
Abstract
Background
Human papillomaviruses (HPVs) are genetically diverse, belonging to five distinct genera: Alpha, Beta, Gamma, Mu and Nu. All papillomaviruses have double stranded DNA genomes that are thought to evolve slowly because they co-opt high-fidelity host cellular DNA polymerases for their replication. Despite extensive efforts to catalogue all the HPV species that infect humans, it is likely that many still remain undiscovered. Here we use the sequences of ten novel Gammapapillomaviruses (Gamma-PVs) characterized in previous studies and related HPVs to analyse the evolutionary dynamics of these viruses at the whole genome and individual gene scales.
Results
We found statistically significant incongruences between the phylogenetic trees of different genes which imply gene-to-gene variation in the evolutionary processes underlying the diversification of Gamma-PVs. We were, however, only able to detect convincing evidence of a single recombination event which, on its own, cannot explain the observed incongruences between gene phylogenies. The divergence times of the last common ancestor (LCA) of the Alpha, Beta, Mu, Nu and Gamma genera was predicted to have existed between 49.7–58.5 million years ago, before splitting into the five main lineages. The LCA of the Gamma-PVs at this time was predicted to have existed between 45.3 and 67.5 million years ago: approximately at the time when the simian and tarsier lineages of the primates diverged.
Conclusion
Consequently, we report here phylogenetic tree incongruence without strong evidence of recombination.
Similar content being viewed by others


Background
Human papillomavirus (HPV) is a member of the Papillomaviridae family [1, 2] which was once part of the larger family of Papovaviridae which was split into Polyomaviridae and Papillomaviridae by the International Committee on Taxonomy of Viruses (ICTV) [3]. There are currently 29 genera in the Papillomaviridae family named according to the Greek alphabet from alpha to omega and following exhaustion of the alphabet the term dyo-(Greek for second time) coined to accommodate the extra genera e.g. dyo-deltapapillomaviruses [1]. HPVs are distributed over 5 genera (Alpha, Beta, Gamma, Mu and Nu). The other papillomavirus (PV) genera are from other mammals (20), birds (3) and reptiles (1) [1]. Below the genus level are species and below the species level are types [2]. The ICTV is responsible for nomenclature of viruses down to species level, and below species level, the International HPV Reference Centre in Stockholm Sweden assigns unique HPV type numbers after the complete genome has been sequenced, cloned and confirmed by the Centre [4]. The recognition of a novel HPV type by the International HPV Reference Centre and the scientific community is based on availability of the full cloned genome, with the L1 gene sequence greater than 10% different or < 90% similar from any previously described type [1].
HPVs are thought to evolve slowly because they replicate by co-opting high fidelity host cellular DNA polymerases that have an error rate of about 4.3 × 10− 5 substitutions per nucleotide site per year [5]. It is therefore generally assumed that HPVs have co-evolved with their hosts [6,7,8]. However, several host factors may affect HPV evolutionary rates over time; for example, the cellular protein APOBEC3 cytidine deaminase may result in nucleotide compositional biases that contribute to cytosine containing codons accruing more frequently that thiamine containing codons. Selection pressures due to cellular or humoral immune responses may also differ between genes and result in these genes displaying different substitution rates. Further, the cellular polymerases of different host species may differ in their degree of fidelity such that virus lineages infecting different hosts might display different substitution rates [9].
It has been estimated that PVs appeared about 250–150 million years ago [10] in the Mesozoic age (age of reptiles/dinosaurs) [6]. Whereas the most recent common ancestor (MRCA) of the Alpha, Beta, Gamma, Mu and Nu HPVs has been inferred to have existed between 30 and 50 MYA [11], the MRCA of the present day Gamma-PVs has been inferred to have existed between 15 and 30 MYA [11]. The evolutionary rate of PVs has been estimated from feline-PVs to be about 1.95 × 10− 8 nucleotide substitutions per site per year [12].
