
Abstract
Background
WUSCHEL-related homeobox (WOX) genes encoding plant-specific homeobox (HB) transcription factors play important roles in the growth and development of plants. To date, WOX genes has been identified and analyzed in many polyploids (such as cotton and tobacco), but the evolutionary analysis of them during polyploidization is rare. With the completion of genome sequencing, allotetraploid Brassica napus and its diploid progenitors (B. rapa and B. oleracea) are a good system for studying this question.
Results
In this study, 52, 25 and 29 WOX genes were identified in allotetraploid B. napus (2n = 4x = 38, AnCn), the An genome donor B. rapa (2n = 2x = 20, Ar) and the Cn genome donor B. oleracea (2n = 2x = 18, Co), respectively. All identified WOX genes in B. napus and its diploid progenitors were divided into three clades, and these genes were selected to perform gene structure and chromosome location analysis. The results showed that at least 70 and 67% of WOX genes maintained the same gene structure and relative position on chromosomes, respectively, indicating that WOX genes in B. napus were highly conserved at the DNA level during polyploidization. In addition, the analysis of duplicated genes and transposable elements (TEs) near WOX genes showed that whole-genome triplication (WGT) events, segmental duplication and abundant TEs played important roles in the expansion of the WOX gene family in B. napus. Moreover, the analysis of the expression profiles of WOX gene pairs with evolutionary relationships suggested that the WOX gene family may have changed at the transcriptional regulation level during polyploidization.
Conclusions
The results of this study increased our understanding of the WOX genes in B. napus and its diploid progenitors, providing a rich resource for further study of WOX genes in these species. In addition, the changes in WOX genes during the process of polyploidization were discussed from the aspects of gene number, gene structure, gene relative location and gene expression, which provides a reference for future polyploidization analysis.
Similar content being viewed by others


Background
The superfamily of HB transcription factors is a large family with many members in eukaryotes [1]. A common feature of proteins in this superfamily is that they contain a homeobox domain that consists of 60–66 amino acids folded into a helix-turn-helix structure, which can be recognized by specific DNA to regulate target gene expression at a precise moment [1, 2]. In this HB superfamily, WOX genes encode plant-specific HB transcription factors [3]. Previous studies have found that WOX genes are present in the genomes of many plants, ranging from lower plants, such as green algae, to higher plants, such as angiosperms [2]. The number of WOX genes varies from plant to plant, and the WOX gene family has gradually expanded with the continuous evolution of plants; for example, there is just one WOX gene in unicellular green algae, three in moss, six in Selaginella, and 15 in Arabidopsis [4]. The WOX gene family can be divided into three clades, the ancient clade, intermediate clade and WUS clade, by phylogenetic analysis of WOX genes in different plants [5, 6]. The WOX genes of lower plants belong only to the ancient clade, while those of higher plants belong to all three clades [7].
Studies have shown that WOX genes play crucial roles in the growth and development of plants, such as stem cell regulation [8], embryo patterning [9], and flower development [10]. The functions and characteristics of WOX genes have been well studied in the typical model plant Arabidopsis thaliana, in which 15 WOX genes have been identified [6, 7]. WUS, which is expressed in the ovule, anther, and shoot apical meristem, has been shown to act a pivotal part in central meristem maintenance [11]. Overexpression of WOX1 causes abnormal meristem in A. thaliana [12]. WOX2 and WOX8 have a critical function in early embryo patterning, and they are expressed in the zygote and then confined to the apical and basal cell, respectively [13, 14]. WOX3 was found to participate in the formation of lateral and marginal regions of vegetative and floral organs [15]. WOX4, in coordination with PXY, works in auxin-dependent cambium stimulation to regulate lateral plant growth [16]. WOX5 was demonstrated to act as a vital regulator in the root apical meristem, which is necessary for forming the correct root pattern [17]. WOX6/PFS2 was shown to regulate ovule development and affect ovule patterning [18]. WOX7, which is expressed in lateral roots, was confirmed to inhibit the development of lateral roots in a sugar-dependent manner [19]. STIMPY/WOX9 integrates developmental signals and cell cycle regulation to maintain cell division and prevent inappropriate differentiation in roots [20]. WOX11 was determined to be involved in a process in which some vascular cambium initially is converted to new lateral root founder cells [21]. WOX11 and its homolog WOX12 were found to participate in de novo root organogenesis [22]. WOX13 promotes replum formation and regulates fruit patterning during fruit development [23]. WOX14 promoted vascular cell lignification by increasing the accumulation of bioactive gibberellin (GA) in the inflorescence stems of Arabidopsis [24].
Brassica napus, an allotetraploid of the Brassica genus, is a considerable oil crop planted worldwide. According to previous studies, the WGT event occurred in ancestors of the Brassica genus ~ 15.9 million years ago (MYA) [25]. Then, diploid B. rapa (2n = 20, Ar) and B. oleracea (2n = 18, Co) were successively formed ~ 4.6 MYA [25]. Finally, B. napus (2n = 4x = 38, AnCn) was formed by natural hybridization and polyploidization of B. rapa and B. oleracea ~ 7500 years ago [25]. The genomes of B. napus (cv. Darmor-bzh), B. rapa (cv. Chiifu-401-42) and B. oleracea (var. capitata-02-12) have already been sequenced and assembled [25,26,27]. Thus, the natural allotetraploid B. napus and its diploid progenitors (B. rapa and B. oleracea) were always used to study the scientific problems associated with polyploidization. So far, WOX genes has been identified and analyzed in many polyploids (such as cotton [5] and tobacco [6]), but the evolutionary analysis of them during polyploidization is rare. Therefore, we described WOX genes systematically in B. napus and its diploid progenitors and hoped to find insights regarding WOX genes during polyploidization. This study included several parts, including the identification of the WOX gene family and gene structure, conserved domain analysis, phylogenetic tree analysis, chromosomal localization analysis, synteny and duplicated gene analysis, and expression pattern analysis.
Results
Identification and characterization of WOX genes
To identify the putative WOX genes in B. napus and its diploid progenitors, 15 WOX protein sequences of Arabidopsis were acquired and used as query sequences to search against the BRAD database [28] using the BLASTp program [29]. As a result, 28, 24 and 62 genes were selected as original candidate genes in B. rapa, B. oleracea and B. napus, respectively. Then, the syntenic genes were searched in the BRAD database by inputting the gene IDs of the WOX genes in Arabidopsis, which is a supplement for the first method. As a result, an additional five genes were also identified as WOX genes in B. oleracea. Then, three public protein databases (Pfam, SMART and CDD database) were used to search the HB domain in protein sequences encoded by candidate WOX genes, and proteins that did not contain the complete conserved HB domain were removed. Finally, 25, 29 and 52 genes were identified as WOX genes in B. rapa, B. oleracea and B. napus, respectively. It was clear that the total WOX genes in two diploid progenitors, B. rapa and B. oleracea, was higher than that in the allotetraploid B. napus, which indicated that a gene loss event might have occurred in the WOX gene family of B. napus during polyploidization.
