
Abstract
Background
Genome sequencing technologies have been improved at an exponential pace but precise chromosome-scale genome assembly still remains a great challenge. The draft genome of cultivated G. arboreum was sequenced and assembled with shotgun sequencing approach, however, it contains several misassemblies. To address this issue, we generated an improved reassembly of G. arboreum chromosome 12 using genetic mapping and reference-assisted approaches and evaluated this reconstruction by comparing with homologous chromosomes of G. raimondii and G. hirsutum.
Results
In this study, we generated a high quality assembly of the 94.64 Mb length of G. arboreum chromosome 12 (A_A12) which comprised of 144 scaffolds and contained 3361 protein coding genes. Evaluation of results using syntenic and collinear analysis of reconstructed G. arboreum chromosome A_A12 with its homologous chromosomes of G. raimondii (D_D08) and G. hirsutum (AD_A12 and AD_D12) confirmed the significant improved quality of current reassembly as compared to previous one. We found major misassemblies in previously assembled chromosome 12 (A_Ca9) of G. arboreum particularly in anchoring and orienting of scaffolds into a pseudo-chromosome. Further, homologous chromosomes 12 of G. raimondii (D_D08) and G. arboreum (A_A12) contained almost equal number of transcription factor (TF) related genes, and showed good collinear relationship with each other. As well, a higher rate of gene loss was found in corresponding homologous chromosomes of tetraploid (AD_A12 and AD_D12) than diploid (A_A12 and D_D08) cotton, signifying that gene loss is likely a continuing process in chromosomal evolution of tetraploid cotton.
Conclusion
This study offers a more accurate strategy to correct misassemblies in sequenced draft genomes of cotton which will provide further insights towards its genome organization.
Similar content being viewed by others

Background
A high-quality genome sequence of species is a prerequisite to provide an inclusive access to complete genes catalog, different regulatory elements controlling their functions, and provides a framework for exploring genomic variations. During the early stages of genome sequencing, capillary technique was used to sequence the free-living organisms, starting with simple microbial genomes [1] followed by plant genomes including Arabidopsis thaliana [2], Oryza sativa [3] and Carica papaya [4]. Afterwards, many other complex plant genomes have been sequenced [5,6,7,8] using next-generation sequencing techniques (NGS). In current era, long-read sequencing (LRS) holds the promises due to its long-reads lengths [9], and many complex plants genome have been sequenced by this technique [10, 11].
In contrast to significant improvement of sequencing techniques, genome assembling continues to encounter many challenges [12, 13]. Particularly, complex and large plant genomes have remained a great challenge for de novo assembly due to its large genome size [14], high ploidy level [15], high rate of repeat elements [16], complex gene contents and high transposon’s activities [17]. One of the most difficult problems during de-novo genome assembly is the ordering and orientation of scaffolds to reconstruct the pseudo-chromosomes. A vigorous de novo assembly of chromosomes requires good quality physical and genetic maps [18, 19], optical maps [20], Hi-C sequence data [21] and genome collinearity and synteny [22] to anchor and orient the scaffolds to reconstruct the chromosomes. However, lack of good genetic or physical maps for most of the newly sequenced species makes difficult the accurate ordering of scaffolds into chromosomes. In this situation, good quality sequenced and assembled “reference genome” of closely related species would guide to an alternative approach which is referred as reference-assisted chromosome assembly. Orientation of scaffolds into chromosomes by reference-assisted chromosome assembly helps to exploit the benefits of assembled chromosomes without adding further efforts of sequencing or map construction [23].
Cotton (Gossypium spp.) is an important natural fiber and edible oil crop, mainly grown in subtropical and temperate areas of the world. Tetraploid genome of cotton is complicated by the presence of two sub-genomes (AT and DT) in its nucleus which were derived from diploid A-genome (G. arboreum) and D-genome (G. raimondii) progenitors. Diploid A genome is about 2-fold larger than D progenitor genome, and AT sub-genome is more stable in G. hirsutum than DT sub-genome [24]. Furthermore, G. arboreum possesses valuable and unique traits such as early maturity, tolerance to biotic and abiotic stresses and great fiber strength, providing a valuable germplasm resource for improving modern tetraploid cotton cultivars [25]. Therefore, existence of high quality reference draft genome sequence of G. arboreum is an essential task for tracing the origin of genome segments and interference of homoeology i.e. genes and RNA-seq [26] in tetraploid cotton.
Previously, genome of cultivated diploid cotton G. arboreum (Shixiya1) was sequenced and assembled using whole-genome shotgun approach which contained a total of 1694 Mb length including 41,330 protein coding genes and 1145 Mb long terminal repeats (LTR)-type retrotransposons [27]. Subsequently, genome sequence of tetraploid cotton G. hirsutum [28] was released which showed a conserved gene order with the A cotton genome (G. arboreum) [27]. However, another sequenced version of G. hirsutum genome [8] reported unobvious collinearity with the sequenced genome of G. arboreum [27], which is mainly due to numerous misassemblies in G. arboreum genome [27]. For instance, several scaffolds belong to different chromosomes were present in one pseudo-molecule of G. arboreum. Several previous studies reported that draft sequenced genome of G. arboreum [27] contained errors and mis-assemblies [8, 29, 30], however this draft genome did not undergo precise quality improvement to correct errors. So, knowing how to assemble this genome accurately, how to best make use of the highly fragmented assemblies and how to perform these applications at the lowest cost are important in today’s funding environment [31]. Here, we demonstrated an initial more accurate effort to reassemble chromosome 12 (A_A12) of G. arboreum using NGS data from previous study [27] without adding any other sequencing efforts, as its homolgous chromosomes of allotetraploid cotton contain important genes related to male sterility, fiber quality and gland development [32,33,34]. The advantage of selecting chromosome 12 also includes that it do not show any translocation [8, 35] in diploid and tetraploid cotton species. Subsequently, reassembled G. arboreum chromosome A_A12 was compared using collinear and syntenic analysis, whole chromosome alignment and dotplotting with its homologous chromosomes 12 of G. raimondii (D_D08) and G. hirsutum (AD_A12 and AD_D12) as well as previously assembled G. arboreum chromosome 12 (A_Ca9) [27] to support the more accuracy of reconstructed chromosome. Furthermore, we performed different comparative analysis such as gene loss, identification and mapping of transcription factor-related genes within homologous chromosomes 12 (A_A12, D_D08, AD_A12 and AD_D12) of three cotton species including G. arboreum, G. raimondii and G. hirsutum.