The discovery of numerous new PVs using deep sequencing methods has begun to shed further light on the evolutionary history of this virus family. However, unravelling the evolutionary history of these viruses is potentially complicated by both inter-gene phylogenetic incongruence and recombination [13]. It has been observed that the nucleotide and encoded amino acid sequences of the E and L genes have evolved slightly differently in terms of evolutionary rate and selection pressures [14, 15]: a factor that could contribute to phylogenetic incongruence between the E and L gene trees. As a consequence of this, no single gene tree will accurately and adequately represent the evolutionary history of complete papillomavirus genomes [13]. Recombination events between different PVs may provide an additional explanation for gene-to-gene phylogenetic incongruence.
The biological plausibility of PV recombination is occasioned by the genetic diversity of PVs and the high frequencies of observed HPV co-infections [16]. However, the study of PV recombination has been hampered by technical difficulties associated with the accurate alignment of highly diverse PV gene sequences [17]. One of the most commonly used approaches to recombination detection is the use of the various recombination analysis tools implemented within the RDP4 software package [18]. During recombination detection, RDP4 rigorously tests the quality of sequence alignments to guard against the detection of false-positive recombination signals that arise due to sequence misalignment [19].
We report here on the use of sequences of ten novel Gamma-PVs characterized in a previous studies [20, 21], and related HPVs to analyse the evolutionary dynamic of these viruses at the whole genome and individual gene levels. Specifically, we use phylogenetic tree incongruence tests to identify incongruence and direct recombination tests to determine whether observed phylogenetic incongruences might be linked to recombination. We further use the novel sequences to estimate the likely times of the MRCA of the Gamma-PVs.
Methods
Source of sequence data
Sequences were downloaded from the Papillomavirus episteme (PAVE) database (https://pave.niaid.nih.gov accessed on 27/01/2018). The sequences for HPV211-HPV216 and HPV219-HPV222 were obtained from our group [20, 21] . The novel HPV DNA sequences were deposited in Genbank under the following accession numbers: HPV211 MF509816, HPV212 MF509817, HPV213 MF509818, HPV214 MF509819, HPV215 MF509820, and HPV216 MF509821. The GenBank accession numbers for HPV219, HPV220, HPV221, HPV222 genomes are MH172376, MH172377, MH172378, MH172379, respectively. PVs are classified by the International Committee on Taxonomy of Viruses (ICTV) into distinct species, but the nomenclature of types can be done by specific working groups. Some of the viruses used in this study are pending classification by the ICTV, but were provisionally grouped into specific genera and types by the HPV reference centre in Sweden (http://www.nordicehealth.se/hpvcenter/reference_clones/).
Phylogenetic incongruence testing
The Shimodaira-Hasegawa test [22] using W-IQ-TREE [23]
We used clustal alignments [24,25,26] of E1, E2, E4, E7, L1 and L2 HPV genes from 80 Gamma-HPVs (including the ten novel types) to compute the log-likelihoods of phylogenetic trees in W-IQ-TREE, which is a fast online phylogenetic tool for maximum likelihood analysis (http://iqtree.cibiv.univie.ac.at) [23]. The tool test tree topology estimates model parameters such as substitution rates and optimizes tree branch lengths to lessen computational usage. We used default settings of the W-IQ-Tree, including best fit model [27] and ultra-fast bootstrap analysis (1000 alignments) [28] to run tree topology analysis including the Kishino-Hasegawa (KH) test [29], Shimodaira-Hasegawa (SH) test [22] and approximately unbiased (AU) test [30] to test if there is a difference in evolutionary patterns amongst the different HPV genes.
Recombination analysis
Eighty complete Gamma-PV genomes that are representative of all currently known Gamma-PVs were obtained from the PAVE database, including the ten novel HPV types recently discovered and genomically characterised by our group [20, 21] . All genomes were linearized at the first nucleotide position of their E6 genes except for Gamma species 6 viruses that lack E6 and for which the start was shifted to the first nucleotide of their E7 genes. We then constructed an alignment containing the 80 Gamma-PVs using MUSCLE. This alignment was analysed using RDP v4.95 [18] (with default settings) which implements analysis of recombination using several methods: RDP [18], BOOTSCAN [31], CHIMAERA [32], GENECONV [33], MAXIMUM X2 [34] and SISCAN [35].