These identified WOX genes in B. napus and its diploid progenitors were named, i.e., from BrWUSa to BrWOX14b in B. rapa, BoWUSa to BoWOX14c in B. oleracea and BnAWUSa to BnAWOX14e in B. napus, according to the homologous relationship with corresponding WOX genes in Arabidopsis (Additional file 1: Table S1). The last lowercase letter in the name represents the degree of homology to the corresponding gene in Arabidopsis, with ‘a’ representing the highest homology, followed by ‘b’, and so on. In B. napus, the capital letters A and C following ‘Bn’ represent the An and Cn subgenomes, respectively. The length of the WOX protein sequences ranged from 133 (BnAWOX14e) to 397 (BnCWOX9a) amino acids in B. napus. In addition, the physical and chemical characteristics of a total of 106 WOX proteins were analyzed and provided, including the molecular weights (MW), theoretical PI values, instability index (II), grand average of hydropathicity (GRAVY) and aliphatic index (Additional file 1: Table S1). The average values of these physical and chemical characteristics were approximately equal to each other in B. napus and its diploid progenitors upon calculation.
Phylogenetic analysis and gene structure analysis
WOX proteins from typical monocots (rice) and dicots (Arabidopsis) were used as reference proteins to construct the WOX phylogenetic tree, where WOX proteins in rice were identified by the same methods mentioned above. Therefore, the unrooted phylogenetic tree was constructed based on a total of 135 WOX protein sequences, including 15 in Arabidopsis, 25 in B. rapa, 29 in B. oleracea, 52 in B. napus and 14 in rice members (Fig. 1). Evidently, the phylogenetic tree showed that WOX proteins were classified into three clades, which were the ancient clade, intermediate clade and WUS clade. According to statistical analysis, the number of WOX proteins in the WUS clade (70) was greater than the sum of proteins in the ancient clade (26) and the intermediate clade (39). Hence, the WUS clade was the largest clade of the WOX proteins in these five species. Notably, WOX proteins in B. napus and its diploid progenitors were related to their corresponding homologs in Arabidopsis or rice in each clade, which suggested that the evolutionary relationship of WOX transcription factors is very close in these species.

Phylogenetic tree of WOX proteins in B. rapa, B. oleracea, B. napus, Arabidopsis and rice. This tree could be divided into the ancient clade (yellow), intermediate clade (red) and WUS clade (green). This phylogenetic unrooted tree was constructed using MEGA7.0 with 1000 bootstrap replicates and only those values greater than 50% are displayed
For exploring more characteristics about WOX proteins in each clade, WOX protein sequences within the three clades were selected separately to build three phylogenetic trees (Fig. 2). The protein sequences in each clade were similar to each other, whether they were proteins among allotetraploid B. napus or its diploid progenitors. The ancient clade consisted of WOX13 and WOX14, while the intermediate clade consisted of WOX8, WOX9, WOX11, and WOX12, and the WUS clade consisted of WUS and WOX1–7. Interestingly, the homolog of WOX10 could not be found either in B. napus or its diploid progenitors. In addition, exon/intron structures were analyzed to show the structural diversity of WOX genes in different clades and to explore whether the gene structure changed during the polyploidization process (Fig. 2). The results showed that most of the genes had three exons in both the ancient clade and intermediate clade, while 21 genes had two exons, and 19 genes had three exons in WUS clade. By comparison, we found that the gene structure of WOX genes from the WUS clade was significantly more conserved than that of the other two clades during allotetraploid B. napus formation. Six out of eight kinds of WOX genes in the WUS clade had the same gene structure, namely, WOX1, WOX2, WOX3, WOX4, WOX5 and WOX7, whether they were from allotetraploid B. napus or its diploid progenitors. In addition, if two genes that came from the allotetraploid and one of its two diploid progenitors branched at the same final level in the phylogenetic tree, they may have a direct evolutionary relationship. Statistical analysis showed that a total of 33 pairs of WOX genes were found that may have direct evolutionary relationships in these three phylogenetic trees (Table 1). Five out of seven pairs of WOX genes (approximately 71%) in the ancient clade, six out of eleven pairs (approximately 55%) in the intermediate clade and 12 out of 15 pairs (80%) in WUS clade had the same number of exons (Table 1). Thus, 23 out of the 33 gene pairs (approximately 70%) maintained the same gene structure during the formation of B. napus. Therefore, WOX genes were conserved at the DNA level during polyploidization. Furthermore, the location of the HB domain was visualized to facilitate the analysis of the changes in the domain’s position between different clades or different species (Fig. 2). The HB domain of many WOX proteins was located in the N-terminus of the protein in both the intermediate and WUS clades but was located in the middle part of the protein in the ancient clade. We could also see that the length and position of the HB domain were generally conserved. In addition, the MEME website was used to predict conserved motifs in WOX proteins (Fig. 2), and the results showed that at most nine motifs were found in WOX proteins, and only motif 1 was found in every WOX. In general, the WOX gene family in B. napus and its diploid progenitors was very conserved at the DNA and protein level, which might be related to the important function of the WOX genes in these species.

Characterizations of the identified WOXs in B. napus and its diploid progenitors. The characterizations include intron/exon structure (yellow background), domain location (blue background) and conserved motif location (purple background). a the characterizations of the WOX genes in the ancient clade. b the characterizations of the WOX genes in the intermediate clade. c the characterizations of the WOX genes in WUS clade. The WOX genes in B. napus were marked by the red circle
Conserved amino acid sequences within the homeobox domain
The WOX gene family is a plant-specific gene family, of which the typical characteristic is that every WOX protein encoded has a completely conserved HB domain [1, 2]. To study the sequence of the conserved HB domains and the degree of their conservation in different Brassicaceae species, multiple sequence alignment was used to generate the protein sequence logos in B. rapa, B. oleracea, B. napus and Arabidopsis (Fig. 3). The sequence logos showed that the amino acids and their distribution in the HB domain were remarkably similar in these four plants. The HB domain contained one loop, one turn and three helix structures and consisted of 57 amino acids, which was consistent with previous research results [5]. Amino acids in the helix structure were more conserved than those in the loop and turn structure, and the most conserved region was helix3, in which ten highly conserved amino acids were contained, such as I, N, Y, and F. In short, the HB domain was still highly conserved in both B. napus and its diploid progenitors.

