
Abstract
Background
The Djallonke sheep is well adapted to harsh environmental conditions, and is relatively resistant to Haemonchosis and resilient to animal trypanosomiasis. The larger Sahelian sheep, which cohabit the same region, is less well adapted to these disease challenges. Haemonchosis and Trypanosomiasis collectively cost the worldwide animal industry billions of dollars in production losses annually.
Results
Here, we separately sequenced and then pooled according to breed the genomes from five unrelated individuals from each of the Djallonke and Sahelian sheep breeds (sourced from Ghana), at greater than 22-fold combined coverage for each breed. A total of approximately 404 million (97%) and 343 million (97%) sequence reads from the Djallonke and Sahelian breeds respectively, were successfully mapped to the sheep reference genome Oar v3.1. We identified approximately 11.1 million and 10.9 million single nucleotide polymorphisms (SNPs) in the Djallonke and Sahelian breeds, with approximately 15 and 16% respectively of these not previously reported in sheep. Multiple regions of reduced heterozygosity were also found; 70 co-localised within genomic regions harbouring genes that mediate disease resistance, immune response and adaptation in sheep or cattle. Thirty- three of the regions of reduced heterozygosity co-localised with previously reported genes for resistance to haemonchosis and trypanosomiasis.
Conclusions
Our analyses suggest that these regions of reduced heterozygosity may be signatures of selection for these economically important diseases.
Similar content being viewed by others


Background
The Djallonke sheep is recognised for its natural ability to withstand a harsh, hot and humid tropical climate, where it is faced with the challenges of persistent drought, diseases and feed scarcity [1, 2]. Adaptation is probably a consequence of natural selection over several millennia [3,4,5]. Genomic regions adjacent to loci under adaptive selection over time are usually characterised by low heterozygosity [6]. The most important livestock diseases are trypanosomiasis and haemonchosis [7,8,9,10]. The natural ability of the Djallonke to survive and remain productive under trypanosome challenge with very low mortality and without the aid of trypanocidal drugs is referred to as trypanotolerance [11]. Trypanosomiasis in sub Saharan Africa is estimated to cause annual losses of more than 4.5 billion dollars (US$) through direct and indirect production costs [12, 13]. Development of trypanotolerance is considered to be the most economical and sustainable option for combating African trypanosomiasis [9, 14, 15]. The potential of this trypanotolerant trait in mitigating the disease in Africa has recently been reviewed [16]. However, because Djallonke sheep have a relatively small mature body weight (between 20 kg to 30 kg [17];) farmers often cross-breed them with the larger, but more disease susceptible, Sahelian breed.
In spite of the importance of the Djallonke and Sahelian sheep to the region, genetic studies are scarce. There are no records of whole genome variant characterisation in either the Djallonke or Sahelian breeds, besides our preliminary report at the International society of animal genetics conference [18]. The objectives of this study are: i) to identify and document variants in each breed and ii) to use these to investigate putative candidate genetic regions in both breeds.
Methods
Animals
Four ewes and one ram of the Djallonke breed (DJ) were selected from the National Open Nucleus Breeding Station (ONBS) dedicated for Djallonke Sheep in Ejura in the Ashanti region (longitude 01o 28′W and latitude 06o 41′ N). Five Sahelian (SA) ewes were selected from the National Sheep Breeding Station in Pong Tamale (longitude 00o 54′W and latitude 09o 38′ N). All sheep were reproductively mature (13–24 months old) and were chosen in consultation with the management of the breeding stations to represent unrelated animals that were true to breed type (phenotypically similar to the breed ideal). The management relied on stock records for determination of relatedness among sheep on two breeding stations. Approximately 9 ml of blood was collected via the jugular vein into disodium EDTA vacutainers. All sampled sheep were monitored on farm for at least 24 h post sampling and no adverse effect was recorded. No sheep were sacrificed during this study. The samples were transported at 0o – 4o C to the laboratory, centrifuged at 800 x g for 3 min at room temperature (15-25 °C) with the rotor bucket brake off. The buffy coat was used immediately for genomic DNA extraction or was stored at -20 °C. Genomic DNA was extracted from each of the buffy coat samples using the Zymo Quick-gDNA™ MiniPrep DNA purification Kit (according to the manufacturer’s protocol). DNA quality and concentration were assessed using agarose gel electrophoresis (1% in 1xTAE) and by Nanodrop spectrophotometry.
Library construction and sequencing
For each individual, 100 ng of DNA was sheared using the Covaris S2 System, to generate a broad range of DNA fragments with sizes from 100 to 1000 bp. The DNA fragments were ligated to T-overhang adaptors with the NEB Next Ultra kit (New England Biosciences). Each animal had a unique barcode (Ion Xpress Barcodes, Life Technologies). Fragments of approximately 300-330 bp were size-selected using the E-gel system (Invitrogen), and recovered fragments were further purified using AMPure XP SPRI beads (Beckman). Equimolar amounts of each library were combined and amplified using an Ion Chef system (ThermoFisher Scientific) via emulsion PCR, then sequenced on an Ion Proton™ system (ThermoFisher Scientific) using a PI chip. Genomic DNA from the ten individuals was separately sequenced, and the sequencing reads were then pooled by breed. After filtering and trimming, an average of 10 and 13% of the reads were excluded due to low quality, and 26 and 28% excluded due to polyclonality for the Djallonke and Sahelian samples, respectively. Coverage analysis was performed on a total (post QC) of 73 Gbp of sequenced data, comprising 404,755,012 pooled reads (average read length 185.4 nucleotides) and 57.6 Gbp comprising 303,136,043 pooled reads (average read length 176.6 nucleotides) for Djallonke and Sahelian sheep, respectively. The genome coverage depth obtained was calculated as being 27.90x and 22.01x for the Djallonke and the Sahelian respectively, and covered 97% of sheep reference assembly v3.1. All the variants were submitted to the European variant archive of the European Bioinformatics Institute with the accession number PRJEB15642.