Construction of a time-scaled HPV phylogeny
We sought to infer the probable divergence times of our newly characterised HPV types from currently known HPVs. The complete L1 nucleotide sequences from 214 PV sequences were selected for analysis (Table 1). Two avian PVs: FcPV (Fringilla coelebs, the common chaffinch), PePV (Psittacus erithacus, the grey parrot); one turtle PV: CcPV1 (Caretta caretta, the loggerhead turtle) and one bovine PV (BPV1) were also included in the analysis as outgroups. We performed a Bayesian evolutionary molecular clock analysis using BEAST v1.8.4 [36] with a GTR + I + G nucleotide substitution model and an uncorrelated lognormal relaxed clock model. A fixed mean substitution rate for HPVs was applied based on estimated evolutionary rates inferred from prior studies that investigated the divergence times of feline PVs (1.95 × 10− 8 nucleotide substitutions per site per year) [12]. The Markov Chain Monte Carlo (MCMC) analysis was run for 100,000 million generations with sampling every 10,000 generations. The final MCMC sampling chains were visually assessed for convergence and good mixing using Tracer v1.7.1 [37]. A Maximum Clade Credibility (MCC) tree was generated after discarding the first 1000 trees that were obtained prior to the burn-in period of the chains.
Results
Phylogenetic incongruence among novel Gamma-PVs gene trees
To determine if the phylogenetic trees for different Gamma-PV genes were congruent, we used a more conclusive test, the SH test [22]. The null hypothesis of the SH test states that the difference between trees (branch length, topology or likelihoods) is zero. The observed differences (deltaL values) are significantly greater than zero and the null hypothesis was rejected, thus declaring that the trees are significantly different i.e. incongruent (p < 0.05). Table 2 shows the results of the SH test using W-IQ-Tree, indicating that there is substantial phylogenetic incongruence between the late and early genes of Gamma-PVs as shown by the p-values (p-SH).
Recombination analysis
The Gamma-PV whole genome alignments contained a total of only three plausible recombination events, namely: events 1, 2 and 3. These events were all detectable by two or more different recombination detection methods with a p-value cut-off < 0.05. However, event 1 and event 2 had no phylogenetic support and were therefore disregarded. Event 3 suggests that HPV4 and its near relatives all share evidence of the same ancestral recombination event [21] Table 3.
The time-scale of Gamma-PVs evolution
A fixed mean substitution rate for HPVs was applied based on estimated evolutionary rates inferred from of feline PVs (1.95 × 10− 8 nucleotide substitutions per site per year) [19]. The divergence times of the MRCA of HPV was predicted to have occurred 53.9 MYA (95% HPD 49.7–58.5), before splitting into the five main potential ancestors (Alpha, Beta, Mu, Nu and Gamma genera). The MRCA of the present-day Gamma-PVs was predicted to have occurred approximately 49.8 MYA (95% HPD 45.3–67.5). The novel HPV 212 was predicted to have diverged from its closest relative, HPV144, about 7.6 MYA (95% HPD 5.2–10.4), divergence times from the MRCA of the remaining nine novel Gamma-PVs are shown in Table 4. The predictions lie between 7.6 to 19.9 MYA.
It has been reported that between 10 and 20 MYA, several hominoid precursors lived in Africa, Europe and Asia [40] and the timing and spatiotemporal patterning of the disappearance of Neanderthal human precursors has only been radiocarbon dated to a limit of 50,000 years ago [41]. Thus, our MRCA prediction suggests that all the current HPV species diverged from what are currently their nearest relatives before the origin of humans.