Sequence logos about the homeobox domain. a The logo in Arabidopsis. b The logo in B. rapa. c The logo in B. oleracea. d The logo in B. napus. This domain contained a total of 57 amino acids, including one loop, one turn and three helix structures
Chromosomal localization and orthologous gene analysis of WOX genes
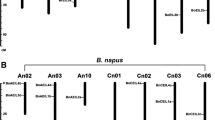
The positions of the identified WOX genes were drafted to chromosomes by using MapInspector software. Ultimately, 25 WOX genes were located on 10 chromosomes in B. rapa (Fig. 4a). Evidently, there is only one WOX gene on chromosome Ar04, three on Ar02 and Ar09, four on Ar03 and Ar05, and two genes on each of the remaining five chromosomes. Twenty-four WOX genes were located on nine chromosomes in B. oleracea, and the other five genes were located on different scaffolds because they had not been assembled into chromosomes (Fig. 4b). Five genes were distributed in chromosome Co02, but in contrast, only one gene was in Co01, Co06 and Co08. Forty-three WOX genes were located on 18 instead of 19 chromosomes in B. napus, and the other nine genes were located on scaffolds (Fig. 4c). It is worth mentioning that no single gene was located on chromosome Cn06 in B. napus. Comparison of the gene distribution of B. napus with B. rapa and B. oleracea showed the important result that many WOX genes retained their relative position in Ar and An, but in contrast, only a few genes retained their relative position in Co and Cn during the formation of B. napus. For example, each pair of chromosomes contained WOX genes with the same number and same location, such as Ar01-An01, Ar04-Ar04, Ar06-Ar06, Ar07-Ar07 and Ar08-Ar08, and other chromosome pairs contained WOX genes with different numbers but similar locations. However, only one chromosome pair, Co02-Cn02, contained WOX genes with the same number and same location. Statistical analysis shows that 21 out of 25 WOX genes (84%) were positioned on the assembled chromosomes in B. rapa, while 12 out of 24 (50%) in B. oleracea maintained their relative position during the formation of B. napus. In combination with previous studies, there are two possible reasons for this result. One possibility is that the Cn subgenome had more abundant TEs than the An subgenome [27]. The presence of TEs in the genome could cause the rearrangement of chromosomal sequences, which affects the genomic structure, such as deletion, inversion, and translocation [30]. The other possibility is that the Cn subgenome underwent more active homologous exchanges (HEs) than the An subgenome during polyploidization [27]. HEs refers to the replacement of some chromosomal regions with duplicated copies of the corresponding fragments of the homologous subgenome [31], and this event was found to occur frequently between the two subgenomes of B. napus during the hybridization and polyploidization process [27].

Chromosome distribution of WOXs in B. rapa (a), B. oleracea (b) and B. napus (c & d). Partial WOX genes in B. oleracea and B. napus located in unassembled scaffolds and these genes were not shown in this figure. The number of chromosomes was indicated at the top of each chromosome. The scale on the left is in megabases (Mb)
Synteny and duplicated gene analysis of WOX genes
Synteny analysis of WOX genes in B. napus and its diploid progenitors was performed to visualize the locus relationship of homologous WOX genes among two genomes (Ar & Co) and two subgenomes (An & Cn). As shown in Fig. 5, two genes linked to each other by one line were syntenic genes, and genes linked by lines of the same color represented the same kind of WOX gene, such as WOX1 and WOX2. Thus, we can see that many chromosomes in all four genomes/subgenomes (Ar, Co, An and Cn) were connected by the same colored line, which indicated that these genomes/subgenomes were evolutionarily related and the WOX genes were so important that most of them were preserved during polyploidization. In addition, WOX genes were evenly distributed in these four genomes/subgenomes (Fig. 5). Moreover, the synteny analysis indicated that the syntenic WOX gene pairs were widely distributed on the genomes of B. napus and its diploid progenitors.

Genome-wide synteny analysis for WOX genes among B. rapa, B. oleracea, B. napus and Arabidopsis. BRA01 to BRA10 represented the ten chromosomes in B. rapa. BOC01 to BOC09 represented the nine chromosomes in B. oleracea. BNA01 to BNA10 and BNC01 to BNC09 represented the ten and nine chromosomes in the An and Cn subgenomes in B. napus, respectively. The orthologous and paralogous WOX genes were mapped onto the chromosomes and linked by each other. Different colored lines in the inner part represent different kinds of WOX genes
Moreover, to explore whether Darwinian positive selection affected the evolution of the WOX genes in B. napus and its diploid progenitors, BLASTn [32] and syntenic gene search in BRAD database [33] were used to identify duplicated genes among them. As a result, 13, 10 and 38 segmental duplicated WOX gene pairs in the B. rapa, B. oleracea and B. napus genomes were found respectively. Then, the nonsynonymous (Ka), synonymous (Ks) and Ka/Ks ratios were calculated to estimate the selection pressure among duplicated WOX gene pairs. Ka/Ks = 1 means that genes were undergoing a neutral evolutionary process; Ka/Ks > 1 or Ka/Ks < 1 indicate that genes were selected positively or undergoing purified selection, respectively [34]. The Ka/Ks values of all duplicated WOX gene pairs in B. napus and its diploid progenitors were below one (Additional file 2: Table S2), except one duplicated gene pair (BnAWOX11b & BnCWOX11b) had no Ka/Ks value in B. napus because these two genes had the same sequence.
Transposable element analysis of WOX proteins
TEs are widely distributed in the genome, and many transposons are located near the host genes [35]. To investigate whether TEs were involved in the expansion of the WOX gene family, we identified the TEs located 2000 bp upstream and downstream of the WOX genes using the homolog search method [5]. Compared to the TEs near the WOX gene family in cotton [5], there were more TEs in both B. napus and its diploid progenitors (Table 2). After analysis, 402, 202 and 235 TEs were found in B. napus, B. rapa and B. oleracea, respectively. Thus, a conclusion can be drawn that as early as the formation of the diploid progenitors of B. napus, the WOX gene family has undergone significant expansion due to the presence of abundant TEs. The three most abundant types of TEs in order of abundance are DNA transposon, LTR retrotransposon and non-LTR retrotransposon. Two types of TEs, Ginger/TDD and R1, were located downstream of BnAWOX3b and BnCWOX12a, respectively, but these two TEs were not detected near the WOX genes in the diploid progenitors. As shown in Table 2, there are 21 kinds of DNA transposons near the WOX genes. The most abundant ones are EnSpm/CACTA, MuDR, hAT, Helitron, Mariner/Tc1 and Harbinger. LTR retrotransposons near the WOX genes mainly contained four types, Gypsy, Copia, BEL and DIRS. Furthermore, there were 18 kinds of non-LTR retrotransposons near the WOX genes of selected species, and the most abundant kind was the L1 type. Statistical analysis shows that 23, 14 and 18 L1-type transposons were found near the WOX genes of B. napus, B. rapa and B. oleracea, respectively, and the number of L1-type transposons was much higher than the number of other non-LTR retrotransposons in these species. Compared to TEs, simple repeats were less abundant in B. napus and its diploid progenitors. Specifically, there were two simple repeats in B. napus, but only one in B. rapa and none in B. oleracea.