Mapping and pre-processing of reads
Base calling, de-multiplexing, quality control (QC) and alignment pre-processing [19] was completed using Torrent Suite 4.6 on a Torrent Server (ThermoFisher Scientific). Briefly, polyclonal and uniformly low-quality reads were removed, and the remaining reads were trimmed from the 3′ end only. Mapping was also performed within Torrent Suite 4.6, using the Torrent mapping alignment program (TMAP). Individual libraries were mapped to the sheep reference genome Oar v3.1 (University of California, Santa Cruz (UCSC)). For each of the sheep breeds, all individual BAM files were merged and sorted using SAMtools v0.1.19-44,428 cd [20], and coverage analysis was performed for both the individual and combined datasets through automated plugins in TorrentSuite 4.6. Duplicate reads were removed using Picard Tools v1.122.
Variant calling pipeline
Genome Analysis Tool Kit version 3.2.2 (GATK) RealignerTargetCreator and IndelRealigner were used to produce realignments of the pooled BAM files for each breed. GATK HaplotypeCaller was used in GVCF mode to call intermediate genome-wide variants separately for the pooled DJ genomes and pooled SA genomes, producing two pooled genomic variant call format (gvcf) files. GATK GenotypeGVCFs was then used to perform a joint genotyping of the two pooled gvcf files with minimum standard confidence thresholds for both calling and emitting variants set at 30 to produce a composite pooled variant call format (vcf) file (Pooled-Sheep VCF). This analysis was selected to ensure good quality variant calling and reduce false discovery rates. Finally, VCFtools (v0.1.15) [21] was used to extract individual Djallonke and Sahelian samples from the composite joint genotyped vcf into separate vcf files, which were used for downstream analyses.
Genetic relationship matrix
To determine genetic relatedness, a genomic relationship matrix (GRM) was computed on a composite vcf file that contained all the 10 vcf files generated from both the Djallonke and the Sahelian samples, using the Genome-wide Complex Trait Analysis (GCTA) software [22, 23]. Furthermore, Principal Components analysis (PCA) was used to compare the autosomal genomes of all individual samples to determine population substructure for each breed [24], and assess the genetic relatedness using the SNPRelate implemented in R CRAN (http://cran.r-project.org) [25].
Detection of regions of reduced heterozygosity
HomSI (Homozygosity Stretch Identifier) was used to identify regions of reduced heterozygosity in both genomes [26]. The Djallonke genome was designated as the index case and compared against the Sahelian as the unaffected case for input settings for the HomSI analysis. Analysis of runs of homozygosity in Djallonke and Sahelian using HomSI, with the stringent settings of 5 Mb window size and 10 kb sliding size, allowed the capturing of a wide spectrum of different lengths of homozygosity throughout the genome [26,27,28,29]. Integrative Genomics Viewer (IGV 2.3.46, www.BroadInstitute.org) was used to view vcf and BAM file tracks aligned to the sheep reference genome Oar v3.1, selecting regions based on genomic coordinates of regions of contrasting reduced heterozygosity identified by HomSI in order to identify candidate genes. Prominent regions were investigated using IGV for the specific genes of interest [30, 31] and were used to identify candidate genes within the region. For every prominent region of low heterozygosity in the HomSI output, the co-localised candidate gene or genes were inferred from the available Ensembl (Release 85 and 86) annotated sheep reference assembly (version 3.1) or from the conserved synteny for other mammalian genomes from the Ensembl genome database [32,33,34]. The approach presented here for investigating genetic evidence of trypanotolerance and resistance to haemonchosis was by directly linking regions with contrasting heterozygosity in this dataset to the reported candidate genes in the database for Animal Quantitative Trait Loci (Animal QTLdb) [35] and other previously published genetic association studies for the two traits. There have been several previous genomic investigations of resistance to nematode infection including H. contortus in multiple sheep breeds and these results were compared with our Djallonke and Sahelian sheep results [36,37,38,39,40]. In contrast, there has been no previous genomic investigation of trypanosomiasis in any sheep breed; therefore, comparison was made with the reported trypanotolerance associated candidate genes in Ndama cattle [41,42,43,44,45].
Annotation and functional analysis of genomic variants
As there were unequal numbers of males and females used between the two breeds, for the purpose of a balanced comparison, autosomal chromosomes were extracted from each of the pooled vcf files using VCF tools v0.1.15. Known SNPS were annotated in the vcf files with SnpSift v4.2 (annotate command) using the Ensembl Release-85 Variation reference vcf for Ovis aries as the database. (ftp.ensembl.org/pub/release-85/variation/vcf/ovis_aries/) [32]. SnpEff v4.2 (Cingolani et al., 2012) was used for functional annotation of identified autosomal SNPs in both the Djallonke and Sahelian genomes based on the Ensembl sheep genome assembly Oar_v3.1.82. Pairwise comparison of Genomic SNPs and INDELS for Sahelian and Djallonke sheep was computed using the BEDTools suite v. 2.26.0 [46].
Results
Genetic relationship matrix and principal component analysis
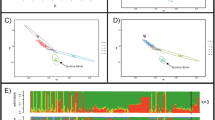
The GRM computed for these datasets supports the assumption that the ten individual animals sampled were unrelated. Additional file 1shows the GRM output for this analysis. The PCA computed for the 10 individual datasets from the two sheep breeds showed distinct clustering for the two breeds (Fig. 1).

PCA of Autosomal Genomes for 5 Djallonke (DJ) and 5 Sahelian (SA) sheep showing distinct clustering of the two breeds
Comparison of genomes
On average, there was one variant every 191 base pairs (bp) in the Djallonke sheep and 1 variant every 193 bp in the Sahelian sheep (Table 2). Transition to transversion ratios were similar in the Djallonke (2.47) and Sahelian (2.48) sheep (Table 1). The estimated missense to silent SNP ratios were also similar between the Djallonke and Sahelian sheep (0.69 and 0.68 respectively). Similarly, the two datasets had equal insertion to deletion ratios (0.38). Similar proportions of SNPs, insertions and deletions were observed for Djallonke (86%: 4%: 10%) and Sahelian (87%: 4%: 9%) sheep. Approximately 84% of variants in both Djallonke and Sahelian sheep were present in the Ensembl Variation database (release 85) hence approximately 16% are unidentified variants (Table 1). Analysis of only SNPs, however, revealed that approximately 94% were present in the database (Table 1), indicating that approximately 6% of SNPs were previously unreported in sheep.
