
Abstract
Background
Mycoplasma pneumoniae is a common cause of respiratory tract infections in children and adults. This study applied high-throughput whole genome sequencing (WGS) technologies to analyze the genomes of 30 M. pneumoniae strains isolated from children with pneumonia in South Korea during the two epidemics from 2010 to 2016 in comparison with a global collection of 48 M. pneumoniae strains which includes seven countries ranging from 1944 to 2017.
Results
The 30 Korean strains had approximately 40% GC content and ranged from 815,686 to 818,669 base pairs, coding for a total of 809 to 828 genes. Overall, BRIG revealed 99% to > 99% similarity among strains. The genomic similarity dropped to approximately 95% in the P1 type 2 strains when aligned to the reference M129 genome, which corresponded to the region of the p1 gene. MAUVE detected four subtype-specific insertions (three in P1 type 1 and one in P1 type 2), of which were all hypothetical proteins except one tRNA insertion in all P1 type 1 strains. The phylogenetic associations of 30 strains were generally consistent with the multilocus sequence typing results. The phylogenetic tree constructed with 78 genomes including 30 genomes from Korea formed two clusters and further divided into two sub-clusters. eBURST analysis revealed two clonal complexes according to P1 typing results showing higher diversity among P1 type 2 strains.
Conclusions
The comparative whole genome approach was able to define high genetic identity, unique structural diversity, and phylogenetic associations among the 78 M. pneumoniae strains isolated worldwide.
Similar content being viewed by others

Background
M. pneumoniae is an important cause of respiratory tract infections in children and adults, ranging from mild upper respiratory infections to life-threatening conditions [1]. M. pneumoniae infections are more common among children 5 years of age or older than among younger children [2]. Mild upper respiratory infections are common with a considerable portion of asymptomatic patients, but 3 to 10% develop pneumonia with a wide spectrum of radiologic findings [3,4,5]. Extrapulmonary abnormalities are an important part of M. pneumoniae diseases both in diagnosis and treatment. The spectrum of manifestations includes extrapulmonary symptoms such as skin rash, hemolytic anemia, arthritis, and neurologic abnormalities [1].
P1 adhesin (P1), a 170-kD surface protein located at the tip-like structure of virulent M. pneumoniae, mediates its cytoadherence to the surface of respiratory epithelial cells [6]. As P1 adhesin protein plays a critical step in the infection process, studies regarding the genetics of M. pneumoniae focused mainly on P1 types and subtypes [7, 8]. P1 typing was the only available tool that could be applied in the past to determine genotype. Although P1 typing can separate M. pneumoniae into two types and additional six variants, it did not always convey information regarding epidemiologic characteristics or clinical severity. New genetic analysis techniques, such as multilocus variable-number tandem-repeat analysis (MLVA) and multilocus sequence typing (MLST), have been applied to M. pneumoniae [9, 10].
Despite the evolution of molecular microbiology and advanced classifications beyond P1 typing, research to understand the entire genome structures of M. pneumoniae in regard to molecular epidemiology has remained much behind that of other bacteria such as Streptococcus pneumoniae and Escherichia coli. Recent advances in molecular microbiology and bioinformatics have made it possible to analyze M. pneumoniae through high-throughput sequencing technologies such as Illumina dye sequencing, pyrosequencing, and single-molecule real-time (SMRT) sequencing [11]. The whole genome of M. pneumoniae is ≈820 kb and has up to 700 coding operons [12]. The comparably small genome size and limited number of operons are challenges in the genomic investigation of M. pneumoniae.
This study aims to analyze genomes of 30 M. pneumoniae strains isolated from children with pneumonia in South Korea during two epidemics from 2010 to 2016 and compare with a global collection of 48 M. pneumoniae strains which includes seven countries ranging from 1944 to 2017.
Results
Strain characteristics
The strains were isolated from nasopharyngeal samples obtained from children with pneumonia. Thirty-seven and 45 M. pneumoniae strains were collected in 2010–12 and 2014–16, respectively. Thirty M. pneumoniae strains were chosen for the current study (Additional file 1). Eighteen strains and twelve strains were selected from 2010 to 12 and 2014–16 epidemic years, respectively. Twenty-four (80.0%) P1 type 1 strains, five (16.7%) P1 type 2c strains and a P1 type 2a strain (3.3%) were included. Five sequence types (STs) were included: ST1 (n = 2, 6.7%), ST3 (n = 20, 66.7%), ST14 (n = 5, 16.7%), ST17 (n = 2, 6.7%), and ST33 (n = 1, 3.3%).