Gene expression pattern analysis of WOX genes
To gain insights into the putative biological functions of WOX genes, we investigated their expression patterns in four tissues (leaves, stems, flowers and siliques) of B. napus and its two diploid progenitors based on our RNA-seq data (Additional file 3: Table S3). As shown in Fig. 6, we found that WOX genes are widely expressed in these four tissues, suggesting that WOX genes have multiple biological functions and operate in different tissues. In addition, the expression of WOX genes from different clades had different characteristics, and the specific characteristics were as follows: The genes of the intermediate clade were generally not expressed in all tissues, except BrWOX9a and BnCWOX9a, which were expressed in flowers; In the ancient clade, most of the WOX13 homologous genes were widely expressed in the four tissues and had a high expression level, and WOX14 homologous genes were not expressed in flowers; In the WUS clade, only the homologous genes of WOX4 were widely expressed in the four tissues. WOX3 and WOX6 homologous genes were not significantly expressed in all four tissues except BnCWOX3a, BoWOX3a, BnAWOX6a and BoWOX6b, which were detected in flowers. In addition, the homologous genes of WOX1 generally were not expressed in stems of B. napus or its two diploid progenitors. In brief, the most active genes were the genes in the ancient clade; conversely, the least active genes were those in the intermediate clade in B. napus and its two diploid progenitors, which suggested that WOX genes in the ancient clade play important roles in the process of growth and development of B. napus and its two diploid progenitors.

Expression patterns of WOXs in four major tissues of B. napus and its diploid progenitors. a The expression pattern of WOXs in B. rapa. b The expression pattern of WOXs in B. oleracea. c The expression pattern of WOXs in B. napus. The scale is FPKM
To explore whether the expression pattern of the WOX genes in the four tissues changed in allotetraploid B. napus and its two diploid progenitors, we selected the previous 33 pairs of genes that may have evolutionary relationships for analysis. As shown in Table 3, the fragments per kilobase of exon per million reads mapped (FPKM) values of the 33 gene pairs with potential direct evolutionary relationships were collected. As a result, we found that there was no direct relationship between the same gene structure and the same expression trend in these gene pairs. For example, 13 out of the 23 (approximately 56%) gene pairs with the same gene structure had absolutely different expression trends in the four tissues, such as BoWOX13a & BnCWOX13c and BoWOX4a & BnCWOX4b. However, 4 out of 10 (40%) gene pairs with distinct gene structures had the same expression trend, such as BoWOX12b & BnCWOX12a and BrWOX11b & BnAWOX11d. These results suggested that many WOX genes have no obvious changes at the DNA level, but most of the genes presented different characteristics at the expression level, which might be caused by the changes in gene expression regulation in the process of polyploidization.
Bias expression analysis of WOX genes
To explore the expression bias of WOX genes in allotetraploid B. napus in different tissues, bias analysis was performed based on FPKM. The 33 previously selected WOX gene pairs could be grouped according to homology, so there were 12 groups of WOX genes, such as WOX14, WOX3 and WUS. Since two groups of genes (WOX11 & WOX5) were present in only one diploid progenitor, the bias analysis cannot be performed on these genes. Therefore, we selected the remaining 10 groups of WOX genes for bias analysis.
The expression bias of the WOX genes showed different characteristics in different tissues. As shown in Additional file 4: Table S4, in flowers, the expression of 9 groups of WOX genes were biased towards B. rapa, and only the expression of WOX12 was biased towards B. oleracea. In leaves, the expression of 7 groups of WOX genes was biased towards B. rapa, and only the expression of WOX14 was biased towards B. oleracea; additionally, the expression of two other groups of genes (WOX6 & WOX8) had no obvious bias. In stems, the expression of 6 groups of WOX genes was biased towards B. rapa, and the expression of two groups of genes (WOX4 & WOX14) was biased towards B. oleracea; additionally, two groups of genes (WOX6 & WOX8) had no obvious bias. In siliques, the expression of 4 groups of WOX genes was biased towards B. rapa, while 5 groups of WOX genes were biased towards B. oleracea, and only WOX6 had no obvious bias.
Hence, the expression bias of WOX genes in stems and leaves was largely identical to each other, except that the expression of WOX4 was biased towards B. rapa in leaves, while biased towards B. oleracea in stems. The expression of WOX6 had no bias in stems, leaves and siliques but was biased towards B. rapa in flowers. The expression of WOX14 was biased towards B. oleracea in stems, leaves and siliques. Three groups of genes (WUS, WOX3 and WOX13) were biased towards B. rapa in all four tissues. In general, the results showed that the expression of WOX genes in B. napus was biased towards B. rapa in stems, leaves and flowers, while they had no obvious bias in siliques.
Discussion
As an important gene family in plants, WOX genes encode WOX proteins to regulate cell division and differentiation, thereby influencing plant growth and development [8,9,10]. Previous studies on the WOX gene family have been performed in many plants, including cotton [5], tabacco [6], maize [7], watermelon [36]. A recent study just analyzed the WOX gene family and their stress- and hormone-responsive patterns in B. napus [37], our study focused more on the changes in WOX genes during the polyploidization process. Compared to their study [37], less but more accurate WOX genes were identified in B. napus and its diploid progenitors in current study. In addition, B. rapa, B. oleracea and B. napus were a nice group to explore polyploidization-related issues in Brassica, but current studies on polyploidization mainly focused on comparative analysis of transcriptomes [38]. Thus, it was necessary to identify the WOX gene family completely, analyze the changes in this gene family during polyploidization and provide a reference at the genome level for follow-up polyploidization studies. Hence, we identified and analyzed the WOX gene family in B. napus and its diploid progenitors (B. rapa and B. oleracea) in this study, with the aim of understanding the evolution of this gene family during the natural polyploidization of allotetraploid B. napus.
The WOX gene family expanded in B. napus
The allotetraploid species B. napus is an important global oil crop producing biofuels and industrial compounds [39]. B. napus was formed ~ 7500 years ago by allopolyploidy, a process in which hybridization happened between ancestors B. rapa (Asian cabbage or turnip) and B. oleracea (Mediterranean cabbage), and then the chromosomes were doubled [27].