The distribution of variants by chromosome
The distribution of the variants (i.e. the sum of SNPs, insertions and deletions) was similar across all the autosomes, with chromosomes 11 and 26 having the lowest and highest frequencies for variants in both breed genomes (Table 2), respectively. A total of 12,821,836 and 12,654,761 variants were identified in Djallonke and Sahelian sheep, respectively, with 12,556,638 (96.30%) shared between the two genomes. In total, 324,760 (2.49%) variants were specific to the Djallonke breed, whereas 158,085 (1.21%) variants were specific to the Sahelian breed. Therefore, the total number of variants identified for the two breeds is 13,039,483. Analysis with BEDTools intersect indicated that 242,572 SNPs and 82,609 indels were specific to the Djallonke, whereas 120,652 SNPs and 37,762 indels were unique to the Sahelian breed.
Distribution of autosomal SNPs by genomic region
Most SNPs in both sheep breeds were intergenic or intronic (Table 3), with approximately 1% located in the remaining genic regions (i.e. untranslated regions (UTR), exons, and splice sites). Although the Djallonke sheep had a higher number of SNPs than the Sahelian sheep, the ratios of the SNPs in these three regions (intergenic, intronic and “other” genic regions including exons) were similar for Djallonke (68.78%: 30.04%: 1.18%) and Sahelian sheep (68.81%: 30.02%: 1.17%). A comparison of the “other” genic category revealed similar proportions of synonymous, non-synonymous, splice site, UTR and miscellaneous variants for each breed (Table 3).
Regions of homozygosity
Approximately 2.5 Gbp of autosomal chromosomal DNA was resolved into about 50,000 detection windows for each breed. HomSI analysis, identified regions having reduced heterozygosity (Fig. 2; blue) of various sizes (1 to > 100 kb) across genic and intergenic regions within both breeds. Seventy of these reduced heterozygosity regions co-localised with known candidate genes. There were also several genic and intergenic regions that showed reduced heterozygosity but did not contain any known putative candidate gene. In addition, there were regions for which the Djallonke show complete fixation for one allele (blue) and the Sahelian showing complete fixation for the alternative allele (white) e.g. TRHDE (Fig. 2).

Comparison of the HomSI analysis of three genes associated with adaptive traits for Djallonke and Sahelian sheep genomes
Putative regions for tolerance to trypanosomiasis
Eight regions of reduced heterozygosity were observed to be co-localised with previously reported trypanotolerance associated genes (Table 4) [42, 47, 48]. Six of the eight reported genes (CTSS, ARHGAP15, INHBA, STX7, RAB35, CD19) were co-localised with regions of reduced heterozygosity in the Djallonke genome only, and the other two (SCAMP1, TICAM1) were co-localised with regions of reduced heterozygosity in both Djallonke and Sahelian genomes (Fig. 3). The Djallonke sheep show longer runs of reduced heterozygosity (blue) than the Sahelian sheep at the INHBA and RAB35 gene regions, but both breeds show reduced heterozygosity across approximately 2-kb (16,922 kb–16,924 kb) of the TICAM1 gene (Fig. 3). The Sahelian breed shows increased heterozygosity (orange) between 9279 kb to 9286 kb in the SCAMP1 region. In contrast, the Djallonke shows reduced heterozygosity within the same region (blue).

Comparison of the HomSI analysis of four Trypanotolerance associated genes for Djallonke and Sahelian sheep genomes
Putative regions for resistance to Haemonchosis
There were 25 regions of reduced heterozygosity that co-localised with previously reported candidate gene for Haemonchosis resistance (Table 5). Twenty-one of the regions had reduced heterozygosity in the Djallonke sheep. The remaining four regions (MHCII-DRB1, PIK3CD, MUC15, IL17RB) had reduced heterozygosity in both Djallonke and Sahelian sheep. Figure 4 shows three regions associated with resistance to Haemonchus contortus infection: the IFNG gene, the CHIA gene, and the SUGT1 gene. In each case, only the Djallonke sheep displayed reduced heterozygosity.

Comparison of the HomSI analysis of three Haemonchotolerance associated genes for Djallonke and Sahelian sheep genomes
Putative regions for adaptation to tropical conditions
There were also genomic regions of reduced heterozygosity that were co-localised with genes known to be associated with immune responses and natural adaptation (Fig. 2). A total of 37 candidate genes fell within these reduced heterozygosity genomic regions in the Djallonke sheep, including 14 that were shared with the Sahelian sheep (Table 6). Three of the gene regions associated with adaptive selection (MSRB3 gene, APC2 gene, and TRHDE gene) are shown in Fig. 2. Differences between these genes were observed with respect to the polymorphism patterns. The Djallonke sheep have reduced heterozygosity (blue) at the region of the MSRB3 gene whilst the Sahelian was more polymorphic (Fig. 2). Over much of the TRDHE gene region, the two sheep breeds were fixed for alternative alleles. Both sheep breeds, however, showed reduced heterozygosity for the same allele between coordinates 41,267,000 and 41,272,000, encompassing 7 exons (exons 16 to 22) of the APC2 gene.
Discussion
A comparison of the genomes of Djallonke and Sahelian sheep in this study has shown that Djallonke sheep have a genetic variant every 191 base pairs while the Sahelian sheep have a genetic variant every 193 base pairs. Approximately 16% of the variants had not been previously reported in sheep. The two breeds also had similar ratios of transitions to transversions (2.5), missense to silent mutations 90.7) and insertions to deletions (0.4). These breeds also had similar proportions of SNP to indels. The distribution of variants across the autosomal chromosomes was also similar; in both breeds chromosome 11 had the lowest frequency of variants while chromosome 26 had the highest frequency.