Results
Re-assembling of G. arboreum chromosome 12 (A_A12)
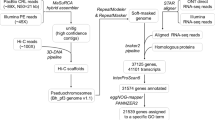
Here, we combined genetic mapping and reference-assisted approaches (Fig. 1) to reassemble G. arboreum chromosome A_A12.

Schematic diagram for reassembling of G. arboreum chromosome 12 (A_A12). Each rectangle corresponded to procedures applied for chromosome reassembling steps. Genotypic data of 24,569 SNP markers used in previous study [27] was first filtered out for construction of linkage groups, which were then assigned to 13 chromosomes of G. arboreum. Afterwards, linkage group belong to G. arboreum chromosome 12 was used for re-assembling. We checked the alignments of scaffolds belonging to G. arboreum chromosome 12 for following levels: (i) Alignment of G. arboreum scaffolds (obtained by the genetic map) to G. raimondii scaffolds [7], (ii) Orientation of G. raimondii (obtained from the previous step) and G. arboreum scaffolds along G. raimondii chromosome (D_D08) [36], and (iii) adjacency of G. arboreum scaffolds within G. hirsutum chromosome (AD_A12) [8]
Genetic map construction for re-assembling
Initially, 3735 high quality markers were selected out of 24,569 SNPs used in previous study [27] for construction of linkage map. A total of 3544 loci were classified into 13 linkage groups at LOD 06 with a total length of 1599.8 cM. Linkage groups 01 and 02 contained more number of markers as compared to others, while linkage group 13 enclosed lowest number of markers (Additional File 1: Fig. S1, Additional File 2: Table S1). Afterwards, chromosomes names were assigned to 13 linkage groups of G. arboreum according to the available mapped markers data of G. hirsutum and G. raimondii which gave the similar good results (Additional File 2: Table S2 and Table S3). However, we did not get same results in case of using mapped marker data of G. arboreum (Additional File 2: Table S4), provided first evidence of misassembles in sequenced genome of G. arboreum [27]. After assigning chromosomes names to 13 linkage groups, linkage group belong to G. arboreum chromosome 12 (A_A12) was used for further reassembling because it contains important genes for different traits and had no translocation. Final linkage group of G. arboreum chromosome A_A12 comprised of 189 markers, distributed within 64 scaffolds and spanned 146.63 cM genetic distance (Additional File 1: Fig. S1, Additional File 2: Table S1).
Reference assisted approach for reassembling
After construction of genetic map which served as a backbone for subsequent reassembling steps, we assessed G. arboreum chromosome A_A12 against two criteria: adjacency of scaffolds and gene integrity via BLAT and gene wise BLASTN approaches (Fig. 1). We checked scaffolds and gene integrity according to three steps: (i) Alignment of G. arboreum scaffolds (obtained by genetic map) to G. raimondii scaffolds [7], (ii) Orientation of G. raimondii (obtained from previous step) and G. arboreum scaffolds along G. raimondii chromosome D_D08 [36], and (iii) adjacency of G. arboreum scaffolds within G. hirsutum chromosome AD_A12 [8].
Based on linkage map and reference assisted approaches, we also identified inter-chromosomal mis-assemblies in 08 scaffolds of G. arboreum having a total of 19.79 Mb length (Additional File 2: Table S5). The final assembly of G. arboreum chromosome A_A12 comprised of 144 scaffolds (N50 = 912 kb) with 94.64 Mb length (Table 1, Additional File 1: Fig. S2).
Gene contents of G. arboreum chromosome A_A12
We generated an updated list of protein coding genes of reconstructed G. arboreum chromosome A_A12 which showed a total of 3361 predicted protein coding genes with an average transcript size of 1263 bp and a mean of 4.7 exons per gene (Table 1). The Cotton_A_14584 gene contained the largest CDS (14,331 bp) with 13 exons, while smallest CDS (90 bp) was enclosed by Cotton_A_37648 with 02 exons. Out of 3361 predicted genes, 2456 have predicted functional description. Gene density is 36 per Mb in G. arboreum chromosome A_A12 which is lower than in G. raimondii chromosome (53 per Mb of chromosome) [36]. Almost similar difference in gene density was reported between A12 and D12 chromosomes of G. hirsutum (29.4 vs 50 per Mb of chromosome) [8] and G. barbadense (33 vs 55.2 per Mb of chromosome), respectively [37].
Collinear and syntenic relationship
Comprehensive search of synteny and collinearity was carried out using BLASTP search comparing G. arboreum chromosome A_A12 with its corresponding homologous chromosomes of G. raimondii (D_D08) [36] and G. hirsutum (AD_A12 and AD_D12) [8]. Results indicated that the corresponding homologous chromosomes 12 of different Gossypium species possess a good syntenic relationship (Fig. 2a-c) such as 25 and 18 collinear blocks (with ≥5 genes per block) were aligned with G. raimondii (D_D08) and G. hirsutum (AD_A12) chromosomes (Additional File 2: Table S6), respectively. Overall gene order and collinearity was also highly conserved (Fig. 3 and Fig. 4a-c, Additional File 1: Fig. S3 and Fig. S4) between re-assembled G. arboreum chromosome A_A12 with its homologous chromosomes of G. raimondii [36] and G. hirsutum [8]. However, this collinearity was not apparent (Fig. 5a-b, Additional File 1: Fig. S5) with previously assembled G. arboreum chromosome (A_Ca9) [27], mainly due to; (i) mistakes in ordering of scaffolds (ii) many scaffolds belong to G. arboreum chromosome A_A12 were not present in it and, (iii) many scaffolds from other chromosomes were anchored and oriented in G. arboreum chromosome A_A12.