Genome assembly
The characteristics of the assemblies and the background information are found in Table 1. The resulting contigs were mapped to the M129 reference genome and joined via PCR. The thirty genomes had all contigs joined to form a single, continuous (circular) contig. Following assembly and editing, the genomes underwent automated gene annotation. With approximately 40% GC content and ranging from 815,686 to 818,669 bp, the genomes coded for a total of 809 to 828 genes.
Overall comparison
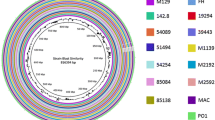
The 30 sequenced genomes were aligned to the reference M129 genome using BLAST Ring Image Generator (BRIG). Overall, the genomes were 99% to > 99% identical. The similarity dropped to approximately 95% in the type 2 strains, which corresponded to the area of the p1 gene (Fig. 1).

Overall sequence identity of the 30 sequenced strains with the reference M129 genome. Solid coloration indicates > 99% identity and transparent grey indicates approximately 95% identity. Location in the reference genome is indicated by numeration on the inside of the ring. GC content in the reference genome is indicated by the black bar graphs between the genomic coordinates and the colored rings (bars pointing toward the outside of the circle indicate high GC content)
Genomic structural comparison
For the detection of large chromosomal rearrangements, deletions, and duplications, MAUVE was applied to the 30 sequenced genomes with 6 reference genomes. All genomes fell into three locally collinear blocks (LCBs), which are conserved segments. The three LCBs were in the same order without any rearrangement. MAUVE detected four subtype-specific insertions (Fig. 2): three type 1-specific insertions (M129 numbering; 169–170 kb, 178–179 kb, and 558–560 kb) and a type 2-specific insertion (M129 numbering; 708 kb). The subtype-specific insertions were manually annotated. Type 1 insertions were all annotated as hypothetical proteins (MPN130, MPN137, MPN138, and MPN457–459) except for the tRNA gene (MPNt26) positioned at 558635 to 558,723 (M129 numbering). The proteins of the type 2 insertion (6 kbp) were annotated as hypothetical proteins without exception (BIX66_03340, 03345, 03350, 03355, and 03360).

Whole genome alignment of the 30 sequenced strains with 6 reference sequences using MAUVE. Regions colored in MAUVE are conserved across all strains. a Two 1 Kbp (approximate) insertions are noticed in the P1 type 1 groups at 169–170 Kb and 178–179 Kb. b A 2 Kbp (approximate) insertion is noticed in the P1 type 1 groups at 558–560 Kb. c A 6 Kbp (approximate) insertion is noticed in the P1 type 2 groups at 708 Kb. All positions are based on M129 reference strain
SNP and indel analysis
SNPs and indels were compared for the identification of sequence level differences against the reference genome. The results are shown in Table 2. As expected, P1 type 1 strains showed fewer variant numbers (140–455) than P1 type 2 strains (1778–1796), showing a clear distinction.
Proteins and functional analysis
The Protein Family Sorter tool at Pathosystems Resource Integration Center (PATRIC) allows selection of a set of genomes of interest and examination of the distribution of protein families across genomes. An interactive heatmap viewer provides a comprehensive view of the distribution of the protein families across multiple genomes, with clustering and anchoring functions to show relative conservation of synteny and to identify lateral transfers. Based on gene annotation from PATRIC, a heatmap of all proteins was produced along with the reference genome M. pneumoniae M129 (Fig. 3). Unsurprisingly, when genomes were classified into P1 types 1 and 2, distinction between the genomes was apparent. Nevertheless, most of the genomes that showed different expressions were hypothetical proteins with uncertain significance.

Heatmap of protein families of 30 sequenced genomes with reference genome M. pneumoniae M129. Cell color represents the number of proteins from a specific genome in a given protein family. Note that P1 types 2 (10–1385, 11–174, 11–949, 11–1384, 14–637 and 16–734) are distinguishable from P1 types 1
Phylogenetic associations
Thirty genomes were aligned with MAFFT, and a phylogenetic tree was generated (Additional file 2). The phylogenetic tree was divided into two clades in accordance with the P1 typing. In general, the STs of the 30 strains were consistent with the phylogenetic relationship.
