob Agents Chemother. 2005; 49(11):4485–91. https://aac.asm.org/content/49/11/4485.
Partridge SR, Tsafnat G, Coiera E, Iredell JR. Gene cassettes and cassette arrays in mobile resistance integrons. FEMS Microbiol Rev. 2009; 33(4):757–84. https://doi.org/10.1111/j.1574-6976.2009.00175.x.
Yang C, Yang Y, Che Y, Xia Y, Li L, Xiong W, Zhang T. Bioprospecting for β-lactam resistance genes using a metagenomics-guided strategy. Appl Microbiol Biotechnol. 2017; 101(15):6253–60. https://doi.org/10.1007/s00253-017-8343-0.
Boulund F, Pereira MB, Jonsson V, Kristiansson E. Computational and statistical considerations in the analysis of metagenomic data In: Nagarajan M, editor. Metagenomics: Perspectives, Methods, and Applications. Cambridge, Massachusetts: Academic Press: 2017.
Berglund F, Marathe NP, Österlund T, Bengtsson-Palme J, Kotsakis S, Flach C-F, Larsson DJ, Kristiansson E. Identification of 76 novel B1 metallo- β-lactamases through large-scale screening of genomic and metagenomic data. Microbiome. 2017; 5(1):134. https://doi.org/10.1186/s40168-017-0353-8.
Berglund F, Österlund T, Boulund F, Marathe NP, Larsson DJ, Kristiansson E. Identification and reconstruction of novel antibiotic resistance genes from metagenomes. Microbiome. 2019; 7(1):52. https://doi.org/10.1186/s40168-019-0670-1.
Poirel L, He C, Nordmann P. Chromosome-encoded Ambler class D β-lactamase of Shewanella oneidensis as a progenitor of carbapenem-hydrolyzing oxacillinase. Antimicrob Agents Chemother. 2004; 48(1):348–51. https://doi.org/10.1128/aac.48.1.348.
Poirel L, Liard A, Nordmann P, Mammeri H. Origin of plasmid-mediated quinolone resistance determinant QnrA. Antimicrob Agents Chemother. 2005; 49(8):3523–5. https://doi.org/10.1128/aac.49.8.3523.
Ebmeyer S, Kristiansson E, Larsson DJ. PER extended-spectrum β-lactamases originate from Pararheinheimera spp. Int J Antimicrob Agents. 2019; 53(2):158–64. https://doi.org/10.1016/j.ijantimicag.2018.10.019.
Ebmeyer S, Kristiansson E, Larsson D. CMY-1/MOX-family AmpC β-lactamases MOX-1, MOX-2 and MOX-9 were mobilized independently from three aeromonas species. J Antimicrob Chemother. 2019; 74(5):1202–6. https://doi.org/10.1093/jac/dkz025.
Larouche A, Roy PH. Effect of attC structure on cassette excision by integron integrases. Mob DNA. 2011; 2(1):3. https://doi.org/10.1186/1759-8753-2-3.
Will S, Joshi T, Hofacker IL, Stadler PF, Backofen R. LocARNA-P: accurate boundary prediction and improved detection of structural RNAs. RNA. 2012; 18(5):900–14. https://doi.org/10.1261/rna.029041.111.
Seshadri R, Kravitz SA, Smarr L, Gilna P, Frazier M. CAMERA: A Community Resource for Metagenomics. PLoS Biol. 2007; 5(3):75. https://doi.org/10.1371/journal.pbio.0050075.
Meyer F, Paarmann D, Souza MD, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinforma. 2008; 9:386. https://doi.org/10.1186/1471-2105-9-386.
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2009; 37(D1):26–31. https://doi.org/10.1093/nar/gkn723.
Mitchell A, Bucchini F, Cochrane G, Denise H, Ten Hoopen P, Fraser M, Pesseat S, Potter S, Scheremetjew M, Sterk P, Finn RD. EBI metagenomics in 2016-an expanding and evolving resource for the analysis and archiving of metagenomic data. Nucleic Acids Res. 2016; 44(D1):595–603. https://doi.org/10.1093/nar/gkv1195.
Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR. HMMER web server: 2015 update. Nucleic Acids Res. 2015; 43(W1):30–8. https://doi.org/10.1093/nar/gkv397.
Mungall CJ, Bada M, Berardini TZ, Deegan J, Ireland A, Harris MA, Hill DP, Lomax J. Cross-product extensions of the Gene Ontology. J Biomed Inform. 2011; 44(1):80–6. https://doi.org/10.1016/j.jbi.2010.02.002.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST plus: architecture and applications. BMC Bioinforma. 2009; 10(421):1. https://doi.org/10.1186/1471-2105-10-421.
Österlund T, Jonsson V, Kristiansson E. HirBin: high-resolution identification of differentially abundant functions in metagenomes. BMC Genomics. 2017; 18(1):316. https://doi.org/10.1186/s12864-017-3686-6.
Tara Oceans Consortium C, Tara Oceans Expedition P. Registry of all stations from the Tara Oceans Expedition (2009-2013). PANGAEA. 2015. https://doi.pangaea.de/10.1594/pangaea.842237.