Syntenic relationship between corresponding homologous chromosomes of different Gossypium species. Syntenic relationship between homologous chromosomes 12 of; aG. raimondii (D_D08) and G. arboreum (A_A12), bG. hirsutum (AD_A12) and G. arboreum (A_A12), and cG. hirsutum (AD_D12) and G. arboreum (A_A12). Syntenic blocks were required to match at least five genes per block after masking repeat regions. Good syntenic relationship was found when comparing the homologous chromosomes of G. raimondii (D_D08) and G. hirsutum (AD_A12 and AD_D12) with reassembled chromosome of G. arboreum (A_A12)

Collinearity of reassembled G. arboreum chromosome (A_A12) with 26 chromosomes of G. hirsutum. Collinear relationship of reassembled G. arboreum chromosome (A_A12) with 26 chromosomes of G. hirsutum was determined by MCScan. After masking the repeat regions, collinearity analysis of G. arboreum chromosome A_A12 was carried out with all 26 chromosomes of G. hirsutum. Results indicated good collinear relationship of reassembled G. arboreum chromosome A_A12 with its corresponding homologous chromosomes 12 (AD_A12 and AD_D12) of G. hirsutum as compare to others chromosomes. G. arboreum chromosome 12 was shown by ‘A_A12’ while, chromosomes belong to At and Dt sub-genomes of G. hirsutum were indicated by ‘AD_A’ and ‘AD_D’

Dotplot representation between homologous chromosomes of different cotton species. A BLASTP search (with an E-value cutoff of 1 × 10− 5) was performed to identify orthologous genes. Afterwards, dotplots representation among homologous chromosomes of three cotton species was carried out by MCScan. aG. arboreum chromosome A_A12 (Y-axis) vs G. raimondii chromosome D_D08 (X-axis), bG. arboreum chromosome A_A12 (Y-axis) vs G. hirsutum chromosome AD_A12 (X-axis), and cG. arboreum chromosome A_A12 (Y-axis) vs G. hirsutum chromosome AD_D12 (X-axis)

Syntenic relationship with previously assembled chromosome 12 of G. arboreum (A_Ca9). Previously assembled chromosome 12 (A_Ca9) of G. arboreum was used to explore the syntenic relationship with a re-assembled G. arboreum chromosome A_A12 and, bG. hirsutum chromosome AD_A12. Syntenic blocks were required to match at least five genes per block. Results indicated poor syntenic relationship of G. arboreum chromosome A_Ca9 with these two chromosomes
Identification of orthologous gene pairs
We identified 2382 and 2603 orthologous gene pairs within homologous chromosomes (AD_A12 and AD_D12) of G. hirsutum and subsequent ancestral diploid A_A12 and D_D08 chromosomes (Additional File 2: Table S7). A total of 2485 ortholog pairs were identified between diploid A_A12 and D_D08 chromosomes.
Gene loss
Gene order was generated among the homologous chromosomes 12 of three Gossypium species by quartet alignments in MCScan [38]. Flanking gene method has been used to find gene loss in the syntenic blocks. Homologous chromosomes of allotetraploid cotton have greater gene loss; 26 genes were lost from AD_A12 and 22 from AD_D12 chromosomes (Table 2). In contrast, 13 and 09 genes were absent from A_A12 and D_D08 chromosomes of G. arboreum and G. raimondii, respectively (Table 3).
Identification and mapping of transcription factor (TF) related genes
Firstly, we generated an updated list of putative TF related genes of G. arboreum chromosome A_A12 using PlantTFDB [39]. This led to the identification of 266 putative members from 40 TF families, representing 8% of the protein-coding genes (Additional File 2: Table S8). There was more enrichment of ERF (35) related genes on chromosome A_A12 followed by bHLH (24), MYB (19), C2H2 (15) and WRKY (13). We also identified TF members of these five major families (ERF, bHLH, MYB, C2H2 and WRKY) in homologous chromosomes 12 of G. raimondii and G. hirsutum (Additional File 2: Table S9) to observe the influence of allopolyploidy on these genes. Comparative physical mapping of these genes on homologous chromosomes 12 of diploid and tetraploid cotton species revealed good collinear relationships among most of the TF-related genes (Fig. 6a-e). In particular, the chromosomal distribution of TF members in AD_A12 and AD_D12 chromosomes were more similar to their diploid progenitor’s chromosomes (A_A12 and D_D08). Moreover, TF encoding genes were not evenly distributed within the chromosomes. In general, the central region of chromosomes contained less number of TF-related genes, while comparatively high densities of TF members were found in bottom section of chromosomes.