All 78 strains, including strains from this study and NCBI, were aligned and phylogenetic tree was constructed and visualized (Fig. 4). In general, the strains in this study were scattered throughout the entire phylogenic tree, along with the expansion of certain clades. Trees were divided into two major clades in accordance with the P1 typing. Each P1 type was divided into another two clades. Clade 1 formed the largest clade. It included strains of ST3 from the current study and global collections. Strains with ST20, ST17 and ST19 were included in Clade 1. Clade 2 was consisted of ST1 strains, exclusively. This clade harbored a subclade which consisted of strains from China in year 2015 and 2016. Clade 2 also included the M129 reference strain. Major ST of Clade 3 was ST14 with one each of ST2, ST15 and ST33 strain. Clade 4 showed high proportion of ST2 strains with a subclade which included four ST2 strains from USA and a ST2 strain from Japan. Overall, Clade 1 showed the most heterogenicity in terms of both the origin and the time of strain collected.

Phylogenetic tree based on whole genome alignment of the 30 sequenced strains with 48 M. pneumoniae genomes accessed from NCBI. The tree was built through 500 bootstraps using the maximum composite likelihood approach based on neighbor-joining algorithms. Branch length designates actual distance. Bootstrapping values over 50 are represented on the tree. Blue colored strains are from this study and red colored strains are the 6 references. Strains are grouped into four distinct clades. ST, sequence type
Comparative genomics with global strains-MLST
For the comparative genome analysis of global strains, 48 genomes of M. pneumoniae were accessed from NCBI. Typing of P1 types and MLST types was performed (Additional file 1). An eBURST diagram was constructed based on the 30 strains from this study, 48 global strains from NCBI, and previously reported STs from PubMLST (http://pubmlst.org/mpneumoniae/).
The eBURST diagram showed two clonal complexes with two singletons of ST12 and ST22 (Fig. 5). The founder ST of CC1 was identified as ST3 with no double locus variants (DLVs). The founder ST of CC2 was recognized as ST2 with multiple subgroup founders (ST7, ST14 and ST24), multiple single locus variants (SLVs) and DLVs. In the eBURST diagram of global strains, ST3 and ST1, and ST2 and ST14 were the main STs from CC1 and CC2, respectively. Strains from this study (colored in red) constituted a considerable proportion of ST3 from CC1 and ST14 from CC2. There were several other STs that were previously reported, but not included in the investigation of this study.

Mycoplasma pneumoniae sequence type (ST) relationship by eBURST analysis including 30 strains from this study, 48 strains from NCBI, and previously reported STs from PubMLST (http://pubmlst.org/mpneumoniae/). Two main CCs were defined with two singletons (ST12 and ST22). ST3 and ST2 were the predicted founder of each CC. The size of each circle correlates with the number of isolates of each ST. STs in gray are previously reported, but not included in the investigation of this study. CC, clonal complex
Discussion
M. pneumoniae is known as an organism ‘difficult-to-culture’ [1]. Thus, unlike ordinary bacterial pathogens, the aid of molecular biology in the diagnosis of M. pneumoniae is critical [13]. As the burden of disease caused by this organism is considerable and patients may experience diverse extrapulmonary clinical manifestations, M. pneumoniae has drawn the attention of many researchers. Nevertheless, in addition to the molecular diagnosis of M. pneumoniae by the P1 adhesin, P1 typing has been the sole method for classification for decades [14]. However, because the size of the M. pneumoniae genome is short compared to that of other bacteria and because the P1 adhesin is the only diverse part of the whole genome, researchers continued to focus on the P1 adhesin. Despite these efforts, P1 was not sufficient for the explanation of epidemics or for the explanation of clinical severity [15, 16].