The transition to transversion ratio obtained for Djallonke (2.47) and Sahelian (2.48) are similar to the expected values observed for other mammalian genomes: 2.26 for cattle whole genome [49], 2.13 for human intergenic SNPs [50], and 2.81 for human exonic SNPs [51]. These comparable ratios support the reliability of the sequenced datasets in this study, and they are therefore expected to contain low numbers of false positives (Type 1 errors) caused by random sequencing errors. This is further underscored by the high sequencing coverage statistics obtained for both genomes (i.e. > 97% and > 20x), which is suitable for “high-confidence” variant calling [52]. The advantage that sequencing has over medium or high-density SNP genotype datasets, is that it provides higher resolution and power for the detection of selection signatures over relatively short distances [53, 54]. For instance, the Illumina Ovine 50KSNP BeadChip and Illumina Bovine HD 800KSNP BeadChip only provide a SNP density of approximately 1 SNP for every 5 million and 3 million bases, respectively. Furthermore, the use of markers on breeds that were not included in the training set for the marker development introduces further possible ascertainment bias into the analysis.
The proportion of SNPs in the intergenic, intronic and the remaining genic regions including exons for the two genomes are similar to the proportions recorded in Korean cattle breeds [49]. The exonic regions, although containing the least number of SNPs, represent the most important subset of SNP, because they are more likely to be associated with changes in protein sequence, structure and function than intronic and intergenic SNPs [55]. In particular, population-specific, rare exonic SNPs have been shown to be the most consequential determinants of fitness traits in humans [56]. Fixed non-synonymous SNPs, which are described as SNPs for which only one allele (of a given locus) is present in a population, are of major interest in identifying breed or population specific traits [49].
The high number of novel variants identified: 2,057,096 (16.03%) in the Djallonke breed and 1,983,296 (15.67%) in the Sahelian sheep confirms that these breeds are an important genetic resource for world sheep diversity. More than 0.5 million SNPs in each of the two sheep breeds are probably novel. There were also high numbers of breed specific variants; 242,572 SNP and 82,609 indels in the Djallonke and 120,652 SNP and 37,762 indels in the Sahelian breed. These breed specific variants could facilitate the sustainable management of these breeds and aid in confronting future emerging livestock diseases as well other global challenges, such as the uncertain consequences of climate change [57]. Recent reports indicate that most of the indigenous African livestock breeds are endangered [58] and might become extinct.
The HomSI scan permitted the identification of regions of reduced heterozygosity in greater detail than other sliding window algorithms such as the “Integrated haplotype homozygosity score (iHS)” [59, 60] and “the composite of likelihood ratio (CLR)” statistics [53, 61]. Furthermore, iHS detects only “ongoing sweeps” and CLR detects only “completed sweeps” in a target genome. Additionally, selection sweeps identified using HomSI are of higher resolution in comparison to the other methods (with sliding windows of 10,000 versus 50,000 base pairs). Runs of reduced heterozygosity were identified in reported candidate gene regions and may have resulted from selective sweeps. There were 70 regions of identified to have relatively reduced heterozygosity that co-localised with previously reported candidate genes for tolerance to trypanosomiasis, resistance to haemonchosis or adaptation to tropical conditions.
Five of the eight candidate trypanotolerance genes previously reported in a peripheral blood mononuclear cell gene expression study in experimentally infected trypanotolerant Ndama cattle [48] fell within the regions of reduced heterozygosity identified in this study (STX7, SCAMP1, RAB35, CD19, CTSS). We identified putative selection signatures that co-localised with four of these five genes in Djallonke sheep, but the fifth candidate gene (SCAMP1) had similar values of heterozygosity in both Djallonke and Sahelian sheep. It is possible that Sahelian sheep may have also undergone some selection for trypanotolerance. Interestingly a sixth candidate gene, the INHBA gene, also fell within a region of reduced heterozygosity in the Djallonke. The INHBA candidate loci is the most significantly associated trypanotolerant loci reported in the Animal QTLdb to date [35]. The INHBA gene was identified through fine mapping analysis of four a priori identified trypanotolerant associated loci in 360 Ndama cattle under natural infection conditions [42]. The INHBA gene has been shown to regulate the differentiation of hematopoietic cells in mammals [62,63,64,65]. This is consistent with the hypothesised mechanisms of trypanotolerance, because the trait is strongly associated with the host’s capacity to control anaemia [4, 42, 66]. The last two trypanotolerance candidate genes, ARHGAP15 and TICAM1, fell within regions of reduced heterozygosity. These two genes were previously identified in a combined transcriptomic and selective sweep analysis of infected trypanotolerant Ndama and Boran cattle [43]. These genes co-localised within regions of reduced heterozygosity in the Djallonke dataset, whereas only TICAM1, but not ARHGAP15, was co-localised with a region of reduced heterozygosity in the Sahelian dataset.
Previous studies on trypanotolerance have used a lower density of molecular markers [41, 42, 47, 48], and the confidence limits of the reported candidate loci are quite large [16]. Comparison of the Djallonke and Sahelian sheep revealed several putative selective sweeps of varying sizes (down to 2 Kilo-bases resolution). Although trypanotolerance is a complex quantitative trait and controlled by many genes, it is highly unlikely that all of the variants captured in these regions are causative variants. It is more likely that some variants are in linkage disequilibrium with the causal variants and hitch-hiked over time [67].