Chromosomal mapping of the TF-related genes on homologous chromosome 12 of three cotton species. Physical mapping of five major TF-related family members including (a) MYB, (b) C2H2, (c) WRKY, (d) bHLH, (e) ERF was performed in homologous chromosome 12 of G. arboreum (A_A12), G. raimondii (D_D08) and G. hirsutum (AD_A12 and AD_D12). Genes in the positive and negative strands were represented by blue and red colors, while lines signified the collinear genes
Discussion
Chromosome-scale assemblies of sequenced plant genomes facilitated the discovery of important features of genome evolution. However, a consistent method for chromosome assembling from NGS data continues to present a serious constraint. Cultivated G. arboreum is important diploid cotton specie that contains important traits such as resistance to biotic and abiotic stresses [40, 41]. Previously, draft genome of G. arboreum has been sequenced and assembled [27] using 193.6 Gb of high-quality sequence reads. However, it contained several errors in ordering and orienting of scaffolds into pseudo-molecules [8, 30]. To address this problem, we re-constructed G. arboreum chromosome A_A12 by combining genetic mapping and reference assisted approaches. Initially, a high density genetic map of G. arboreum was constructed using 3735 good quality SNP markers from previous study [27], consisted of 3544 SNP loci and spanned 1599.8 cM in 13 linkage groups. Subsequently, linkage group belong to G. arboreum chromosome A_A12 was proceed for reassembling using reference assisted approach as it contains important genes for different traits [32,33,34], and do not contain any translocation [8, 35]. Final assembly of G. arboreum chromosome A_A12 comprised of 144 scaffolds and spanned 94.64 Mb length, which is almost twice the size (57.13 Mb) of its homologous chromosome (D_D08) of G. raimondii [36]. These results were consistent with chromosome size difference between the homologous chromosome 12 of At (87.4 Mb) and Dt (59.1 Mb) subgenome of G. hirsutum [8]. Similarly, tetraploid genome of G. barbadense [37] contained A12 and D12 chromosomes of the103.3 Mb and 58.2 Mb, respectively.
Further, both G. arboreum and G. raimondii chromosomes (A_A12 and D_D08) contained 3361 and 2990 genes, resulted lower gene density (36 vs 53 per Mb of chromosome) in A_A12 chromosome than D_D08 [36]. Similar difference in gene density was observed between the A12 and D12 chromosomes of G. hirsutum [8] and G. barbadense [37]. This lower gene density in chromosome A_A12 than D_D08 is mainly due to the presence of more repetitive elements. Previously, several studies also reported that larger genome size of G. arboreum relative to G. raimondii was mainly due to the presence of repetitive elements [42, 43]. Additionally, G. arboreum genome contained [27] high percentage of transposable elements as compared to G. raimondii [7, 36].
Polyploidization is often followed by whole genome duplication that is illustrated by genome reorganization and immense gene loss [44,45,46]. This process has been observed in different plants i.e. wheat [47], Brassica [48] and maize [49]. Though, some other plants including Arabidopsis [50] and cotton [51] do not illustrate various changes in their genome sequences. In current study, synteny and collinearity, whole chromosomal alignment and homologous gene dotplotting showed highly conserved syntenic and collinear relationship among homologous chromosomes of G. hirsutum, G. raimondii and reassembled G. arboreum chromosome, depicting preservation of very similar genomic structure since their divergence [52, 53]. Previous studies also reported highly conserved collinear relationship among different cotton species, which is also consistent to our results [8, 54]. This is possibly because actual progenitors which may form stable cultivated allotetraploid were lost or unstable tetraploid was eliminated by natural selection during early generations. However, this synteny was not apparent with previously assembled chromosome of G. arboreum (A_Ca9) [27]. In addition, homologous gene dotplotting with G. arboreum chromosome A_Ca9 also showed unobvious collinear relationship, confirming various mistakes in ordering and anchoring of scaffolds. Previous report [8] also showed unobvious collinearity between the homologous chromosomes of G. hirsutum and G. arboreum, which was consistent to our result.
Differential gene loss is an important factor during genome evolution which affects synteny between corresponding regions of different chromosomes [55,56,57], and can lead to immediate loss of gene function. In current study, we found a higher rate of gene loss in homologous chromosomes of tetraploid (AD_A12 and AD_D12) than diploid (A_A12 and D_D08) cotton. These results were consistent with the previous reports [8, 28], suggesting gene loss is probably an enduring process in chromosomal evolution of tetraploid cotton.
Transcription factors play a significant role in plant growth and development, secondary metabolism, organ morphogenesis and resistance against different stresses in cotton [58,59,60]. Several previous reports computed genome-wide analysis of TF-related genes in different cotton species and compared their physical location on different chromosomes [61,62,63,64]. In current study, distribution of TF-related genes showed that homologous chromosomes of G. raimondii (D_D08) and G. arboreum (A_A12) contained almost similar number of TF genes with minimum deviation, and they had good collinear relationship with each other. For Instance, 13 WRKY genes were identified on each of re-constructed G. arboreum A_A12 and G. raimondii D_D08 chromosomes with high collinearity. Recent study also reported highly conserved collinearity among TF-related genes of four Gossypium species [65]. In contrast, another study using previously assembled G. arboreum genome [27] identified different number of WRKY genes and their unobvious collinearity in G. arboreum and G. raimondii chromosomes 12, respectively [63]. Furthermore, distribution of TF encoding genes was not even within the corresponding homologous chromosome of three cotton species which is likely due to sequence exchange through recombination mispairing [66].
Conclusion
In conclusion, we generated an improved reassembly of G. arboreum chromosome A_A12 using NGS data of previous study [27] by combining genetic mapping and reference assisted approaches. This study provides an initial more accurate strategy for correcting mis-assemblies in sequenced genome of G. arboreum which can also be applied to improve chromosome-scale assemblies of large and complex plant genomes without having good genetic or physical maps.
Methods
Genomes and markers data
Sequenced genome data of G. arboreum [27] including scaffolds, predicted annotated genes and genotypic data of 24,569 SNP markers as well as scaffolds data of G. raimondii [7] was obtained from Institute of Cotton Research, Chinese Academy of Agricultural Sciences, Anyang, China. Chromosomal and genes annotation data of G. hirsutum [8] and G. raimondii [36] was downloaded from the CottonFGD (https://cottonfgd.org/). Meanwhile, sequence data of previous mapped markers of G. hirsutum and G. raimondii for each chromosome was downloaded from COTTONGEN (https://www.cottongen.org/find/markers).