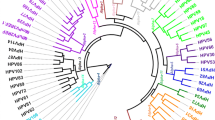
The MRCA of the Gamma-6 species was predicted to have existed 22 MYA (95% HPD 17.7–27.3) earlier than that of most other Gamma-PVs species. The MRCA of all other Gamma-species viruses and the Gamma-6 viruses was predicted to have occurred 46.8 MYA (95% HPD 42.9–51.3). The rest of the node divergence times are shown in Fig. 1 with the 95% highest posterior densities.

Molecular divergence times of PVs. A fixed mean substitution rate for HPVs was applied based on estimated evolutionary rates inferred from a study that investigated the times to the MRCA based of feline papillomaviruses (1.95 × 10− 8 nucleotide substitutions per site per year) [12]. Classification was based on [1, 2]. Posterior support values and exact divergence estimates in million years (with 95% highest posterior density) for the nodes corresponding to the 10 HPV types are presented in Table 2, the novel types are indicated in red
Discussion
Phylogenetic incongruence and recombination
An analysis of phylogenetic trees generated from different genes of the ten novel South African Gamma-PVs and their closest known relatives indicated differences, ranging from slight to major, between the branch lengths and branching orders of phylogenetic trees constructed from different genes. Differences between internal branches of phylogenetic trees constructed using different parts of the same HPV genomes have been described extensively elsewhere [6, 42, 43]. These differences could imply the occurrence of recombination, as has been previously reported [44]. Different PV genes have been shown to have different evolutionary rates, with an overall rate of 1.95 × 10− 8 and a range of 1.76 × 10− 8 to 2.69 × 10− 8 substitutions/site/year [13]. The PV genome has a modular nature, which is evident today in the different evolutionary rates of the different genes, the new genes E5, E6 and E7 encoding oncoproteins diverge faster than the old four genes E1, E2, L2 and L1 [15]. The four old proteins are by themselves able to fulfil the basic tasks of replication, regulation, stabilisation and viral DNA packaging leading to vegetative release of viral progeny from host cells [45]. The acquisition of the new oncogenes has introduced two PV phylogenies, high risk Alpha-PVs cluster together according to the phylogeny of these oncogenes (E5, E6, E7) but they do not cluster together according to the phylogeny of the capsid proteins (L1 and L2) [42]. Additionally, the number of mutations are higher in the new oncogenes than in most of the PV genes [42]. It is thus proposed that the history of PVs took place in different stages, the first stage represents the initial appearance of a prototype-PV with the cardinal genes (E1, E2, L2 and L1) found in all PVs. Then second stage involved further divergent evolutionary processes that led to the acquisition of the E5, E6 and E7 oncoproteins, and these newly acquired proteins evolved about two times faster than the core region of the genome [15]. It has been observed that the genes and encoded amino acid sequences of viral early and late proteins have evolved differently in terms of evolutionary rate and selection pressure [14, 15], and hence the incongruence between early and late trees. PV recombination events may provide a clue to the phylogenetic incongruence, but recombination is not the only explanation for this incongruence.
We did not, however find strong evidence of recombination among Gamma-HPV types. From the 80 full Gamma-PVs (70 known HPV types and the ten novel types) included in the analysis only one strongly supported recombination event was detected. Recombination in Gamma-PVs has been reported elsewhere [46]. In that study, seven potential recombination events were reported using separate analyses of individual genes (rather than analysing full genomes). We detected few Gamma-PVs recombination events. Varsani et al. (2006) [19] reported 529 potential recombination events, yet only ten were true events. The phenomenon is not exclusive to Gamma-PVs, in Alphapapillomaviruses recombination events have been well described [16, 47].
The difficulty of detecting recombination events in PVs relates to the technical difficulty of aligning highly divergent DNA sequences [6, 16, 19]. Another factor is that most recombination detection methods are only designed to detect recombination events when one or both of the parental sequences have close relatives represented in the dataset being analysed [13]. Therefore, as more PVs are discovered it becomes more likely that sequences closely related to the parents of recombinants will be included in recombination analyses. We also only included Gamma-PVs, in our recombination analysis dataset and our analysis was therefore only powered to detect for evidence of intra-genus recombination. However, this is probably not a major issue since the only convincing evidence of recombination in PVs that has so far been published has been between PVs in the same genus [47]. We have previously reported that the ten novel Gamma-PVs are under no positive selection pressure but rather purifying selection (dn/ds < 1) ([21].