Gene ontology (GO) revealed that the 25 haemonchosis associated regions contain genes involved in multiple biological processes such as immune response and chemotaxis (MHCII-DRB1, IL20RA, IL17RB, FCER2, HRH1), response to pain and tissue homeostasis (RELN, SOX9), and protein coding, binding, methylation and phosphorylation (ATP2B1, SOX9, MUC15, UBE2N, LRP8, RELN, NSUN2, LAMC1, ABCB9, PIK3CD, SUGTI, PAK4) [32, 33]. Other functions of the identified candidate genes include calcium binding and transport (LRP8, LAMC1) and carbohydrate metabolism (CHI3L2, CHIA) [32, 33].
Six of the genes falling within regions of reduced heterozygosity identified in this study (LRP8, ATP2B1, LAMC1, SOX9, MUC15, UBE2N) were also associated with resistance to H. contortus infection in a recent GWAS study using a backcross population of Red Maasai and Dorper sheep under natural infection conditions [38]. A further six genes (CHI3L2, CHIA, DENND2D, RELN, NSUN2, and HRH1) were among the previously reported top 1% of candidate genes for resistance and susceptibility to gastrointestinal nematodes in divergent populations of Romney and Perendale sheep [37]. Two of the genes (IL20RA, PIK3CD) were associated with resistance to experimental challenge with H. contortus [36]. In a gene expression study of deliberately infected Chinese Hu sheep, four genes (ABCB9, SUGT1, PAK4, FCER2) were found to contribute to the key immunological responses [39]. More recently, five of the genes (AREG, KIT, IL17RB, CXCL12, CLCR6) were also found to be up regulated in H. contortus resistant Canarian hair sheep [40]. Three of the 23 genes (IL20RA, PIK3CD, RELN) have also been associated with resistance to other gastrointestinal nematodes such as Trichostrongyle species, Teladorsagia circumcincta and other Nematodirus species [36, 38].
A total of 37 regions with reduced heterozygosity contained genes associated with adaptive responses. Some of the genes were involved in immune functions (e.g. IL12RB2, ALCAM, APC2, IL1R, 1IL7), homeostasis (e.g. HSPA1A, ATP12A, PDK2, NF1, ABCG2), melanogenesis/ thermotolerance (GNAI3, LMLN, PLB1, MITF) and cellular and digestive metabolism (GLB1, SUCLG2, TRHDE, OLR1) [31, 68, 69]. These genes are plausible candidates for resistance to disease, heat tolerance, or the ability to exist on low quality diets in the harsh, hot and humid climatic conditions faced by these sheep breeds.
Twelve of the 37 low heterozygosity regions (NPR2, ABCG2, FGF5, MSRB3, PDK2, NF1, NFATC2, OR2AG1, PRLR, ABHD2, MITF, GLB1) were also reported in the top 0.1% of candidate genes identified in a previous genome-wide study for signatures of recent selection in 74 different sheep breeds selected from various regions of the world [68]. Fourteen of the 37 regions (GNAI3, LMLN, BMP4, TRHDE, ALK, IL1R1, IL7, ATP12A, PCDH9, PLCB1, ELF2, PGRMC2, ALDH1A3, and SUCLG2) were also among the genes recently reported as candidate adaptive genes in indigenous Egyptian sheep and goat breeds [31].
More recently, in a study of natural local environmental adaptation, ten regions (IL12RBB2, ALCAM, SYNJ1, KRIT1, TSPAN12, APC, OLR1, IFTM21, and BATF2) were among those reported as being important for adaptation in Dall sheep (Ovis dalli dalli) [69]. This approach combined targeted resequencing of a priori identified candidate adaptive genes of immunity and metabolism in domestic sheep (O. aries) and bighorn sheep (Ovis canadensis) to develop a panel of SNP markers [69]. As with Djallonke sheep, Dall sheep have undergone many centuries of natural selection with limited human intervention. In contrast to the tropical climatic conditions for Djallonke and Sahelian sheep, the Dall sheep breed evolved under Arctic and sub-Arctic climatic challenges, and hence the common swept regions have direct bearing to only immune functions and not climatic adaptation.
The many shared adaptive signatures of selection between the Djallonke and Sahelian sheep in this study can be attributed to common selection pressures due to their shared environment over several centuries. Historical admixture has been reported in Djallonke sheep populations in different regions of sub-Saharan Africa [70, 71]. The high number of shared variants (96%) also supports the possibility of migration between the breeds. Introgression from a breed with a high frequency of a homozygous region may reduce heterozygosity in the recipient breed.
Conclusions
A whole genome analysis of the Djallonke and Sahelian sheep breeds identified over 1 million novel genomic variants. This large number of novel variants suggests that the two sheep breeds represent unique genetic resources, and hence are important for world sheep diversity. The considerable number of breed-specific SNPs identified in Djallonke and Sahelian sheep could aid the sustainable management of each breed. The results also appear to support previous reports of genetic regions associated to trypanotolerance, resistance to H. contortus infection and adaptation to a harsh tropical climate. The genomic evidence of trypanotolerance, inferred from conserved orthologues of trypanotolerant Ndama cattle, suggests evidence of similar adaptive selection response for a common disease in two different ruminant species. However, a more comprehensive genetic study in a larger dataset coupled with clinical parasitology will be required to a make any definitive statement.
Availability of data and materials
The data has been generated from this study has been release to the European variant archive of the European Bioinformatics Institute with the accession number PRJEB15642. The data has also been shared with the International Sheep Genomics Consortium.
Abbreviations
- Animal QTLdb:
-
Animal Quantitative Trait Loci
- CLR:
-
composite of likelihood ratio
- DJ:
-
Djallonke
- GATK:
-
Genome Analysis Tool Kit
- GCTA:
-
Genome-wide Complex Trait Analysis
- GRM:
-
Genomic relationship matrix
- HomSI:
-
Homozygosity Stretch Identifier
- iHS:
-
Integrated haplotype homozygosity score
- PCA:
-
Principal component analysis
- SA:
-
Sahelian
- SNP:
-
Single nucleotide polymorphism
- UTR:
-
Untranslated regions
References
Traoré A, Notter DR, Soudre A, Kaboré A, Álvarez I, Fernández I, Sanou M, Shamshuddin M, Periasamy K, Tamboura HH, et al. Resistance to gastrointestinal parasite infection in Djallonké sheep. Animal. 2017;11:1–9.