SNP markers selection
Markers data of 24,569 SNPs [27] was filtered out to obtain good quality linkage map of G. arboreum. Firstly, Chi-square test was executed to find markers diverging from Mendelian segregation patterns. Markers were excluded from analysis when they displayed very significant distortion (P < 0.01) from expected segregation ratio, also when they had more than 30% missing genotypic data. We identified markers with more than 95% similarity, and only one such marker was used for linkage map analysis.
Genetic map construction
Linkage groups were constructed by JoinMap 4.0 [67] using F2 generation from previous study [27]. Markers were allocated to linkage groups by independence logarithm of odds (LOD) of 2.5–50.0 with a step of 1.0. Linkage groups were generated using LOD thresholds of 6.0 and maximum recombination thresholds of 0.4. We used a maximum likelihood mapping algorithm for calculation efficiency of marker order [68] if linkage group contained more than 500 markers. However, the scope of corresponding linkage groups (3000–6000 cM) exceeded JoinMap 4.0. Therefore, linkage length was divided by 100 for the presentation of genetic map [69]. In other linkage groups having less than 500 markers, a linear regression algorithm and the Kosambi mapping function [70] was used to convert recombination frequencies into centiMorgan (cM) map distances. Final linkage map was drawn using Mapchart 2.2 [71].
Assign chromosomes names to linkage groups
To assign chromosomes names to each linkage group, sequence data of mapped markers for each chromosome of G. hirsutum and G. raimondii was obtained from COTTONGEN (https://www.cottongen.org/find/markers). Then a BLAST search was made using the marker sequence data of G. hirsutum and G. raimondii as a query and G. arboreum scaffolds corresponding to SNP markers of each linkage group as a database.
Initial alignment of G. arboreum scaffolds
All scaffolds belonging to 189 SNP markers of G. arboreum chromosome A_A12 were pairwise aligned with the G. raimondii scaffolds [7] by BLAST-Like Alignment Tool (BLAT). The resulted alignments were required to have score values showing the length and similarity of aligned regions, while only best BLAT hit was counted from the alignments. Afterward, each of the pairwise alignment was validated by anchoring the protein coding genes of G. raimondii scaffolds [7] within G. arboreum scaffolds by BLASTN. If a gap between two coordinated scaffolds was > 100 kb then the corresponding region of D scaffolds was extracted to align it with the scaffolds of G. arboreum (as a database) by BLAT followed by gene wise BLASTN. This step is repeated until maximum number of G. arboreum scaffolds were aligned with the G. raimondii scaffolds [7].
Final alignment of G. arboreum scaffolds
Next, all G. raimondii and G. arboreum scaffolds [7] obtained by initial alignment were separately pair-wise aligned with another version of G. raimondii chromosome (D_D08) [36] via BLAT and gene wise BLASTN. Unlocated and unplaced scaffolds of G. arboreum were excluded from the assembly. Again, if a gap between two coordinated scaffolds was more than 100 kb, the corresponding nucleotide sequence of G. raimondii chromosome (D_D08) was extracted and used as a query to align it with G. arboreum scaffolds by BLAT and BLASTN. Eventually, all resulted scaffolds were further confirmed by arranging them on the homologous chromosome (AD_A12) of G. hirsutum [8].
Correction of assembly using genetic map and syntenic relationship
The linkage map of G. arboreum chromosome A_A12 and its synteny with the homologous chromosome of G. raimondii [36] and G. hirsutum [8] was used to find false joins within the scaffolds and to anchor the scaffolds into chromosome. Scaffolds were broken if they enclosed a false join based on genetic map and syntenic relationship. Then, corrected scaffolds were arranged to generate chromosome A_A12 of G. arboreum.
Gene contents of G. arboreum chromosome A_A12
An AGP (a golden path) file that records the position of protein-coding genes for each scaffold of G. arboreum [27] was obtained from Institute of Cotton Research, Chinese Academy of Agricultural Sciences, Anyang, China. We generated an updated list of genes and proteins for re-assembled G. arboreum chromosome A_A12 by arranging the genes and proteins of each scaffolds in their respective order. Putative functional description of all genes was explored by CottonFGD (https://cottonfgd.org/search/).
Syntenic and collinear analysis
Syntenic blocks between corresponding homologous chromosomes of G. arboreum (A_A12), G. hirsutum (AD_A12 and AD_D12) [8] and G. raimondii (D_D08) [36] were identified by MCScan [38] with default parameters. After removing multiple matches and tandem duplications, syntenic blocks having more than five gene pairs were identified.
Identification of orthologous gene sets
All protein sequences of corresponding homologous chromosomes 12 of each cotton species (G. arboreum, G. raimondii and G. hirsutum) were compared by BLASTP (e-value < 1 × 10− 5). Genes were classified into ortholog clusters with OrthoMCL against OrthoMCL database proteins [72]. Multiple sequence alignment of G. arboreum, G. raimondii and G. hirsutum protein coding sequences was performed with ClustalW [73]. Based on the orthologous gene sets between homologous chromosomes of G. arboreum (A_A12), G. raimondii (D_D08) [36], and two sub-genomes of G. hirsutum (AD_A12 and AD_D12) [8], synonymous and non-synonymous substitutions per site among three cotton species were calculated by Synonymous Non-synonymous Analysis Program (SNAP) [74].
Gene loss
Gene-loss events were depicted using flanking gene method from the synteny table generated by MCScan [38]. For instance, given flanking genes X, Y and Z in order, if gene Y is present in the corresponding homologous chromosomes 12 of three Gossypium genomes, but missed in chromosome of other one genome, then gene Y is referred as a lost gene. However, both X and Z genes are essentially to be present in homologous chromosome (A_A12, D_D08, AD_A12 and AD_D12) of all four Gossypium genomes.