In the present study, a total of 52 WOX genes were identified in B. napus, which is the largest number reported in plants to date [5,6,7, 40]. The quantitative advantage demonstrated that the WOX gene family of B. napus has undergone significant expansion during the formation of this species. Each Brassicaceae genome underwent a whole-genome duplication (WGD) event ~ 35 MYA ago [41, 42]. Additionally, after their divergence from the Arabidopsis lineage, the Brassica genome experienced a Brassica-lineage-specific WGT event ~ 15.9 MYA [26, 43]. Therefore, it is apparent that the Brassica genome underwent paleopolyploidization [42, 43]. Polyploidization is the leading cause of the increase in the number of WOX genes. The polyploidization is a significant event in the evolution of flowering plants, which may play an important part in the adaptation of plants to new living environments [44]. In addition, segmental or tandem duplication also gives rise to an increase in the number of genes [5, 45, 46]. Segmented duplication often occurs in plants because most plants have undergone polyploidization events and thus retain a large number of duplicated chromosomal blocks in their genome [45]. For example, segmental duplication is the primary force for Hsf gene expansion in sesame [47]. In our study, 30 out of 52 WOX genes were involved in segmental duplication, that is, approximately 58% of the WOX genes have experienced segmental duplication events, suggesting that this event played an important role in the expansion of the WOX gene family. Tandem duplicated genes were defined as an array of at least two homologous genes within 100 kb [48]. Tandem duplication provides a means to amplify important adaptive resistance genes [46]. Unfortunately, we did not find any tandem duplicated genes in this study, but it is undeniable that it is still a basic factor for gene expansion. Additionally, abundant TEs can also lead to the expansion of the WOX gene family. Studies have shown that the proliferation of TEs leads to genome expansion in Corydoradinae catfishes [49]. In this study, 402, 202 and 235 TEs were found near WOX genes in B. napus, B. rapa and B. oleracea, respectively. Compared with a previous study [5], the numbers of TEs were very large, so the conclusion can be drawn that the WOX gene family has undergone significant expansion due to the presence of abundant TEs. After gene duplication, the new gene would be redundant with the pre-existing gene, and this redundancy has been considered an essential driving force in the process of evolutionary innovation [46]. At present, some models can be used to understand gene duplication events, such as neofunctionalization and DDC subfunctionalization, which provide a novel theoretical framework for further study of this process [46].
Gene loss events occurred in the WOX gene family of B. napus
Gene loss always occurs when the genomic sequence rearranges after hybridization and chromosome doubling [50]. Although the number of WOX genes in B. napus is very large, gene loss also occurred during polyploidization, which was determined from the comparison of the number of WOX genes in B. napus (52) with the sum of the gene numbers in its two progenitors (54).
B. napus, a typical allotetraploid, is an ideal model to study natural polyploidy [27]. The ancestor of Brassica underwent a WGT event after separation from Arabidopsis, so theoretically, the genes in Arabidopsis should have three homologous genes in diploid Brassica. However, in this study, only two of 15 and three of 15 AtWOX genes had three orthologous genes in the B. rapa and B. oleracea genomes, respectively. Furthermore, nine of 15 AtWOX genes had two orthologous genes in both the B. rapa and B. oleracea genomes. The remaining genes had zero or one orthologous gene in the B. rapa and B. oleracea genomes. Hence, the conclusion can be drawn that most WOX genes experienced gene loss after the noted WGT event in the formation of B. rapa and B. oleracea. Furthermore, B. napus was formed by natural hybridization and polyploidization of B. rapa and B. oleracea. Fourteen of fifteen AtWOX genes had less than six orthologous genes in the B. napus genome. However, only one AtWOX gene had six orthologous genes in B. napus, and these six genes were evenly distributed in the An and Cn subgenomes, which indicated that most WOX genes also experienced gene loss during the formation of B. napus.
Although gene loss events have happened in B. napus and its diploid progenitors, B. napus still had more WOX genes in its genome compared to the allotetraploid Nicotiana tabacum [6] and Gossypium hirsutum [5].
The WOX gene family is highly conserved at the DNA and protein level in B. napus
WOX proteins belong to a plant-specific branch in the superfamily of HB transcription factors [2]. Previous research has found that the HB domain may have already diverged before the separation of animals, plants and fungi [51]. The HB domain of the WOX proteins contains one loop, one turn and three helix structures [5]. It has been reported that the HB domain of the WOX genes contains some highly conserved amino acids in these three helix structures; for instance, it contains Q, L and E in helix 1, P, I and L in helix 2, and I, N, V, F, Y, W, F, Q, N and R in helix 3 [28]. Moreover, previous studies have demonstrated that the amino acids in the loop and turn structure are less conserved [5]. Evidently, our results regarding the HB domain of the WOX proteins in B. napus and its diploid progenitors were consistent with the above conclusions. According to previous studies, WOX genes can be divided into three clades, namely, the ancient clade, intermediate clade and WUS clade [5, 6], as was observed in our present study (Fig. 1).
After analysis, WOX genes, whether in allotetraploid B. napus or its diploid progenitors, were highly conserved at the DNA and protein levels, including the gene structure of WOX genes, conserved amino acids in the HB domain, and types of motifs. Specifically, it was calculated that at least 70% of WOX genes maintained the same gene structure during the formation of allotetraploid B. napus. Furthermore, according to our statistical analysis, 67% of WOX genes maintained their relative positions on the chromosomes during evolution. In summary, WOX genes were highly conserved at the DNA level during the polyploidization process from diploid to allotetraploid B. napus.
It is generally believed that B. napus (2n = 4x = 38, AnCn) was reunited by the hybridization of an A-genome material B. rapa (2n = 20, Ar) with a C-genome material B. oleracea (2n = 18, Co), followed by a chromosome doubling event [27]. Our study showed that the distribution features of the WOX genes on four genomes/subgenomes (Ar, Co, An and Cn) were roughly consistent with the above hypothesis, which confirmed that WOX genes in B. napus were highly conserved during hybridization and polyploidization.
The expression features of the WOX gene family were changed in B. napus compared with its diploid progenitors
A previous study has shown that some WOX genes are expressed at low levels in reproductive tissues and are not expressed in their vegetative tissues in cotton [5]. In our study, most of the WOX genes were also expressed at low levels or even were not expressed in the four tested tissues. The reason for this might be that the WOX genes are often expressed at some restricted locations, such as embryos or quiescent centers in roots. Segmental duplication is an important way to increase diversity at the DNA level. After duplication, genes may still function as before or may acquire only part of the function of the previous gene, namely, subfunctionalization; moreover, it is also possible for genes to obtain new functions, namely, neofunctionalization [52]. In B. napus, the differential expression pattern of these duplicated genes indicated that they might have undergone functional divergence after duplication. For example, the expression pattern of BnCWOX13e, which is the duplicated gene of BnCWOX13a, is completely different from that of BnCWOX13a, which indicated that BnCWOX13e might only have acquired some of the functions of the original gene or might have acquired new functions. Of course, there are still some duplicated gene pairs that maintain the previous expression patterns, such as BnAWOX4b & BnCWOX4b, and BnCWOX3a & BnAWOX3c. These genes may have maintained their previous functions.