Recent advances in molecular microbiology have widened the scope of the implementation of sophisticated techniques, such as MLVA and MLST [9, 10]. New classifications developed by such new technologies have expanded P1 classification with enhanced distinction. Nevertheless, epidemics still cannot be clearly explained by the new technologies, and there are reports that chest X-rays are the most predictive clue in the course of infection regardless of the molecular genetics [4]. Nevertheless, attempts to utilize molecular biology by using MLVA or MLST have shown useful insights in understanding epidemiology of M. pneumoniae. A recent study from Korea demonstrated high proportion of ST3 in a 16-year period [17]. ST3 was also frequently identified in Japan during the similar period, but ST19 was prevalent among macrolide-resistant strains in Japan, while ST19 has never been identified in Korea [18]. A study from China, which applied MLVA on 835 samples from different regions, has also found regional differences in genotype distribution [19].
Although not extensively applied, high-throughput technologies have been applied to the investigation of M. pneumoniae. A study conducted by Xiao et al. analyzed 15 M. pneumoniae genomes obtained by Illumina sequencing, including 11 clinical isolates and 4 reference strains (20). Although approximately 1500 SNP and indel variants exist between type 1 and type 2 strains, an overall high degree of sequence similarity was found among the strains (> 99% identical to each other). The study concluded that the M. pneumoniae genome is extraordinarily stable over time and geographic distances across the globe, with a striking lack of evidence of horizontal gene transfer.
One of the most recent NGS studies performed by Diaz et al. demonstrated WGS analysis of 107 M. pneumoniae isolates, including 67 newly sequenced isolates, using the Pacific BioSciences RS II and/or Illumina MiSeq sequencing platforms [21]. Population structure analysis done by this study supported the existence of six distinct subgroups. Although this study included the largest collection of M. pneumoniae isolates ever, only a few strains were included from Asian regions where the unique epidemiologic features (for example, high rate of macrolide-resistance among M. pneumoniae) are noticed.
Comparative genome analysis was performed using BRIG, MAUVE, and MAFFT. The genomes were classified mainly by the legendary P1. BRIG clearly distinguished P1 types 1 and 2, but no further information could be found, as separate genes could not be visualized [22]. MAUVE utilizes LCBs, which are conserved segments that appear to be internally free from genome rearrangements [23]. The result from MAUVE showed that large rearrangements (e.g., plasmids, phage or resistance genes) were not observed among M. pneumoniae. Specific insertions were noted in both P1 types. Nevertheless, the translated proteins of the inserted genes were generally hypothetical proteins with the exception of a tRNA. This is consistent with a previous report by Xiao et al., but the two insertions at 169–170 kb and 178–179 kb have not been described previously [20]. The heatmap generated by PATRIC confirmed the P1 classification by differences in protein production. This is consistent with additional studies that applied NGS technology [24, 25].
The SNP approach is widely used in the study of antimicrobial resistance and genetic diversity and is not limited to M. pneumoniae [26,27,28]. This study is consistent with previous studies investigating SNPs within M. pneumoniae [20, 21]. Variant calling against M129 of P1 subtypes showed substantially fewer variants compared to P1 type 2 in both nonsynonymous SNPs and total variants.
The two phylogenetic trees constructed and visualized in this study revealed notable findings. First, the phylogenetic relatedness of the 30 strains demonstrated strong correlation according to the P1 type. Each ST type was generally grouped by the same branch. Nevertheless, when global strains were considered together, there were a few exceptions which suggests the associations demonstrated by the phylogenetic tree do not fully correlate with the ST type. Examples include ‘Kor/2011/11–1384/P1_2a/ST33’ strain which is placed along with ST14 strains or ‘Kenya/2010/K27/P1_2/ST16’ strain which is placed along with ST2 strains. Second, when eBURST analysis and the phylogenetic associations with global strains are considered together, the correlation of two methods for comparative genomics were apparent. Clade 2 from the phylogenetic tree stands for ST1 strains of CC1 in the MLST analysis. It is highly probable that Clade 1 takes the rest of the strains in CC1. Contiguous strains of the CC2 which includes ST2, ST15, ST14 and ST33 are consistent with strains from Clade 3. In another direction, contiguous strains of the CC2 which includes ST2, ST4, ST7 and ST16 is consistent with strains from Clade 4. Even though not apparent in the phylogenetic analysis probably due to the genetical proximity of the strains, eBURST analysis shows ST3 as the founder strain of the CC1. We assume that despite the fact that M129, one of the ST1 strains, is used as reference strain, it is more convincing that ST1 strains may have evolved from the ST3 strains. The strain ‘USA/1960/P1_1428/P1_1/ST3’ which is the earliest known strain of the P1 type 1 strains also supports this idea.