Identification and mapping of transcription factor related genes
Transcription factor (TF) related genes were identified by searching all protein sequences of re-assembled G. arboreum chromosome A_A12 using Plant Transcription Factor Database, PlantTFDB [39]. Afterwards, only top five putative TF-related genes including ERF, bHLH, MYB, C2H2 and WRKY were used for further analysis. The Hidden Markov Model (HMM) profiles of gene domains were obtained from Pfam [75] for gene family identification. HMMER 3.0 [76] search was used to confirm the putative TF-related genes in homologous chromosomes 12 of G. arboreum, G. raimondii and G. hirsutum. Chromosomal position of all TF-related genes was resolved by BLASTN searches against chromosomes of G. arboreum (A_12), G. raimondii (D_D08) [36] and G. hirsutum (AD_A12 and AD_D12) [8]. All TF-related genes were mapped on the chromosomes using the Mapchart 2.2 [71].
Availability of data and materials
The sequence data of re-constructed G. arboreum chromosome 12 has been deposited at the NCBI Genbank under the accession number CP053561. The other data sets generated in this study are included within the article and supplementary files.
Abbreviations
- A_A12:
-
Re-assembled G. arboreum chromosome 12
- A_Ca9:
-
Chromosome 12 of G. arboreum [27]
- D_D08:
-
Chromosome 12 of G. raimondii [36]
- AD_A12:
-
Chromosome 12 for At subgenome of G. hirsutum [8]
- AD_D12:
-
Chromosome 12 for Dt subgenome of G. hirsutum [8]
- NGS:
-
Next-generation sequencing
- LRS:
-
Long-read sequencing
- cM:
-
Centi-Morgan
- LOD:
-
Logarithm of the odds
- SNP:
-
Single nucleotide polymorphism
- BLAST:
-
Basic local alignment search tool
- BLAT:
-
BLAST-Like alignment tool
- HMM:
-
Hidden markov model
- N50:
-
50% of the assembled nucleotides within scaffolds
- TF:
-
Transcription factor
References
Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269(5223):496–512.
Initiative AG. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408(6814):796–815.
Sasaki T. The map-based sequence of the rice genome. Nature. 2005;436(7052):793–800.
Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KL. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature. 2008;452(7190):991–6.
Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, Jaiswal P, Mockaitis K, Liston A, Mane SP. The genome of woodland strawberry (Fragaria vesca). Nat Genet. 2011;43(2):109–16.
Varshney RK, Chen W, Li Y, Bharti AK, Saxena RK, Schlueter JA, Donoghue MT, Azam S, Fan G, Whaley AM. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotech. 2012;30(1):83–9.
Wang K, Wang Z, Li F, Ye W, Wang J, Song G, Yue Z, Cong L, Shang H, Zhu S. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012;44(10):1098–103.
Zhang T, Hu Y, Jiang W, Fang L, Guan X, Chen J, Zhang J, Saski CA, Scheffler BE, Stelly DM. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotech. 2015;33(5):531–7.
van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. The third revolution in sequencing technology. Trends Genet. 2018;34(9):666–81.
Xia E, Li F, Tong W, Yang H, Wang S, Zhao J, Liu C, Gao L, Tai Y, She G. The tea plant reference genome and improved gene annotation using long-read and paired-end sequencing data. Sci Data. 2019;6(1):1–9.
Girollet N, Rubio B, Lopez-Roques C, Valiere S, Ollat N, Bert PF. De novo phased assembly of the Vitis riparia grape genome. Sci Data. 2019;6(1):1–8.
Schatz MC, Delcher AL, Salzberg SL. Assembly of large genomes using second-generation sequencing. Genome Res. 2010;20(9):1165–73.
Earl D, Bradnam K, John JS, Darling A, Lin D, Fass J, Yu HOK, Buffalo V, Zerbino DR, Diekhans M. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res. 2011;21(12):2224–41.
Pellicer J, Fay MF, Leitch IJ. The largest eukaryotic genome of them all? Bot J of Linn Soc. 2010;164(1):10–5.
Meyers LA, Levin DA. On the abundance of polyploids in flowering plants. Evolution. 2006;60(6):1198–206.
Gore MA, Chia JM, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, Grills GS, Ross-Ibarra J. A first-generation haplotype map of maize. Science. 2009;326(5956):1115–7.
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–5.
Marra M, Hillier L, Waterston RH, Chinwalla A, Wallis J, Sekhon M, Wylie K, Mardis ER, Wilson RK, Fulton R. A physical map of the human genome. Nature. 2001;409:934–41.
Lewin HA, Larkin DM, Pontius J, O'Brien SJ. Every genome sequence needs a good map. Genome Res. 2009;19(11):1925–8.
de Jesus ST, Parise D, Profeta R, Parise MTD, Gomide ACP, Kato RB, Pereira FL, Figueiredo HCP, Ramos R, Brenig B. Re-sequencing and optical mapping reveals misassemblies and real inversions on Corynebacterium pseudotuberculosis genomes. Sci Rep. 2019;9(1):1–11.
Ghurye J, Rhie A, Walenz BP, Schmitt A, Selvaraj S, Pop M, Phillippy AM, Koren S. Integrating hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comp Biol. 2019;15(8):e1007273.
Waterhouse RM, Aganezov S, Anselmetti Y, Lee J, Ruzzante L, Reijnders MJ, Feron R, Berard S, George P, Hahn MW. Evolutionary superscaffolding and chromosome anchoring to improve Anopheles genome assemblies. BMC Biol. 2020;18(1):1–20.
Tamazian G, Dobrynin P, Krasheninnikova K, Komissarov A, Koepfli KP, O’brien SJ. Chromosomer: a reference-based genome arrangement tool for producing draft chromosome sequences. GigaSci. 2016;5(1):38.
Guo W, Cai C, Wang C, Han Z, Song X, Wang K, Niu X, Wang C, Lu K, Shi B. A microsatellite-based, gene-rich linkage map reveals genome structure, function, and evolution in Gossypium. Genetics. 2007;176:527–41.