Furthermore, the expression characteristics of some WOX genes suggested that their function is consistent with that of homologous genes in Arabidopsis. For example, studies have shown that WOX6 regulates the development of ovules [18], and the homologs of WOX6 expressed only in reproductive tissue (flowers) in our data, indicating they may play similar roles in B. napus and its two diploid progenitors; however, this speculation needs to be tested experimentally. Similarly, AtWOX14 was mainly involved in the lignification process in Arabidopsis [24], and we noticed that the homologs of WOX14 were not expressed in flowers, so it is speculated that WOX14 homologous genes also have a similar function to AtWOX14. Moreover, an interesting conclusion was found in our study, which was that there was no significant correlation between gene structure and expression pattern. Specifically, approximately 56% of the gene pairs in B. napus and its diploid progenitors, which were conserved at the DNA level, were changed at the expression level.
In short, our study showed that approximately 70% of the members of the WOX gene family in B. napus maintained their gene structure during the polyploidization process, but approximately 56% of them changed significantly at the expression level, revealing that the WOX gene family has changed during polyploidization.
Conclusions
In this study, 52, 25 and 29 WOX genes were identified in allotetraploid B. napus, the An genome donor B. rapa and the Cn genome donor B. oleracea, respectively. After analysis, whole genome duplication, segmental duplication and abundant TEs were determined to be the three major impetuses for the expansion of the WOX gene family during the process of polyploidization. Moreover, gene loss events have happened in the WOX gene family in B. napus during polyploidization. Additionally, the WOX gene family in B. napus was highly conserved at the DNA and protein level but changed at the expression level during polyploidization. Together, these results can increase our understanding of the evolution of the WOX gene family and provide a reference for future polyploidization analysis.
Methods
Plant materials and transcriptome sequencing
The seeds of B. napus (cv. Darmor), B. rapa (cv. Chiifu) and B. oleracea (cv. Jinzaosheng) were obtained from the Oil Crops Research Institute, Chinese Academy of Agricultural Sciences. These materials were grown in soil under natural conditions (Wuhan, China). Some well-grown inflorescences were bagged to prevent pollen contamination before flowering. Young leaves, inflorescence stems, blooming flowers and siliques (10 DAP) from 6-month-old plants were collected and frozen immediately in liquid nitrogen for transcriptome sequencing. The platform for transcriptome sequencing was Illumina (HiSeq X-Ten).
Identification of WOX genes
The WOX genes in allotetraploid B. napus and its diploid progenitors (B. rapa and B. oleracea) were comprehensively identified by three methods. The first method is the BLASTp search method. Fifteen protein sequences encoded by WOX genes in A. thaliana were acquired from the TAIR database (http://www.arabidopsis.org/), which were used as query sequences to perform the BLASTp search (E-value <1e-5) with all protein sequences of the three selected Brassica species, using the BRAD database (http://brassicadB.org/brad/) [28]. Then, the repeated genes were removed manually, and original candidate genes were obtained. The second method is searching syntenic genes in the BRAD database by inputting the 15 WOX gene IDs of A. thaliana. The third method is used to screen the candidate genes accurately by searching for the conserved HB domain of the corresponding proteins. Three public databases, including NCBI Conserved Domain Database (CDD; https://www.ncbi.nlm.nih.gov/cdd) [53], SMART database (http://smart.embl-heidelberg.de/) [54], and Pfam database (http://pfam.xfam.org/) [55], were used to search the HB domain of candidate sequences, and the domain ID is PF00046, SM000389 and PF00046 in each database, respectively. Sequences not containing the complete conserved HB domain were removed. Finally, all WOX genes were identified in B. napus and its diploid progenitors in currently available data, and the identified genes were named according to the homologous relationship with WOX genes in A. thaliana. The coding sequences of the identified WOX genes and the amino acid sequences of the corresponding WOX proteins in the three selected Brassica species were acquired from the BRAD database. WOX genes of rice were identified using the first and the last methods mentioned above, and all information regarding rice was taken from the open rice database (http://rice.plantbiology.msu.edu/) [56].
Chromosome location and structure of WOX genes
Information about the physical locations of WOX genes in the genomes of B. napus and its diploid progenitors was collected from the BRAD database, and their positions were drafted to chromosomes by using MapInspector software. The structures of the WOX genes were displayed using the gene structure display server 2.0 (GSDS 2.0; http://gsds.cbi.pku.edu.cn//index.php) [57] program to obtain their exon/intron composition information.
Conserved motifs and HB domain analysis of WOX proteins
The conserved motifs were investigated using the MEME tool (http://meme-suite.org/tools/meme) [58], with the number of found motifs as nine and the other parameters as default values. The location diagram of the HB domain of the WOX genes was drawn using IBS 1.0.3 software (http://ibs.biocuckoo.org/download.php) [59], according to the related information from the SMART database. For conserved sequence logo analysis, the conserved HB domain sequences of WOX genes from B. rapa, B. oleracea, B. napus and Arabidopsis were aligned by ClustalX 2.1 [60], and the multiple alignment results were submitted to an online tool, WEBLOGO (http://weblogo.berkeley.edu/logo.cgi) [61], for generating the logos.
Characteristics and phylogenetic relationship analysis of WOX proteins
The WOX protein sequences in B. napus and its diploid progenitors were analyzed for physical and chemical characteristics by the online ProtParam tool of ExPASy (http://weB.expasy.org/protparam/) [62], including the number of amino acids, MW, theoretical pI, II, and GRAVY. The full-length WOX proteins in B. rapa, B. oleracea, B. napus, A. thaliana and rice were aligned using ClustalW. Phylogenetic analysis of WOX proteins was performed using MEGA 7.0.26 [63] with the neighbor-joining (NJ) method based on the Poisson model, and the bootstrap method was used to test the tree with 1000 replicates. An online website iTOL (Interactive Tree of Life, http://itol.embl.de/) [64] was used to annotate the tree.
Duplications and syntenic analysis of WOX genes
Two methods were used to identify the duplicated WOX genes in B. napus and its diploid progenitors. First, the BLASTn program was used with both coverage and identity > 80% were considered as candidate duplicated genes [32]. Second, gene IDs were inputted into the BRAD database to search for their syntenic genes [33]. The common duplicated genes identified by the two methods were considered as the final duplicated genes. DNAsp5 software [65] was applied to calculate the Ks, Ka and evolutionary constraint (Ka/Ks) between the duplicated WOX gene pairs. Then, Ks values were used to estimate the divergence events, and the divergence time of duplicated genes was calculated using the formula T = Ks/2r Mya (Millions of years), where ‘r’ is equal to 1.5 × 10− 8 according to a previous study [66]. The syntenic genes of WOX genes in B. napus and its diploid progenitors were searched on the BRAD database, and then Circos software [67] was used to show the syntenic relationship between them.