In general, the result of the current study is consistent with that of the previous studies [20, 21]. High stability was observed by the small number of SNPs across the genome and lack of rearrangements. The fact that P1 types shown as a major factor for the genetic classification is also consistent with the findings of the current study. Diaz et al. grouped 107 strains from four other studies and their study into three P1 type 1 and two P1 type 2 subgroups based on core protein sequences [21]. Even though there are differences in the methods of tree alignment, construction, and visualization, the subgroups are consistent with the current study, in general. A distinct subgroup designated as 1 N (New) which included four isolates from their study was the only subgroup which did not exist on the current study. When comparisons are made between the different phylogenetic trees, we find that the abundance and heterogenicity of the Clade 1 in the current study and the group 1 U (Ubiquitous) in the study of Diaz et al. as the common finding. We assume that this certain subgroup harbors the most actively evolving strains in global and demands attention in terms of pathogenicity or in accordance with macrolide resistance.
This study has some limitations. First, the number of strains included in the study was small, thus we were not able to interpret the clinical significance of the findings. Second, isolates were chosen from two consecutive outbreaks. Further analysis from sporadic cases and new outbreaks is needed. Nevertheless, this study expanded our understanding of the genome structure of M. pneumoniae through whole genome analysis. Whole genome approach provided more detailed information than traditional molecular typing methods for exploring genomic diversity among M. pneumoniae strains.
Conclusions
The comparative whole genome approach was able to define high genetic identity, unique structural diversity, and phylogenetic associations among the 78 M. pneumoniae strains isolated worldwide.
Methods
M. pneumoniae strains
This study comprised M. pneumoniae strains detected from children with pneumonia at two hospitals during two consecutive outbreaks of M. pneumoniae pneumonia in South Korea in 2010–2012 and 2014–2016. Specimens were obtained from Seoul National University Children’s Hospital (Seoul) and Seoul National University Bundang Hospital (Seongnam). Epidemic periods and the diagnosis of M. pneumoniae pneumonia were defined as previously described [17].
Cultivation
Culture of M. pneumoniae was performed using pleuropneumonia-like organism broth as previously described [4]. Reference strain M129 (ATCC 29342) was used as a positive control for culture.
DNA preparation
DNA was extracted directly from cultivated M. pneumoniae using an extraction kit (DNeasy Kit; QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The p1 gene was amplified by PCR for the confirmation of M. pneumoniae.
MLST analysis and P1 typing
MLST was performed on the M. pneumoniae DNA samples as previously described [10]. P1 subtypes and each subtype variants were determined by sequencing the RepMP2/3 and RepMP4 genes and in comparison with previously published data [29, 30].
Selection of strains for whole-genome analysis
A total of 30 strains were selected for the whole-genome sequencing (WGS) investigation. Thirty-seven M. pneumoniae strains were isolated during the 2010–2012 epidemic. P1 subtype 1 accounted for 71.9% and ST3 was responsible for 62.2%. The remaining 37.8% consisted of ST1, ST14, ST17, and ST33. In contrast, among the 45 isolates detected during the 2014–2016 epidemic, P1 subtype 1 accounted for 50.0% and the ST distribution was 88.9% for ST3 and 11.1% for ST14. In order to include as many different STs as possible, all strains that showed STs other than ST3 (ST1, ST14, ST17, and ST33) were included for WGS analysis. We have randomly selected 20 ST3 strains from each epidemic.
Next-generation sequencing (NGS)
The library for whole genome sequencing was prepared using Truseq Nano DNA Lib Prep Kit (Illumina, San Diego, CA, USA) and sequenced using MiSeq Reagent Kit V2 (Illumina, San Diego, CA, USA) on the Illumina MiSeq desktop sequencer (Illumina, San Diego, CA, USA). Illumina NGS workflows include four basic steps: library preparation, cluster amplification, sequencing and alignment. The NGS library is prepared by fragmenting a genomic DNA sample and ligating specialized adapters to both fragment ends. The library is loaded into a flow cell, and the fragments are hybridized to the flow cell surface. Each bound fragment is clonally amplified through bridge amplification. Sequencing repeats, including fluorescently labeled nucleotides, are added, and the first base is incorporated. The flow cell is imaged, and the emission from each cluster is recorded. The emission wavelength and intensity are used to identify the base. This cycle is repeated ‘n’ times to create a read length of ‘n’ bases. In this study, paired-end 250-bp reads were used with an average depth (coverage) of 442.93 (ranging from 172.95 to 795.39). The average number of reads during the sequencing was 1,445,719 (ranging from 564,516 to 2,596,168). Instead of directly aligning the reads to a reference sequence, de novo assembly was performed.