Goossens B, Osaer S, Ndao M, Van Winghem J, Geerts S. The susceptibility of Djallonke and Djallonke-Sahelian crossbred sheep to Trypanosoma congolense and helminth infection under different diet levels. Vet Parasitol. 1999;85(1):25–41.
Dolan RB. Genetics and trypanotolerance. Parasitol Today. 1987;3(5):137–43.
Naessens J. Bovine trypanotolerance: a natural ability to prevent severe anaemia and haemophagocytic syndrome? Int J Parasitol. 2006;36(5):521–8.
Muigai AWT, Hanotte O. The origin of African sheep: archaeological and genetic perspectives. Afr Archaeol Rev. 2013;30(1):39–50.
Peripolli E, Munari DP, Silva MVGB, Lima ALF, Irgang R, Baldi F. Runs of homozygosity: current knowledge and applications in livestock. Anim Genet. 2016;48(3):255.
Marshall K, Mugambi JM, Nagda S, Sonstegard TS, Van Tassell CP, Baker RL, Gibson JP. Quantitative trait loci for resistance to Haemonchus contortus artificial challenge in red Maasai and Dorper sheep of East Africa. Anim Genet. 2013;44(3):285–95.
Benavides MV, Sonstegard TS, Van Tassell C. Genomic regions associated with sheep resistance to gastrointestinal nematodes. Trends Parasitol. 2016;32(6):470–80.
Goossens B, Osaer S, Kora S, Jaitner J, Ndao M, Geerts S. The interaction of Trypanosoma congolense and Haemonchus contortus in Djallonke sheep. Int J Parasitol. 1997;27(12):1579–84.
Osaer S, Goossens B, Kora S, Gaye M, Darboe L. Health and productivity of traditionally managed Djallonke sheep and west African dwarf goats under high and moderate trypanosomosis risk. Vet Parasitol. 1999;82(2):101–19.
Murray M, Trail JCM. Genetic resistance to animal trypanosomiasis in Africa. Prev Vet Med. 1984;2(1–4):541–51.
Sanni MT, Gbolabo OO, Mufliat AA, Abdulmojeed Y, Christian ONI, Olufunmilayo AA, Adewale OT, Michael OO, Mathew W, Michael IT, et al. Molecular diagnosis of subclinical African Trypanosoma vivax infection and association with physiological indices and serum metabolites in extensively managed goats in the tropics. Open J Vet Med. 2013;03:39.
Stijlemans B, De Baetselier P, Magez S, Van Ginderachter JA, De Trez C. African trypanosomiasis-associated anemia: the contribution of the interplay between parasites and the mononuclear phagocyte system. Front Immunol. 2018;9:218.
Geerts S, Osaer S, Goossens B, Faye D. Trypanotolerance in small ruminants of sub-Saharan Africa. Trends Parasitol. 2009;25(3):132–8.
Namangala B. Contribution of innate immune responses towards resistance to African trypanosome infections. Scand J Immunol. 2012;75(1):5–15.
Yaro M, Munyard KA, Stear MJ, Groth DM. Combatting African animal Trypanosomiasis (AAT) in livestock: the potential role of trypanotolerance. Vet Parasitol. 2016;225:43–52.
Brahi OHD, Xiang H, Chen X, Farougou S, Zhao X. Mitogenome revealed multiple postdomestication genetic mixtures of west African sheep. J Anim Breed Genet. 2015;132(5):399–405.
Yaro M, Munyard KA, Morgan E, Allcock RJ, Stear MJ, Groth DM. P4041 Pooled whole-genome sequencing reveals molecular signatures of natural adaptive selection in Djallonke sheep of Ghana. J Anim Sci. 2016;94(7supplement4)):98–9.
Yuan Y, Xu H, Leung RK-K. An optimized protocol for generation and analysis of ion proton sequencing reads for RNA-Seq. BMC Genomics. 2016;17(1):403.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome project data processing S: the sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82.
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565.
Reich D, Price AL, Patterson N. Principal component analysis of genetic data. Nat Genet. 2008;40:491.
Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28(24):3326–8.
Gormez Z, Bakir-Gungor B, Sagiroglu MS. HomSI: a homozygous stretch identifier from next-generation sequencing data. Bioinformatics. 2014;30(3):445–7.
Bayrakli F, Poyrazoglu HG, Yuksel S, Yakicier C, Erguner B, Sagiroglu MS, Yuceturk B, Ozer B, Doganay S, Tanrikulu B, et al. Hereditary spastic paraplegia with recessive trait caused by mutation in KLC4 gene. J Hum Genet. 2015;60(12):763–8.
Kancheva D, Atkinson D, De Rijk P, Zimon M, Chamova T, Mitev V, Yaramis A, Maria Fabrizi G, Topaloglu H, Tournev I, et al. Novel mutations in genes causing hereditary spastic paraplegia and Charcot-Marie-tooth neuropathy identified by an optimized protocol for homozygosity mapping based on whole-exome sequencing. Genet Med. 2016;18(6):600–7.
Tuncer FN, Gormez Z, Calik M, Altiokka Uzun G, Sagiroglu MS, Yuceturk B, Yuksel B, Baykan B, Bebek N, Iscan A, et al. A clinical variant in SCN1A inherited from a mosaic father cosegregates with a novel variant to cause Dravet syndrome in a consanguineous family. Epilepsy Res. 2015;113:5–10.
Brown EA, Pilkington JG, Nussey DH, Watt KA, Hayward AD, Tucker R, Graham AL, Paterson S, Beraldi D, Pemberton JM, et al. Detecting genes for variation in parasite burden and immunological traits in a wild population: testing the candidate gene approach. Mol Ecol. 2013;22(3):757–73.
Kim ES, Elbeltagy AR, Aboul-Naga AM, Rischkowsky B, Sayre B, Mwacharo JM, Rothschild MF. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity. 2016;116(3):255–64.
Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. Ensembl 2015. Nucleic Acids Res. 2015;43(D1):D662–9.
Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, et al. Ensembl 2016. Nucleic Acids Res. 2016;44(D1):D710–6.
Herrero J, Muffato M, Beal K, Fitzgerald S, Gordon L, Pignatelli M, Vilella AJ, Searle SMJ, Amode R, Brent S, et al. Ensembl comparative genomics resources. Database. 2016;2016:bav096.
Hu ZL, Park CA, Reecy JM. Developmental progress and current status of the animal QTLdb. Nucleic Acids Res. 2016;44(D1):D827–33.
Periasamy K, Pichler R, Poli M, Cristel S, Cetrá B, Medus D, Basar M, KT A, Ramasamy S, Ellahi MB, et al. Candidate Gene Approach for Parasite Resistance in Sheep – Variation in Immune Pathway Genes and Association with Fecal Egg Count. PLoS One. 2014;9(2):e88337.
McRae KM, McEwan JC, Dodds KG, Gemmell NJ. Signatures of selection in sheep bred for resistance or susceptibility to gastrointestinal nematodes. BMC Genomics. 2014;15(1):1–13.
Benavides MV, Sonstegard TS, Kemp S, Mugambi JM, Gibson JP, Baker RL, Hanotte O, Marshall K, Van Tassell C. Identification of novel loci associated with gastrointestinal parasite resistance in a red Maasai x Dorper backcross population. PLoS One. 2015;10(4):e0122797.
Yang Y, Zhou Q-J, Chen X-Q, Yan B-L, Guo X-L, Zhang H-L, Du A-F. Profiling of differentially expressed genes in sheep T lymphocytes response to an artificial primary Haemonchus contortus infection. Parasit Vectors. 2015;8(1):235.
Guo Z, González JF, Hernandez JN, McNeilly TN, Corripio-Miyar Y, Frew D, Morrison T, Yu P, Li RW. Possible mechanisms of host resistance to Haemonchus contortus infection in sheep breeds native to the Canary Islands. Sci Rep. 2016;6:26200.
Hanotte O, Ronin Y, Agaba M, Nilsson P, Gelhaus A, Horstmann R, Sugimoto Y, Kemp S, Gibson J, Korol A, et al. Mapping of quantitative trait loci controlling trypanotolerance in a cross of tolerant west African N’Dama and susceptible east African Boran cattle. Proc Natl Acad Sci U S A. 2003;100(13):7443–8.
Dayo GK, Gautier M, Berthier D, Poivey JP, Sidibe I, Bengaly Z, Eggen A, Boichard D, Thevenon S. Association studies in QTL regions linked to bovine trypanotolerance in a west African crossbred population. Anim Genet. 2012;43(2):123–32.
Noyes H, Brass A, Obara I, Anderson S, Archibald AL, Bradley DG, Fisher P, Freeman A, Gibson J, Gicheru M, et al. Genetic and expression analysis of cattle identifies candidate genes in pathways responding to Trypanosoma congolense infection. Proc Natl Acad Sci U S A. 2011;108(22):9304–9.
Maillard JC, Berthier D, Thevenon S, Piquemal D, Chantal I, Marti J. Efficiency and limits of the serial analysis of gene expression (SAGE) method: discussions based on first results in bovine trypanotolerance. Vet Immunol Immunopathol. 2005;108(1–2):59–69.
Berthier D, Quere R, Thevenon S, Belemsaga D, Piquemal D, Marti J, Maillard JC. Serial analysis of gene expression (SAGE) in bovine trypanotolerance: preliminary results. Genet Sel Evol. 2003;35(Suppl 1):S35–47.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2.
Dayo GK, Thevenon S, Berthier D, Moazami-Goudarzi K, Denis C, Cuny G, Eggen A, Gautier M. Detection of selection signatures within candidate regions underlying trypanotolerance in outbred cattle populations. Mol Ecol. 2009;18(8):1801–13.
O’Gorman GM, Park SD, Hill EW, Meade KG, Coussens PM, Agaba M, Naessens J, Kemp SJ, MacHugh DE. Transcriptional profiling of cattle infected with Trypanosoma congolense highlights gene expression signatures underlying trypanotolerance and trypanosusceptibility. BMC Genomics. 2009;10(1):207.
Choi JW, Choi BH, Lee SH, Lee SS, Kim HC, Yu D, Chung WH, Lee KT, Chai HH, Cho YM, et al. Whole-genome Resequencing analysis of Hanwoo and Yanbian cattle to identify genome-wide SNPs and signatures of selection. Mol Cell. 2015;38(5):466–73.
Gudbjartsson DF, Sulem P, Helgason H, Gylfason A, Gudjonsson SA, Zink F, Oddson A, Magnusson G, Halldorsson BV, Hjartarson E, et al. Sequence variants from whole genome sequencing a large group of Icelanders. Scientific Data. 2015;2:150011.
Guo Y, Long J, He J, Li C-I, Cai Q, Shu X-O, Zheng W, Li C. Exome sequencing generates high quality data in non-target regions. BMC Genomics. 2012;13(1):1–10.
Choi J-W, Liao X, Stothard P, Chung W-H, Jeon H-J, Miller SP, Choi S-Y, Lee J-K, Yang B, Lee K-T, et al. Whole-genome analyses of Korean native and Holstein cattle breeds by massively parallel sequencing. PLoS One. 2014;9(7):e101127.
Boitard S, Boussaha M, Capitan A, Rocha D, Servin B. Uncovering adaptation from sequence data: lessons from genome Resequencing of four cattle breeds. Genetics. 2016;203(1):433.
Yaro M, Munyard KA, Stear MJ, Groth DM. Molecular identification of livestock breeds: a tool for modern conservation biology. Biol Rev Camb Philos Soc. 2017;92(2):993–1010.