Analysis of transposable elements near WOX genes
Methods for detecting TEs include de novo prediction, the homology-based method, the structure-based method, and the comparative genomic method, among which the most common method is the homology-based method [68], which is based on detecting homology to known TEs. To understand whether TEs play roles in the expansion of the WOX gene family, we detected TEs 2000 bp upstream and downstream of WOX genes based on the homology-based method [5]. TEs were identified using the Repeat Masking tool in the known TEs database Repbase (https://www.girinst.org/censor/index.php) [69, 70]. Finally, we calculated the total number and number of various types of TEs present near WOX genes in B. napus and its diploid progenitors.
Transcriptome data analysis and gene expression heatmap of WOX genes
The Illumina RNA-seq data were analyzed to reveal the expression patterns of WOX genes in B. napus and its diploid progenitors. The raw data of RNA-seq reads were deposited in the NCBI database under accession number (SRR7816633-SRR7816668). FPKM values were used to represent the gene expression levels. The heatmap of the expression patterns of WOX genes was generated by MutiExperimentViewer (MeV; version 4.8.1) software.
Abbreviations
- A. thaliana :
-
Arabidopsis thaliana
- B. napus :
-
Brassica napus
- B. oleracea :
-
Brassica oleracea
- B. rapa :
-
Brassica rapa
- BLAST:
-
Basic Local Alignment Search Tool
- FPKM:
-
Fragments per kilobase million
- GA:
-
Gibberellin
- GRAVY:
-
Grand average of hydropathicity
- HB:
-
Homeobox
- HEs:
-
Homologous exchanges
- II:
-
Instability index
- Ka:
-
Non-synonymous substitution
- Ks:
-
Synonymous substitution
- MW:
-
Molecular weight
- MYA:
-
Million years ago
- NJ:
-
Neighbor-joining
- TEs:
-
Transposable elements
- WGD:
-
Whole-genome duplication
- WGT:
-
Whole-genome triplication
- WOX :
-
WUSCHEL-related homeobox
References
Gehring WJ, Affolter M, Burglin T. Homeodomain proteins. Annu Rev Biochem. 1994;63:487–526.
van der Graaff E, Laux T, Rensing SA. The WUS homeobox-containing (WOX) protein family. Genome Biol. 2009;10:248.
Lian G, Ding Z, Wang Q, Zhang D, Xu J. Origins and evolution of WUSCHEL-related homeobox protein family in plant kingdom. Sci World J. 2014;2014:534140.
Mukherjee K, Brocchieri L, Burglin TR. A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol Biol Evol. 2009;26:2775–94.
Yang Z, Gong Q, Qin W, Yang Z, Cheng Y, Lu L, et al. Genome-wide analysis of WOX genes in upland cotton and their expression pattern under different stresses. BMC Plant Biol. 2017;17:113.
Zhou X, Guo Y, Zhao P, Sun MX. Comparative analysis of WUSCHEL-related homeobox genes revealed their parent-of-origin and cell type-specific expression pattern during early embryogenesis in tobacco. Front Plant Sci. 2018;9:311.
Zhang X, Zong J, Liu J, Yin J, Zhang D. Genome-wide analysis of WOX gene family in rice, sorghum, maize, Arabidopsis and poplar. J Integr Plant Biol. 2010;52:1016–26.
Dolzblasz A, Nardmann J, Clerici E, Causier B, van der Graaff E, Chen J, et al. Stem cell regulation by Arabidopsis WOX genes. Mol Plant. 2016;9:1028–39.
Breuninger H, Rikirsch E, Hermann M, Ueda M, Laux T. Differential expression of WOX genes mediates apical-basal axis formation in the Arabidopsis embryo. Dev Cell. 2008;14:867–76.
Costanzo E, Trehin C, Vandenbussche M. The role of WOX genes in flower development. Ann Bot. 2014;114:1545–53.
Laux T, Mayer KF, Berger J, Jurgens G. The WUSCHEL gene is required for shoot and floral meristem integrity in Arabidopsis. Development. 1996;122:87–96.
Zhang Y, Wu R, Qin G, Chen Z, Gu H, Qu LJ. Over-expression of WOX1 leads to defects in meristem development and polyamine homeostasis in Arabidopsis. J Integr Plant Biol. 2011;53:493–506.
Haecker A, Gross-Hardt R, Geiges B, Sarkar A, Breuninger H, Herrmann M, et al. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development. 2004;131:657–68.
Ueda M, Zhang Z, Laux T. Transcriptional activation of Arabidopsis axis patterning genes WOX8/9 links zygote polarity to embryo development. Dev Cell. 2011;20:264–70.
Shimizu R, Ji J, Kelsey E, Ohtsu K, Schnable PS, Scanlon MJ. Tissue specificity and evolution of meristematic WOX3 function. Plant Physiol. 2009;149:841–50.
Suer S, Agusti J, Sanchez P, Schwarz M, Greb T. WOX4 imparts auxin responsiveness to cambium cells in Arabidopsis. Plant Cell. 2011;23:3247–59.
Gonzali S, Novi G, Loreti E, Paolicchi F, Poggi A, Alpi A, et al. A turanose-insensitive mutant suggests a role for WOX5 in auxin homeostasis in Arabidopsis thaliana. Plant J. 2005;44:633–45.
Park SO, Zheng Z, Oppenheimer DG, Hauser BA. The PRETTY FEW SEEDS2 gene encodes an Arabidopsis homeodomain protein that regulates ovule development. Development. 2005;132:841–9.
Kong D, Hao Y, Cui H. The WUSCHEL related Homeobox protein WOX7 regulates the sugar response of lateral root development in Arabidopsis thaliana. Mol Plant. 2016;9:261–70.
Wu X, Dabi T, Weigel D. Requirement of homeobox gene STIMPY/WOX9 for Arabidopsis meristem growth and maintenance. Curr Biol. 2005;15:436–40.
Baesso B, Chiatante D, Terzaghi M, Zenga D, Nieminen K, Mahonen AP, et al. Transcription factors PRE3 and WOX11 are involved in the formation of new lateral roots from secondary growth taproot in A. thaliana. Plant Biol. 2018;20:426–32.
Liu J, Sheng L, Xu Y, Li J, Yang Z, Huang H, et al. WOX11 and 12 are involved in the first-step cell fate transition during de novo root organogenesis in Arabidopsis. Plant Cell. 2014;26:1081–93.
Romera-Branchat M, Ripoll JJ, Yanofsky MF, Pelaz S. The WOX13 homeobox gene promotes replum formation in the Arabidopsis thaliana fruit. Plant J. 2013;73:37–49.