Genome assembly and annotation
NGS reads were assembled de novo using SPAdes [31]. The number of contigs generated ranged from 3 to 8 per strain. These contigs were mapped to the M129 reference genome using the BLAST-like alignment tool (BLAT) and visualized using Integrative Genomics Viewer (IGV) [32,33,34]. This mapping was used to develop PCR primers to join the contigs. High fidelity PCRs and Sanger sequencing were performed using standard methods. Overlapping and joining of the contigs were performed manually with Sequencher version 5.4.6 (Gene Codes Corporation, Ann Arbor, MI, USA). The initial NGS reads were aligned to the de novo assembled genome for the correction of errors. The corrected and completed circular genomes were annotated using Rapid Annotation using Subsystem Technology (RAST) [35].
Comparative genomics
Completed genomes were aligned using BRIG for the overall sequence similarity between the strains [22]. MAUVE was used to detect large chromosomal rearrangements, deletions, and duplications [23]. In the phylogenetic analysis with the 48 global strains downloaded from the National Center for Biotechnology Information (NCBI) were included. MAFFT was applied using the ‘FFT-NS-2’ method for multiple sequence alignment of the strains from the current study and with the global strains. Phylogenetic tree was constructed using the maximum composite likelihood approach based on neighbor-joining algorithms and visualized using Phylo.io (strains from the current study) and MEGA X (with the global strains) [36, 37]. For the phylogenetic tree with the global strains, 500 iterations of bootstrapping analysis were used to generate confidence values. eBURST version 3 software (http://eburst.mlst.net/) was used to estimate the relationships among the strains and to assign strains to a clonal complex (CC) [38].
Single nucleotide polymorphism (SNP) and insertion/deletion (indel) analysis
To call SNPs and indels, completed genomes were first broken into 10-kb “reads” at 1-kb intervals and then aligned to the M129 reference strain (NCBI Accession Number NC_000912) using BWA v0.7.7 [39]. Variant calling was performed using Samtools [40]. The effects of the SNPs and indels in the resulting VCF files were evaluated and annotated using SnpEff v3.3 [41].
Proteins and functional analysis
For the analysis of proteins and functional annotation, PATRIC was used, and a heatmap was generated based on annotations [42]. Gene translation, multiple sequence alignment and visualization of proteins were performed using Clustal Omega [43]. Annotation of any hypothetical genes was performed using a BLAST search against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [44, 45].
References genomes
Six reference genomes were included in each analysis as appropriate (Table 3). M. pneumoniae M129, FH, 309, KCH-402 and K405 are representatives of each P1 type and subtype. M. pneumoniae S355 is included, as this strain is one of the earliest strains that was fully sequenced and expressed macrolide resistance. Two FH strains were downloaded from NCBI, and the genome sequenced with Illumina was used as the reference genome.
Availability of data and materials
All data generated or analyzed during this study are included in this published article. The gene sequences are deposited in NCBI database under the accession numbers CP039761-CP039790.
Abbreviations
- BRIG:
-
BLAST Ring Image Generator
- CC:
-
clonal complex
- DLV:
-
double locus variant
- MLST:
-
multilocus sequence typing
- MLVA:
-
multilocus variable-number tandem-repeat analysis
- PATRIC:
-
Pathosystems Resource Integration Center
- SLV:
-
single locus variant
- ST:
-
sequence type
References
Waites KB, Xiao L, Liu Y, Balish MF, Atkinson TP. Mycoplasma pneumoniae from the respiratory tract and beyond. Clin Microbiol Rev. 2017;30(3):747–809.
Jain S, Williams DJ, Arnold SR, Ampofo K, Bramley AM, Reed C, et al. Community-acquired pneumonia requiring hospitalization among U.S. children. N Engl J Med. 2015;372(9):835–45.