It has been shown elsewhere that different PV genes are under different selection pressures [48] and also that different genes have different evolutionary rates ranging between 2 × 10− 8 and 5 × 10− 9 substitutions per site per year [12, 44]. Thus, no single gene tree will accurately represent the evolutionary history of entire PV genomes [13].
Consequently, we report here phylogenetic tree incongruence with no evidence of recombination. It has been proposed that in such scenarios there is convergent as opposed to divergent evolution [49]. Convergent evolution can be defined as the independent evolution of similar features or characteristics in species of different lineages.
Temporal evolution within Gamma-PVs
We show here that the MRCA of HPV was predicted to have occurred about 50–60 MYA. This is comparable to work done by Chen et al. (2007) [11], and that the MRCA of the Gamma-PVs existed about 45–67 (49.8) MYA. Further, we were able to show that within the Gamma-PV genus, the Gamma-6 species split from the rest of all the other Gamma species about 43–51 (46.8) MYA, while Van Doorslaer and Mcbride (2016) [50] showed that Gamma-6 species last shared a common ancestor with other Gamma-PVs around 60 MYA. Further, we showed that the MRCA of the Gamma-6 species occurred about 22 MYA, which concurs with Van Doorslaer and Mcbride (2016) [50] who reported that the Gamma-6 species MRCA existed 23.4 MYA. Therefore, we hypothesise that the Gamma-6 species lost the E6 gene between 20 and 60 MYA. This suggests that E6 was lost before the evolution of hominoid primates between 10 and 20 MYA [40]. This implies two things: 1) that viruses lacking E6 may infect old world and new world monkeys, suggesting that it could be productive to hunt for these viruses in primates, and 2) that the E6-minus viruses co-evolved with their hosts over a long enough period of time for us to have been able to isolate them from current humans (assuming papillomaviruses species specificity). Chen et al. (2007) [11] had previously predicted the loss of the E6 gene to have occurred about 15–30 MYA, the estimate was based only on the L1 open reading frame (ORF) of nine divergent papillomaviruses. Here, we have estimated this date using 214 papillomavirus L1 nucleotide sequences from all the genera containing HPV sequences.
PVs lacking E6 have also been described in parrots (PePV1), donkeys (EaPV1) and bovines [51]. Presently, 7 known human PVs of the Gamma-6 species lack the E6. The size of E6 (mean 253.5 nt) in the genomes of PVs infecting birds and turtles is about half the size that it is in mammal-infecting PV genomes (mean 438 nt) [51]. The larger size of the mammalian PV E6 accommodates a second E6 zinc finger binding motif domain [52]: a domain that was possibly the result of duplication of an ancestral E6 motif [53]. E6 mediated p53 degradation has been described as one of the hallmarks of HPV mediated carcinogenesis, and the presence of this double motif may explain the increased likelihood of HPVs in causing cancer compared to PVs infecting birds and turtles.
Gamma-PVs lack an E5 gene, which is located between the early genes and the late genes and is thought to have evolved originally from a non-coding region that was integrated between the early and the late genes of an ancestral sequence belonging to the Alphapapillomaviruses lineage [51]. Willem et al. also suggest that integration of E5 in this region promoted an adaptive radiation which yielded E6 and E7 proteins capable of degrading tumour suppressor proteins and facilitating carcinogenesis [51]. This is supported by the fact that 1) E6 and E7 proteins in Alpha PVs (together with E5) have greater oncogenic potential as classified by IARC as compared to the E6 and E7 of Gamma-PVs (that lack E5) and 2) E5 has an evolutionary rate that is approximately twice that of the remainder of the PV genome [54]. The integration of the E5 ORF was predicted to have occurred in an ancestral virus that existed 30–60 MYA (in the Cenozoic era) which eventually gave rise to the Alpha. Mu and Nu lineages; each of which has different cell tropisms and clinical manifestations [6]. Willem et al. [51] inferred the appearance of the E5 oncogene occurred 53–58 MYA: well within the range of that predicted by Bravo et al. [6].