Kantartzi SK, Ulloa M, Sacks E, Stewart JM. Assessing genetic diversity in Gossypium arboreum L. cultivars using genomic and EST-derived microsatellites. Genetica. 2009;136(1):141–7.
Page JT, Huynh MD, Liechty ZS, Grupp K, Stelly D, Hulse AM, Ashrafi H, Van Deynze A, Wendel JF, Udall JA. Insights into the evolution of cotton diploids and polyploids from whole-genome re-sequencing. Genes Genom Genet. 2013;3(10):1809–8.
Li F, Fan G, Wang K, Sun F, Yuan Y, Song G, Li Q, Ma Z, Lu C, Zou C. Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet. 2014;46(6):567–72.
Li F, Fan G, Lu C, Xiao G, Zou C, Kohel RJ, Ma Z, Shang H, Ma X, Wu J. Genome sequence of cultivated upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotech. 2015;33(5):524–30.
Li X, Jin X, Wang H, Zhang X, Lin Z. Structure, evolution, and comparative genomics of tetraploid cotton based on a high-density genetic linkage map. DNA Res. 2016;23(3):283–93.
Fang L, Gong H, Hu Y, Liu C, Zhou B, Huang T, Wang Y, Chen S, Fang DD, Du X. Genomic insights into divergence and dual domestication of cultivated allotetraploid cottons. Genome Biol. 2017;18(1):33.
Schatz MC, Witkowski J, McCombie WR. Current challenges in de novo plant genome sequencing and assembly. Genome Biol. 2012;13(4):243.
Rong J, Pierce GJ, Waghmare VN, Rogers CJ, Desai A, Chee PW, May OL, Gannaway JR, Wendel JF, Wilkins TA. Genetic mapping and comparative analysis of seven mutants related to seed fiber development in cotton. Theor Appl Genet. 2005;111(6):1137–46.
Chen D, Ding Y, Guo W, Zhang T. Molecular mapping of genic male-sterile genes ms15, ms5 and ms6 in tetraploid cotton. Plant Breed. 2009;128(2):193–8.
Cheng H, Lu C, John ZY, Zou C, Zhang Y, Wang Q, Huang J, Feng X, Jiang P, Yang W. Fine mapping and candidate gene analysis of the dominant glandless gene Gl2e in cotton (Gossypium spp.). Theor Appl Genet. 2016;129(7):1347–55.
Gerstel D. Chromosomal translocations in interspecific hybrids of the genus Gossypium. Evolution. 1953:234–44.
Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, Jin D, Llewellyn D, Showmaker KC, Shu S, Udall J. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012;492(7429):423–7.
Liu X, Zhao B, Zheng HJ, Hu Y, Lu G, Yang CQ, Chen JD, Chen JJ, Chen DY, Zhang L. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci Rep. 2015;5:14139.
Wang Y, Tang H, DeBarry JD, Tan X, Li J, Wang X, Lee T, Jin H, Marler B, Guo H. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49.
Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, Gao G. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017;45(D1):D1040–5.
Akhtar K, Haidar S, Khan M, Ahmad M, Sarwar N, Murtaza M, Aslam M. Evaluation of Gossypium species for resistance to cotton leaf curl Burewala virus. Annl Appl Biol. 2010;157(1):135–47.
Zhang L, Li F, Liu C, Zhang C, Wu Z. Isolation and analysis of a drought-related gene from a cotton (Gossypium arboreum) SSH library. Cotton Sci. 2010;22(2):110–4.
Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006;16(10):1252–61.
Hawkins JS, Proulx SR, Rapp RA, Wendel JF. Rapid DNA loss as a counterbalance to genome expansion through retrotransposon proliferation in plants. Proc Nat Acad Sci. 2009;106(42):17811–6.
Otto SP. The evolutionary consequences of polyploidy. Cell. 2007;131(3):452–62.
Soltis PS, Soltis DE. The role of hybridization in plant speciation. Ann Rev Plant Biol. 2009;60:561–88.
Brenchley R, Spannagl M, Pfeifer M, Barker GL, D’Amore R, Allen AM, McKenzie N, Kramer M, Kerhornou A, Bolser D. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature. 2012;491(7426):705–10.
Feldman M, Liu B, Segal G, Abbo S, Levy AA, Vega JM. Rapid elimination of low-copy DNA sequences in polyploid wheat: a possible mechanism for differentiation of homoeologous chromosomes. Genetics. 1997;147(3):1381–7.
Gaeta RT, Pires JC, Iniguez-Luy F, Leon E, Osborn TC. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell. 2007;19(11):3403–17.
Woodhouse MR, Schnable JC, Pedersen BS, Lyons E, Lisch D, Subramaniam S, Freeling M. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homeologs. PLoS Biol. 2010;8(6):e1000409.
Wang J, Tian L, Lee HS, Wei NE, Jiang H, Watson B, Madlung A, Osborn TC, Doerge R, Comai L. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics. 2006;172(1):507–17.
Liu B, Brubaker C, Mergeai G, Cronn R, Wendel J. Polyploid formation in cotton is not accompanied by rapid genomic changes. Genome. 2001;44(3):321–30.
Rong J, Abbey C, Bowers JE, Brubaker CL, Chang C, Chee PW, Delmonte TA, Ding X, Garza JJ, Marler BS. A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics. 2004;166(1):389–417.
Yu Y, Yuan D, Liang S, Li X, Wang X, Lin Z, Zhang X. Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypium hirsutum and G. barbadense. BMC Genom. 2011;12(1):15.
Brubaker C, Paterson A, Wendel J. Comparative genetic mapping of allotetraploid cotton and its diploid progenitors. Genome. 1999;42(2):184–203.