Mansel JK, Rosenow EC 3rd, Smith TF, Martin JW Jr. Mycoplasma pneumoniae pneumonia. Chest. 1989;95(3):639–46.
Yoon IA, Hong KB, Lee HJ, Yun KW, Park JY, Choi YH, et al. Radiologic findings as a determinant and no effect of macrolide resistance on clinical course of Mycoplasma pneumoniae pneumonia. BMC Infect Dis. 2017;17(1):402.
Spuesens EB, Fraaij PL, Visser EG, Hoogenboezem T, Hop WC, van Adrichem LN, et al. Carriage of Mycoplasma pneumoniae in the upper respiratory tract of symptomatic and asymptomatic children: an observational study. PLoS Med. 2013;10(5):e1001444.
Su CJ, Chavoya A, Dallo SF, Baseman JB. Sequence divergency of the cytadhesin gene of Mycoplasma pneumoniae. Infect Immun. 1990;58(8):2669–74.
Su CJ, Chavoya A, Baseman JB. Regions of Mycoplasma pneumoniae cytadhesin P1 structural gene exist as multiple copies. Infect Immun. 1988;56(12):3157–61.
Kenri T, Okazaki N, Yamazaki T, Narita M, Izumikawa K, Matsuoka M, et al. Genotyping analysis of Mycoplasma pneumoniae clinical strains in Japan between 1995 and 2005: type shift phenomenon of M. pneumoniae clinical strains. J Med Microbiol. 2008;57(Pt 4):469–75.
Degrange S, Cazanave C, Charron A, Renaudin H, Bebear C, Bebear CM. Development of multiple-locus variable-number tandem-repeat analysis for molecular typing of Mycoplasma pneumoniae. J Clin Microbiol. 2009;47(4):914–23.
Brown RJ, Holden MT, Spiller OB, Chalker VJ. Development of a multilocus sequence typing scheme for molecular typing of Mycoplasma pneumoniae. J Clin Microbiol. 2015;53(10):3195–203.
Mukhopadhyay R. DNA sequencers: the next generation. Anal Chem. 2009;81(5):1736–40.
Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24(22):4420–49.
Loens K, Ursi D, Goossens H, Ieven M. Molecular diagnosis of Mycoplasma pneumoniae respiratory tract infections. J Clin Microbiol. 2003;41(11):4915–23.
Diaz MH, Winchell JM. The evolution of advanced molecular diagnostics for the detection and characterization of Mycoplasma pneumoniae. Front Microbiol. 2016;7:232.
Jacobs E, Ehrhardt I, Dumke R. New insights in the outbreak pattern of Mycoplasma pneumoniae. Int J Med Microbiol IJMM. 2015;305(7):705–8.
Waller JL, Diaz MH, Petrone BL, Benitez AJ, Wolff BJ, Edison L, et al. Detection and characterization of Mycoplasma pneumoniae during an outbreak of respiratory illness at a university. J Clin Microbiol. 2014;52(3):849–53.
Lee JK, Lee JH, Lee H, Ahn YM, Eun BW, Cho EY, et al. Clonal expansion of macrolide-resistant sequence type 3 Mycoplasma pneumoniae, South Korea. Emerg Infect Dis. 2018;24(8):1465–71.
Ando M, Morozumi M, Adachi Y, Ubukata K, Iwata S. Multilocus sequence typing of Mycoplasma pneumoniae, Japan, 2002-2016. Emerg Infect Dis. 2018;24(10):1895–901.
Xue G, Li M, Wang N, Zhao J, Wang B, Ren Z, et al. Comparison of the molecular characteristics of Mycoplasma pneumoniae from children across different regions of China. PLoS One. 2018;13(8):e0198557.
Xiao L, Ptacek T, Osborne JD, Crabb DM, Simmons WL, Lefkowitz EJ, et al. Comparative genome analysis of Mycoplasma pneumoniae. BMC Genomics. 2015;16:610.
Diaz MH, Desai HP, Morrison SS, Benitez AJ, Wolff BJ, Caravas J, et al. Comprehensive bioinformatics analysis of Mycoplasma pneumoniae genomes to investigate underlying population structure and type-specific determinants. PLoS One. 2017;12(4):e0174701.
Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402.
Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–403.
Zhang J, Song X, Ma MJ, Xiao L, Kenri T, Sun H, et al. Inter- and intra-strain variability of tandem repeats in Mycoplasma pneumoniae based on next-generation sequencing data. Future Microbiol. 2017;12:119–29.
Lluch-Senar M, Cozzuto L, Cano J, Delgado J, Llorens-Rico V, Pereyre S, et al. Comparative “-omics” in Mycoplasma pneumoniae clinical isolates reveals key virulence factors. PLoS One. 2015;10(9):e0137354.
Ramanathan B, Jindal HM, Le CF, Gudimella R, Anwar A, Razali R, et al. Next generation sequencing reveals the antibiotic resistant variants in the genome of Pseudomonas aeruginosa. PLoS One. 2017;12(8):e0182524.
Lee JY, Na IY, Park YK, Ko KS. Genomic variations between colistin-susceptible and -resistant Pseudomonas aeruginosa clinical isolates and their effects on colistin resistance. J Antimicrob Chemother. 2014;69(5):1248–56.
Li SL, Sun HM, Zhu BL, Liu F, Zhao HQ. Whole genome analysis reveals new insights into macrolide resistance in Mycoplasma pneumoniae. Biomed Environ Sci BES. 2017;30(5):343–50.
Spuesens EB, Oduber M, Hoogenboezem T, Sluijter M, Hartwig NG, van Rossum AM, et al. Sequence variations in RepMP2/3 and RepMP4 elements reveal intragenomic homologous DNA recombination events in Mycoplasma pneumoniae. Microbiology. 2009;155(Pt 7):2182–96.
Zhao F, Cao B, Li J, Song S, Tao X, Yin Y, et al. Sequence analysis of the p1 adhesin gene of Mycoplasma pneumoniae in clinical isolates collected in Beijing in 2008 to 2009. J Clin Microbiol. 2011;49(8):3000–3.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64.
Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–92.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6.
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 2014;42(Database issue):D206–14.
Robinson O, Dylus D, Dessimoz C. Phylo.Io: interactive viewing and comparison of large phylogenetic trees on the web. Mol Biol Evol. 2016;33(8):2163–6.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K, Mega X. Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–9.
Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186(5):1518–30.
Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26(5):589–95.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Cingolani P, Platts A, Wang Le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80–92.
Wattam AR, Brettin T, Davis JJ, Gerdes S, Kenyon R, Machi D, et al. Assembly, annotation, and comparative genomics in PATRIC, the all bacterial bioinformatics resource center. Methods Mol Biol. 2018;1704:79–101.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D61.
Acknowledgements
Preliminary results from this study were presented at the IDWeek conference; Washington, DC; October 2–6, 2019.
Funding
This research was supported by the 2017 Seoul National University Hospital Research Fund (0320170230) and the Basic Science Research Program through the National Research Foundation of Korea, which is funded by the Ministry of Education, Science and Technology (NRF-2018R1D1A1A09082098). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. The study sponsors provided research grants to perform the study which was written by Dr. EH Choi.
Author information
Authors and Affiliations
Contributions
JKL, MWS, and EHC conceived the original research idea. JKL and MSH collected and prepared study samples. JKL, MWS, MSH, YY, and SIC conducted the experiments and contributed for raw data analysis. JKL, MWS, DS, JIK, and SSP contributed in the bioinformatics analysis. JKL wrote the first draft of the manuscript and EHC critically revised the manuscript. JKL and EHC completed the manuscript. JKL and MWS contributed equally to this article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The institutional review board of Seoul National University Hospital approved the study protocol (IRB no. H-1012–007–341). Informed consent was exempted because nasopharyngeal aspirates were obtained as a standard of patient care to identify the etiologic agents of acute pneumonia.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
P1 type and MLST type of the 30 strains from this study and 48 strains from NCBI.
Additional file 2:
Phylogenetic tree based on whole genome alignment of the 30 sequenced strains.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lee, J.K., Seong, MW., Shin, D. et al. Comparative genomics of Mycoplasma pneumoniae isolated from children with pneumonia: South Korea, 2010–2016. BMC Genomics 20, 910 (2019). https://doi.org/10.1186/s12864-019-6306-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-6306-9