We also reported the acquisition of E10 in HPV214 [21], which we hypothesised coincided with E6 loss as previously reported [55]. We speculate that if the loss of E6 occurred 20–60 MYA then E10 was acquired a few million years later or that the loss and gain might have occurred concurrently as a modification of E6 to E10. This is supported by the fact that the E10 ORF overlaps with the more conserved portion of the E6 scar [50].
We report divergence times from 7.6 to 20 MYA with most lying well within the origin times of many other known PVs. However, HPV211 of the Gamma-8 species branches earlier from the other five HPV types in this species, i.e., the MRCA of HPV211 and the other 5 Gamma-8 species types is predicted to have occurred 20 MYA. This implies that HPV211 is closer to the ancestral or parental sequence of Gamma-8 species compared to HPV112, HPV119, HPV168, HPV147, and HPV164, and hence, it is more likely to be major/minor parent than it can be a recombinant. The MRCA of HPV222 and the other 3 members of the Gamma-19 species is 19.2 MYA. HPV222 branches from HPV161, HPV162 and HPV166 earlier than the others, also making it closer to the ancestral or parental sequence of the Gamma-19 species.
Conclusion
In this work, we report the evolutionary characterisation of Gamma-PVs including that of the novel HPV types. To get a deeper insight into the evolutionary processes that may influence the diversification of Gamma-PVs, we explored phylogenetic incongruences among different genes of the novel types, attempted to discover potential recombination events between all known Gamma-PVs, and also estimated the time scale for Gamma-PVs evolution. Consequently, we report here phylogenetic tree incongruence without strong evidence of recombination.
Abbreviations
- BPV1:
-
Bovine PV
- CcPV1:
-
Caretta caretta PV
- FcPV:
-
Fringilla coelebs PV
- Gamma-PVs :
-
Gammapapillomaviruses
- HPV:
-
Human papillomavirus
- ICTV:
-
International Committee on Taxonomy of Viruses
- KH:
-
Kishino-Hasegawa
- LCA:
-
Last common ancestor
- MCC:
-
Maximum Clade Credibility
- MCMC:
-
Markov Chain Monte Carlo
- MRCA:
-
Most recent common ancestor
- MYA:
-
Million years ago
- PePV:
-
Psittacus erithacus PV
- PV:
-
Papillomavirus
- SH:
-
Shimodaira-Hasegawa
References
Bernard HU, et al. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401(1):70–9.
de Villiers EM, et al. Classification of papillomaviruses. Virology. 2004;324(1):17–27.
vanRegenmortel, ICTV. ictv report, 2002.
Mühr LSA, Eklund C, Dillner J. Towards quality and order in human papillomavirus research. Virology. 2018;519:74–6.
Korona DA, Lecompte KG, Pursell ZF. The high fidelity and unique error signature of human DNA polymerase epsilon. Nucleic Acids Res. 2011;39(5):1763–73.
Bravo IG, Felez-Sanchez M. Papillomaviruses: viral evolution, cancer and evolutionary medicine. Evol Med Public Health. 2015;2015(1):32–51.
Chen Z, et al. Evolutionary dynamics of variant genomes of human papillomavirus types 18, 45, and 97. J Virol. 2009;83(3):1443–55.
Dube Mandishora RS, et al. Intra-host sequence variability in human papillomavirus. Papillomavirus Res. 2018;5:180–91.
de Oliveira CM, et al. High-level of viral genomic diversity in cervical cancers: a Brazilian study on human papillomavirus type 16. Infect Genet Evol. 2015;34:44–51.