Zhan X, Dixon A, Batbayar N, Bragin E, Ayas Z, Deutschova L, Chavko J, Domashevsky S, Dorosencu A, Bagyura J, et al. Exonic versus intronic SNPs: contrasting roles in revealing the population genetic differentiation of a widespread bird species. Heredity. 2015;114(1):1–9.
Gu W, Gurguis CI, Zhou JJ, Zhu Y, Ko E-A, Ko J-H, Wang T, Zhou T. Functional and structural consequence of rare Exonic single nucleotide polymorphisms: one story Two Tales. Genome Biol Evol. 2015;7(10):2929–40.
Mwacharo JM, Elbeltagy AR, Kim ES, Haile A, Rischkowsky B, Rothschild MF. S0124 Indigenous stocks as treasure troves for sustainable livestock production in the 21st century: Insights from small ruminant genomics. J Anim Sci. 2016;94(7supplement4):12–3.
Mwai O, Hanotte O, Kwon YJ, Cho S. African indigenous cattle: unique genetic resources in a rapidly changing world. Asian Australas J Anim Sci. 2015;28(7):911–21.
Lee KT, Chung WH, Lee SY, Choi JW, Kim J, Lim D, Lee S, Jang GW, Kim B, Choy YH. Whole-genome resequencing of Hanwoo (Korean cattle) and insight into regions of homozygosity. BMC Genomics. 2013;14(1):519.
Gautier M, Foucaud J, Gharbi K, Cezard T, Galan M, Loiseau A, Thomson M, Pudlo P, Kerdelhue C, Estoup A. Estimation of population allele frequencies from next-generation sequencing data: pool-versus individual-based genotyping. Mol Ecol. 2013;22(14):3766–79.
Qanbari S, Pausch H, Jansen S, Somel M, Strom TM, Fries R, Nielsen R, Simianer H. Classic selective sweeps revealed by massive sequencing in cattle. PLoS Genet. 2014;10(2):e1004148.
Johansson BM, Wiles MV. Evidence for involvement of activin a and bone morphogenetic protein 4 in mammalian mesoderm and hematopoietic development. Mol Cell Biol. 1995;15(1):141–51.
Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE, Group TMGD. The mouse genome database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res. 2015;43(D1):D726–36.
Smith CM, Finger JH, Hayamizu TF, McCright IJ, Xu J, Berghout J, Campbell J, Corbani LE, Forthofer KL, Frost PJ, et al. The mouse gene expression database (GXD): 2014 update. Nucleic Acids Res. 2014;42(D1):D818–24.
Bult CJ, Krupke DM, Begley DA, Richardson JE, Neuhauser SB, Sundberg JP, Eppig JT. Mouse tumor biology (MTB): a database of mouse models for human cancer. Nucleic Acids Res. 2015;43(D1):D818–24.
Trail JC, d'Ieteren GD, Maille JC, Yangari G. Genetic aspects of control of anaemia development in trypanotolerant N'Dama cattle. Acta Trop. 1991;48(4):285–91.
Maynard Smith J, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35.
Kijas JW, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M, Servin B, McCulloch R, Whan V, Gietzen K, et al. Genome-wide analysis of the world's sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012;10(2):e1001258.
Roffler GH, Amish SJ, Smith S, Cosart T, Kardos M, Schwartz MK, Luikart G. SNP discovery in candidate adaptive genes using exon capture in a free-ranging alpine ungulate. Mol Ecol Resour. 2016;16(5):1147–64.
Alvarez I, Traore A, Kabore A, Zare Y, Fernandez I, Tamboura HH, Goyache F. Microsatelitte analysis of the Rousse (red Sokoto) goat of Burkina Faso. Small Rumin Res. 2012;105(1–3):83–8.
Alvarez I, Traoré A, Tamboura HH, Kabore A, Royo LJ, Fernández I, Ouédraogo-Sanou G, Sawadogo L, Goyache F. Microsatellite analysis characterizes Burkina Faso as a genetic contact zone between Sahelian and Djallonké sheep. Anim Biotechnol. 2009;20(2):47–57.
Acknowledgements
The authors thank Dr. Abdulrahman Iddriss of University of Development Studies in Ghana for invaluable support during in Ghana. A special thanks to the farm staff and the managers of Ejura and Pong Tamale sheep breeding station in Ghana for facilitating the sampling of blood. We greatly thank CHIRI Bioscience of Curtin University for the use of its facilities.
Funding
MY was funded by a Curtin University International Strategic Research Scholarship (CIRS). This work was part funded by the BBSRC Animal Health Research Grant BB/L004070/1. Neither the BBSRC nor Curtin University played any role in the design of the study, collection, analysis, interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
DMG and KM supervised the project and provided a substantive contribution to all aspects of this work presented, it’s conceptualisation, experimental design, supervision of experiments, performance, analysis and interpretation of the data. MJS provided technical advice on the project concept. MY did all the blood sampling and DNA extractions. MY drafted the manuscript under the direct supervision and advice of DMG, KAM and MJS. RJNA performed the initial pre-processing and mapping of sequenced data with Torrent Suite 4.6 and reviewed the associated method section of manuscript. MY, EM, and KAM performed all the GATK pipeline analyses. MY, EM and KAM did the HomSI analysis and the snpEff annotation. MY did the GO analyses. All the genomes were sequenced in RJNA’s laboratory. All Authors have read and approved the final manuscripts.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was performed according to the Australian Code of Practice for Care and Use of Animals for Scientific Purposes. The Curtin University Animal Ethics approval number is AEC_2014_35.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Shows the details of Genomic relationship matrix output for this analysis for all the individual Djallonke and Sahelian sheep sampled for this study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yaro, M., Munyard, K.A., Morgan, E. et al. Analysis of pooled genome sequences from Djallonke and Sahelian sheep of Ghana reveals co-localisation of regions of reduced heterozygosity with candidate genes for disease resistance and adaptation to a tropical environment. BMC Genomics 20, 816 (2019). https://doi.org/10.1186/s12864-019-6198-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-6198-8