Denis E, Kbiri N, Mary V, Claisse G, Conde ESN, Kreis M, et al. WOX14 promotes bioactive gibberellin synthesis and vascular cell differentiation in Arabidopsis. Plant J. 2017;90:560–72.
Liu S, Liu Y, Yang X, Tong C, Edwards D, Parkin IA, et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun. 2014;5:3930.
Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 2011;43:1035–9.
Chalhoub B, Denoeud F, Liu S, Parkin IA, Tang H, Wang X, et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345:950–3.
Cheng F, Liu S, Wu J, Fang L, Sun S, Liu B, et al. BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 2011;11:136.
Ma J, Yang Y, Luo W, Yang C, Ding P, Liu Y, et al. Genome-wide identification and analysis of the MADS-box gene family in bread wheat (Triticum aestivum L.). PLoS One. 2017;12:e0181443.
McClintock B. The discovery and characterization of transposable elements. The collected papers of Barbara McClintock. New York: Garland; 1987.
Wu Y, Ke Y, Wen J, Guo P, Ran F, Wang M, et al. Evolution and expression analyses of the MADS-box gene family in Brassica napus. PLoS One. 2018;13:e0200762.
Kong X, Lv W, Jiang S, Zhang D, Cai G, Pan J, et al. Genome-wide identification and expression analysis of calcium-dependent protein kinase in maize. BMC Genomics. 2013;14:433.
Yan C, Duan W, Lyu S, Li Y, Hou X. Genome-wide identification, evolution, and expression analysis of the ATP-binding cassette transporter gene family in Brassica rapa. Front Plant Sci. 2017;8:349.
Tang J, Wang F, Hou XL, Wang Z, Huang ZN. Genome-wide fractionation and identification of WRKY transcription factors in Chinese cabbage (Brassica rapa ssp. pekinensis) reveals collinearity and their expression patterns under abiotic and biotic stresses. Plant Mol Biol Report. 2014;32:781–95.
Oliver KR, McComb JA, Greene WK. Transposable elements: powerful contributors to angiosperm evolution and diversity. Genome Biol Evol. 2013;5:1886–901.
Zhang N, Huang X, Bao Y, Wang B, Liu L, Dai L, et al. Genome-wide identification and expression profiling of WUSCHEL-related homeobox (WOX) genes during adventitious shoot regeneration of watermelon (Citrullus lanatus). Acta Physiol Plant. 2015;37:224.
Wang MM, Liu MM, Ran F, Guo PC, Ke YZ, Wu YW, et al. Global analysis of WOX transcription factor gene family in Brassica napus reveals their stress- and hormone-responsive patterns. Int J Mol Sci. 2018;19:3470.
Wang R, Zou J, Meng J, Wang J. Integrative analysis of genome-wide lncRNA and mRNA expression in newly synthesized Brassica hexaploids. Ecol Evol. 2018;8:6034–52.
Allender CJ, King GJ. Origins of the amphiploid species Brassica napus L. investigated by chloroplast and nuclear molecular markers. BMC Plant Biol. 2010;10:54.
Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017;45:D1040–d1045.
Bowers JE, Chapman BA, Rong J, Paterson AH. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003;422:433–8.
Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473:97–100.
Lysak MA, Koch MA, Pecinka A, Schubert I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 2005;15:516–25.
Ramsey J, Schemske DW. Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu Rev Ecol Syst. 1998;29:467–501.
Cannon SB, Mitra A, Baumgarten A, Young ND, May G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;4:10.
Flagel LE, Wendel JF. Gene duplication and evolutionary novelty in plants. New Phytol. 2009;183:557–64.
Dossa K, Diouf D, Cisse N. Genome-wide investigation of Hsf genes in sesame reveals their segmental duplication expansion and their active role in drought stress response. Front Plant Sci. 2016;7:1522.
Kayum MA, Park JI, Nath UK, Saha G, Biswas MK, Kim HT, et al. Genome-wide characterization and expression profiling of PDI family gene reveals function as abiotic and biotic stress tolerance in Chinese cabbage (Brassica rapa ssp. pekinensis). BMC Genomics. 2017;18:885.
Marburger S, Alexandrou MA, Taggart JB. Whole genome duplication and transposable element proliferation drive genome expansion in Corydoradinae catfishes. Proc Biol Sci. 2018;285:20172732.
Paterson AH, Bowers JE, Chapman BA. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc Natl Acad Sci U S A. 2004;101:9903–8.
Bharathan G, Janssen BJ, Kellogg EA, Sinha N. Did homeodomain proteins duplicate before the origin of angiosperms, fungi, and metazoa? Proc Natl Acad Sci U S A. 1997;94:13749–53.
Jain M, Tyagi AK, Khurana JP. Genome-wide identification, classification, evolutionary expansion and expression analyses of homeobox genes in rice. FEBS J. 2008;275:2845–61.
Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45:D200–d203.
Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2015;43:D257–60.
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–85.
Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6:4.
Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31:1296–7.
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME suite: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–8.
Liu W, Xie Y, Ma J, Luo X, Nie P, Zuo Z, et al. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31:3359–61.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82.
Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90.
Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, et al. ExPASy: SIB bioinformatics. Resource portal. Nucleic Acids Res. 2012;40:W597–603.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Koch MA, Haubold B, Mitchell-Olds T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol Biol Evol. 2000;17:1483–98.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Bergman CM, Quesneville H. Discovering and detecting transposable elements in genome sequences. Brief Bioinform. 2007;8:382–92.
Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–20.
Nansheng C. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2004;5:4.10.11–14.10.14.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (31570539, 31370258). The funding body didn’t participate in the design of the study, collection, analysis and interpretation of data or in writing the manuscript.
Availability of data and materials
All data generated or analysed during this study were included in this published article and the additional files.
Author information
Authors and Affiliations
Contributions
JW and ML conceived and designed the study. ML performed the bioinformatics analyses. ML wrote the manuscript. XW provided the experimental materials. ML, RW and ZL were responsible for planting materials. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. The characteristics of WOXs in B. napus and its diploid progenitors with their Arabidopsis orthologs. (DOCX 32 kb)
Additional file 2:
Table S2. Estimated Ka/Ks ratios of duplicated WOX gene pairs in B. napus and its diploid progenitors. (DOCX 25 kb)
Additional file 3:
Table S3. The FPKM values of all WOX genes of B. napus and its diploid progenitors (B. rapa and B. oleracea) in four tissues. (XLSX 21 kb)
Additional file 4:
Table S4. The |log2FC| of WOX genes in four tissues. (XLSX 10 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, M., Wang, R., Liu, Z. et al. Genome-wide identification and analysis of the WUSCHEL-related homeobox (WOX) gene family in allotetraploid Brassica napus reveals changes in WOX genes during polyploidization. BMC Genomics 20, 317 (2019). https://doi.org/10.1186/s12864-019-5684-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5684-3