Smelov V, et al. Beta and gamma human papillomaviruses in anal and genital sites among men: prevalence and determinants. Sci Rep. 2018;8(1):8241.
Chen Z, et al. Human papillomavirus (HPV) types 101 and 103 isolated from cervicovaginal cells lack an E6 open reading frame (ORF) and are related to gamma-papillomaviruses. Virol J. 2007;360(2):447–53.
Rector A, et al. Ancient papillomavirus-host co-speciation in Felidae. Genome Biol. 2007;8(4):R57.
Van Doorslaer K. Evolution of the papillomaviridae. Virology. 2013;445(1–2):11–20.
Harari A, Chen Z, Burk RD. Human papillomavirus genomics: past, present and future. Curr Probl Dermatol. 2014;45:1–18.
García-Vallvé S, Alonso Á, Bravo IG. Papillomaviruses: different genes have different histories. Trends Microbiol. 2005;13(11):514–21.
Angulo M, Carvajal-Rodriguez A. Evidence of recombination within human alpha-papillomavirus. Virol J. 2007;4:33.
Posada D, Crandall KA. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc Natl Acad Sci. 2001;98(24):13757–62.
Martin D, Rybicki E. RDP: detection of recombination amongst aligned sequences. Bioinformatics. 2000;16(6):562–3.
Varsani A, et al. Evidence of ancient papillomavirus recombination. J Gen Virol. 2006;87(Pt 9):2527–31.
Murahwa AT, et al. Complete genome sequences of four novel human Gammapapillomavirus types, HPV-219, HPV-220, HPV-221, and HPV-222, isolated from penile skin swabs from south African men. Genome Announc. 2018;6(25).
Murahwa, A.T., et al., Discovery, characterisation and genomic variation of six novel gammapapillomavirus types from penile swabs in South Africa. Papillomavirus Research, 2019. epub ahead of print(10.1016/j.pvr.2019.02.005).
Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16(8):1114.
Trifinopoulos J, et al. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016;44(W1):W232–5.
Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol. 2011;7:539.
Li W, et al. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015;43(W1):W580–4.
McWilliam, H., et al., Analysis Tool Web Services from the EMBL-EBI Nucleic Acids Res, 2013. 41(Web Server issue): p. W597–600.
Kalyaanamoorthy S, et al. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14(6):587–9.
Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30(5):1188–95.
Kishino H, Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol. 1989;29(2):170–9.
Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst Biol. 2002;51(3):492–508.
Martin DP, et al. A modified Bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res Hum Retrovir. 2005;21(1):98–102.
Martin DP, Williamson C, Posada D. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005;21(2):260–2.
Padidam M, Sawyer S, Fauquet CM. Possible emergence of new Geminiviruses by frequent recombination. Virology. 1999;265(2):218–25.
Smith JM. Analyzing the mosaic structure of genes. J Mol Evol. 1992;34(2):126–9.
Gibbs MJ, Armstrong JS, Gibbs AJ. Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics. 2000;16(7):573–82.
Drummond AJ, et al. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–73.
Rambaut A, et al. Posterior summarization in Bayesian Phylogenetics using tracer 1.7. Syst Biol. 2018;67(5):901–4.
Kishino H, Miyata T, Hasegawa M. Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J Mol Evol. 1990;31(2):151–60.
Strimmer K, Rambaut A. Inferring confidence sets of possibly misspecified gene trees. Proc Biol Sci. 2002;269(1487):137–42.
Andrews P. Evolution and environment in the Hominoidea. Nature. 1992;360(6405):641–6.
Higham T, et al. The timing and spatiotemporal patterning of Neanderthal disappearance. Nature. 2014;512(7514):306–9.
Bravo IG, Alonso Á. Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. J Virol. 2004;78(24):13613–26.
Schiffman M, et al. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology. 2005;337(1):76–84.
Shah SD, Doorbar J, Goldstein RA. Analysis of host–parasite incongruence in papillomavirus evolution using importance sampling. Mol Biol Evol. 2010;27(6):1301–14.
Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004;68(2):362–72.
Bolatti EM, et al. Characterization of novel human papillomavirus types 157, 158 and 205 from healthy skin and recombination analysis in genus gamma-papillomavirus. Infect Genet Evol. 2016;42:20–9.
Narechania A, et al. Phylogenetic incongruence among oncogenic genital alpha human papillomaviruses. J Virol. 2005;79(24):15503–10.
Rodríguez R. M.d.R., et al., Identificación de factores de riesgo para contraer virus del papiloma humano en sexoservidoras. Rev Cuba Obstet Ginecol. 2012;38:244–55.
Castoe TA, et al. Evidence for an ancient adaptive episode of convergent molecular evolution. Proc Natl Acad Sci U S A. 2009;106(22):8986–91.
Van Doorslaer K, McBride AA. Molecular archeological evidence in support of the repeated loss of a papillomavirus gene. Sci Rep. 2016;6:33028.
Willemsen, A. and I.G. Bravo, Origin and evolution of papillomavirus (onco)genes and genomes. bioRxiv, 2018.
Zanier K, et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science. 2013;339(6120):694–8.
Suarez I, Trave G. Structural insights in multifunctional papillomavirus Oncoproteins. Viruses. 2018;10(1):37.
Puustusmaa M, et al. The enigmatic origin of papillomavirus protein domains. Viruses. 2017;9(9):240.
Van Doorslaer K, et al. The papillomavirus episteme: a major update to the papillomavirus sequence database. Nucleic Acids Res. 2017;45(D1):499–506.
Acknowledgements
We thank J. Dillner, C. Eklund and D. Bzhalava from the International HPV Reference Center for receiving and confirming the novel HPV types.
Funding
This study was supported in part by the Poliomyelitis Research Fund (PRF) of South Africa and the National Research Foundation (NRF) of South Africa. This work is based upon research supported by the South African Research Chairs Initiative of the Department of Science and Technology of South Africa and NRF. ATM was a the recipient of a PhD fellowship from the Letten Research Foundation of Norway, Oslo administered by B. Stray-Pedersen. None of the funding organisations were involved in the design of the study, collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
Eighty complete Gamma-PV genome sequences used in the recombination analysis and the 214 PV sequences used for the construction of the time scaled HPV phylogeny are available from the PAVE database (https://pave.niaid.nih.gov), including the ten novel HPV types discovered and genomically characterised by our group [20, 21]. The Shimodaira-Hasegawa test [22] using W-IQ-TREE [23], original output tables are available from the corresponding author upon request. Because of the nature of data formats generated in the recombination analysis RDPv4.5 software has to be installed to view the data, and can be downloaded from http://web.cbio.uct.ac.za/~darren/rdp.html, the files can be made available on request. The time scale raw data set can be visually assessed for convergence and good mixing using Tracer v1.7.1 [37] available for download on https://github.com/beast-dev/tracer/releases/tag/v1.7.1, raw data can also be made available from the corresponding author on request.
Author information
Authors and Affiliations
Contributions
ATM did the laboratory work, the analysis and the writing. FN did the evolutionary analysis using BEAST, TM and HO did phylogenetic tree constructions and the phylogenetic incongruence tests. DM the developer of the RDP4 program did the recombination analysis. ALW conceived the whole concept and ideas for this manuscript and also supervised the group. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval for the main study (from which the ten novel types were discovered) was granted by the Health Research Ethics Committee of the University of Cape Town, Faculty of Health Sciences (HREC reference: 231/2015 and 258/2006). Written consent was obtained from all the study participants. No extra permissions to do the analysis in this paper were necessary.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Murahwa, A.T., Nindo, F., Onywera, H. et al. Evolutionary dynamics of ten novel Gamma-PVs: insights from phylogenetic incongruence, recombination and phylodynamic analyses. BMC Genomics 20, 368 (2019). https://doi.org/10.1186/s12864-019-5735-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5735-9