Hugerth LW, Larsson J, Alneberg J, Lindh MV, Legrand C, Pinhassi J, Andersson AF. Metagenome-assembled genomes uncover a global brackish microbiome. Genome Biol. 2015; 16:279. https://doi.org/10.1186/s13059-015-0834-7.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Antolin M, Artiguenave F, Blottiere H, Borruel N, Bruls T, Casellas F, Chervaux C, Cultrone A, Delorme C, Denariaz G, Dervyn R, Forte M, Friss C, van de Guchte M, Guedon E, Haimet F, Jamet A, Juste C, Kaci G, Kleerebezem M, Knol J, Kristensen M, Layec S, Le Roux K, Leclerc M, Maguin E, Melo Minardi R, Oozeer R, Rescigno M, Sanchez N, Tims S, Torrejon T, Varela E, de Vos W, Winogradsky Y, Zoetendal E, Bork P, Ehrlich SD, Wang J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010; 464(7285):59–65. https://doi.org/10.1038/nature08821.
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012; 490(7418):55–60. https://doi.org/10.1038/nature11450.
Bengtsson-Palme J, Angelin M, Huss M, Kjellqvist S, Kristiansson E, Palmgren H, Larsson DGJ, Johansson A. The human gut microbiome as a transporter of antibiotic resistance genes between continents. Antimicrob Agents Chemother. 2015; 59(10):6551–60. https://doi.org/10.1128/aac.00933-15.
Ilmberger N, Gullert S, Dannenberg J, Rabausch U, Torres J, Wemheuer B, Alawi M, Poehlein A, Chow J, Turaev D, Rattei T, Schmeisser C, Salomon J, Olsen PB, Daniel R, Grundhoff A, Borchert MS, Streit WR. A comparative metagenome survey of the fecal microbiota of a breast-and a plant-fed Asian elephant reveals an unexpectedly high diversity of glycoside hydrolase family enzymes. PLoS ONE. 2014; 9(9):106707. https://doi.org/10.1371/journal.pone.0106707.
Howe AC, Jansson JK, Malfatti SA, Tringe SG, Tiedje JM, Brown CT. Tackling soil diversity with the assembly of large, complex metagenomes. Proc Natl Acad Sci. 2014; 111(13):4904–9. https://doi.org/10.1073/pnas.1402564111.
Kouzuma A, Kasai T, Nakagawa G, Yamamuro A, Abe T, Watanabe K. Comparative metagenomics of anode-associated microbiomes developed in rice paddy-field microbial fuel cells. PLoS ONE. 2013; 8(11):2–11. https://doi.org/10.1371/journal.pone.0077443.
White RA, Power IM, Dipple GM, Southam G, Suttle CA. Metagenomic analysis reveals that modern microbialites and polar microbial mats have similar taxonomic and functional potential. Front Microbiol. 2015; 6(9):966. https://doi.org/10.3389/fmicb.2015.00966.
Acknowledgments
The authors are thankful to Atsushi Kouzuma, Curtis Suttle, Johan Bengtsson-Palme, Kazuya Watanabe, Luisa Hugerth, Rick White, Simon Güllert, and Wolfgang Streit for providing assemblies of their metagenomic data.
Funding
This research was funded by the Swedish Research Council (VR, 2012-05975), Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS, 2011-5025), Chalmers Life Science and Transport Areas of Advance, and the Wallenberg Foundation. The funders did not participate in the design of the study, the analysis and interpretation of data, or in writing the manuscript. Open access funding provided by Chalmers University of Technology.
Author information
Authors and Affiliations
Contributions
The study was designed by EK, MBP and MAF. The method was implemented by MBP under supervision of EK and MAF. The analysis of integron-associated genes was performed by MBP under supervision from EK and MAF. Data generation and pre-processing was done by TÖ, ME and TB. The manuscript was drafted by MBP and EK. All authors edited and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study does has not directly involved humans, animals or plants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
Table S1. The full catalog of integron-associated genes identified in this study. Details for all attC sites and integron-associated genes presented in cassette structure, including sequence and functional annotation. Note that in order to preserve the cassette structure, genes and attC sites redundancy has not been removed.
Additional file 2
Figure S1. Functional annotation of the integron-associated genes using TIGRFAM functional roles. Of the 13,397 integron-associated genes in our catalog 1203 genes matched a TIGRFAM with a known function. Percentages on the plot are given in relation to those numbers
Additional file 3
Table S2. Gene ontology analysis of the integron-associated genes using PFAM families [30]. Out of the 13,397 non-redundant integron-associated genes in our catalog, 3488 matched a PFAM family with a known function, which were in turn mapped to the metagenomics GO slim [78]. Note that the GO terms are organized in decreasing order inside each category, molecular function, biological process and cellular component.
Additional file 4
Table S3. Overrepresentation analysis of each COGs category between integron-associated and non-integron-associated genes found in the metagenomes. For each category, overrepresentation is given as odds ratio and the p-value of Fisher exact test.
Additional file 5
Secondary structural consensus for the 5 distinct clusters of attC sites. The clusters were generated by GraphClust.
Additional file 6
Table S4. Over-representation test using Fisher’s exact test for COG functional categories [28]. Significant p-values are shown in bold.
Additional file 7
Table S5. Over-representation test using Fisher?s exact test for GO-terms found in the metagenomics GO slim [78]. Significant p-values are shown in bold.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Buongermino Pereira, M., Österlund, T., Eriksson, K.M. et al. A comprehensive survey of integron-associated genes present in metagenomes. BMC Genomics 21, 495 (2020). https://doi.org/10.1186/s12864-020-06830-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-06830-5