Paterson AH, Chapman BA, Kissinger JC, Bowers JE, Feltus FA, Estill JC. Many gene and domain families have convergent fates following independent whole-genome duplication events in Arabidopsis, Oryza. Saccharomyces and Tetraodon Trends Genet. 2006;22(11):597–602.
Domazet-Loso T, Tautz D. An evolutionary analysis of orphan genes in Drosophila. Genome Res. 2003;13(10):2213–9.
Krylov DM, Wolf YI, Rogozin IB, Koonin EV. Gene loss, protein sequence divergence, gene dispensability, expression level, and interactivity are correlated in eukaryotic evolution. Genome Res. 2003;13(10):2229–35.
Wang S, Wang JW, Yu N, Li CH, Luo B, Gou JY, Wang LJ, Chen XY. Control of plant trichome development by a cotton fiber MYB gene. Plant Cell. 2004;16(9):2323–34.
Meng X, Li F, Liu C, Zhang C, Wu Z, Chen Y. Isolation and characterization of an ERF transcription factor gene from cotton (Gossypium barbadense L.). Plant Mol Biol Rep. 2010;28(1):176–83.
Chen X, Jin X, Li X, Lin Z. Genetic mapping and comparative expression analysis of transcription factors in cotton. PLoS One. 2015;10(5):e0126150.
Ma J, Liu F, Wang Q, Wang K, Jones DC, Zhang B. Comprehensive analysis of TCP transcription factors and their expression during cotton (Gossypium arboreum) fiber early development. Sci Rep. 2016;6(1):1–10.
Pant P, Iqbal Z, Pandey BK, Sawant SV. Genome-wide comparative and evolutionary analysis of calmodulin-binding transcription activator (CAMTA) family in Gossypium species. Sci Rep. 2018;8(1):1–17.
Ding M, Chen J, Jiang Y, Lin L, Cao Y, Wang M, Zhang Y, Rong J, Ye W. Genome-wide investigation and transcriptome analysis of the WRKY gene family in Gossypium. Mol Genet Genomics. 2015;290(1):151–71.
Lei Z, He D, Xing H, Tang B, Lu B. Genome-wide comparison of AP2/ERF superfamily genes between Gossypium arboreum and G. raimondii. Genet Mol Res. 2016;15(3):15038211.
Liu Z, Fu M, Li H, Chen Y, Wang L, Liu R. Systematic analysis of NAC transcription factors in Gossypium barbadense uncovers their roles in response to Verticillium wilt. Peer J. 2019;7:e7995.
Friedman AR, Baker BJ. The evolution of resistance genes in multi-protein plant resistance systems. Curr Opin Genet Dev. 2007;17(6):493–9.
Van Ooijen J. JoinMap 4. Software for the calculation of genetic linkage maps in experimental populations Kyazma BV, Wageningen, Netherlands; 2006. p. 33.
Haldane J. The combination of linkage values and the calculation of distances between the loci of linked factors. J Genet. 1919;8(29):299–309.
Xiao B, Tan Y, Long N, Chen X, Tong Z, Dong Y, Li Y. SNP-based genetic linkage map of tobacco (Nicotiana tabacum L.) using next-generation RAD sequencing. J Biol Res-Thessaloniki. 2015;22(1):11.
Kosambi D. The estimation of map distances from recombination values. Annals Eugen. 1944;2(172.1):75.
Voorrips R. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93(1):77–8.
Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, Shanmugam D, Roos DS, Stoeckert CJ. Using OrthoMCL to assign proteins toOrthoMCL‐DB groups or to cluster proteomes into new Ortholog groups. Curr Prot Bioinformatics. 2011;35(1):6–12.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Korber B. HIV signature and sequence variation analysis. Computational analysis of HIV molecular sequences. 2000;4:55–72.
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2015;44(D1):D279–85.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37.
Acknowledgements
We would like to thank anonymous reviewers for their valuable suggestions.
Funding
This work was supported by grants from National Natural Science Foundation of China (31621005), and National Key Research and Development Program (2018YFD0100400, 2016YFD0101006). The funding agencies had no role in study design, data collection and analysis, decision to publish and preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
JA and GS conceived and designed the experiments. JA, DZ, HC and QY performed the experiment. JA, QW, YZ, MAA and XF analyzed the data. JA and GS wrote the paper. WM, JZY and GS critically revised the paper. All authors read and approved the final version of manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Fig. S1
Genetic map of G. arboreum genome. Fig. S2 Arrangement of G. arboreum scaffolds within reassembled G. arboreum chromosome 12 (A_A12). Fig. S3 Collinearity among homologous chromosomes 12 of three cotton species. Fig. S4 Alignments of reassembled G. arboreum chromosome A_A12 with the whole genome of G. hirsutum.Fig. S5 Dotplot representation with the previously assembled G. arboreum chromosome.
Additional file 2 Table S1
Genetic map construction of G. arboreum.Table S2 Chromosomes names assignment to each linkage group with respect to previous mapped markers of G. raimondii.Table S3 Chromosomes names assignment to each linkage group with respect to previous mapped markers of G. hirsutum.Table S4 Chromosomes names assignment to each linkage group with respect to previous mapped markers of G. arboreum.Table S5 Statistics for misassembled scaffolds. Table S6 Collinear blocks among the homologous chromosome 12 of different cotton species. Table S7 Orthologous gene pairs between homologous chromosomes 12 of different cotton species. Table S8 TF-related genes in reassembled G. arboreum chromosome A_A12. Table S9 TF-related genes on homologous chromosome 12 of three cotton species. (XLS 1436 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ashraf, J., Zuo, D., Cheng, H. et al. Improved reconstruction and comparative analysis of chromosome 12 to rectify Mis-assemblies in Gossypium arboreum. BMC Genomics 21, 470 (2020). https://doi.org/10.1186/s12864-020-06814-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-06814-5