
Abstract
Background
RNA sequencing is a widely used technology for differential expression analysis. However, the RNA-Seq do not provide accurate absolute measurements and the results can be different for each pipeline used. The major problem in statistical analysis of RNA-Seq and in the omics data in general, is the small sample size with respect to the large number of variables. In addition, experimental design must be taken into account and few tools consider it.
Results
We propose OMICfpp, a method for the statistical analysis of RNA-Seq paired design data. First, we obtain a p-value for each case-control pair using a binomial test. These p-values are aggregated using an ordered weighted average (OWA) with a given orness previously chosen. The aggregated p-value from the original data is compared with the aggregated p-value obtained using the same method applied to random pairs. These new pairs are generated using between-pairs and complete randomization distributions. This randomization p-value is used as a raw p-value to test the differential expression of each gene. The OMICfpp method is evaluated using public data sets of 68 sample pairs from patients with colorectal cancer. We validate our results through bibliographic search of the reported genes and using simulated data set. Furthermore, we compared our results with those obtained by the methods edgeR and DESeq2 for paired samples. Finally, we propose new target genes to validate these as gene expression signatures in colorectal cancer. OMICfpp is available at http://www.uv.es/ayala/software/OMICfpp_0.2.tar.gz.
Conclusions
Our study shows that OMICfpp is an accurate method for differential expression analysis in RNA-Seq data with paired design. In addition, we propose the use of randomized p-values pattern graphic as a powerful and robust method to select the target genes for experimental validation.
Similar content being viewed by others

Background
The sequencing technologies have provided major advances in the understanding of biological mechanisms. Particularly, within these sequencing technologies, the RNA-Seq has contributed to understanding gene expression, changing our view of the transcriptome [1, 2]. The identification of differentially expressed genes, new transcripts, expressed mutations, among others, has allowed a better understanding of human diseases. New biomarkers or therapeutic targets against diseases such as cancer have been proposed using this technology [3].
However, there is no standard pipeline for the analysis of RNA-Seq data. In fact, each step of the analysis admits many options. The reads can be aligned (or mapped) using different tools. Some widely used aligners are STAR [4], Tophat [5] or Bowtie [6]. Then, the matrix of counts is obtained, i.e the estimation of RNA abundance (cDNA) by the number of aligned read over a gene or isoform. These counts can be obtained using software like HTSeq [7] or featureCounts function of the Rsubread package [8]. The differential expression analysis can be done using the widely used edgeR [9], DESeq [10], among others. Besides, the RNA-Seq data results can be different for each pipeline and it is not established which is the best analysis protocol [11].
There are (and will be) many challenges to solve in mapping, read count and statistical analysis. In this sense, the major problem in statistical analysis of RNA-Seq, and in all omics data, is the small sample size with respect to the large number of variables (genes, isoforms, exon, …). It is not rare that just a few samples determine the results i.e. a great variation accounted by a few observations. Additionally, there exists important confounding variables in the differential expression analysis. They are the library size, the gene length and others [11, 12]. It is not rare that a first differential expression analysis provides several candidate genes that are not significant in a posterior experimental validation. Thereby, the RNA-Seq do not provide accurate absolute measurements [12]. In order to solved it, new methods for RNA-Seq data analysis have been developed [13, 14].
In this paper, we propose a new method for the differential expression RNA-Seq analysis with paired design. Our approach proposes to compare the counts within each pair by taking into account library sizes [15]. The p-values for all pairs corresponding to a given gene are aggregated using ordered weighted averages [16]. This aggregated value will quantify the phenotype-expression association from the gene expression profile. These values are used to test differential expression using randomization distributions. Our approach is compared with edgeR [9] and DESeq2 [17] methods for paired samples.
The methodology have been tested using a 68 pairs data set from patients with colorectal cancer. Of these, 50 are obtained from The Cancer Genome Atlas (TCGA) [18] and 18 from PRJNA218851 BioProject [19, 20].
Each pair is composed with a sample from solid tumor and adjacent normal tissue from the same individual. The new methodology has been implemented in the R package OMICfpp and is available at http://www.uv.es/ayala/software/OMICfpp_0.2.tar.gz.
Methods
Data
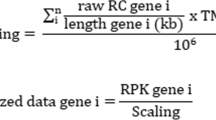
A colorectal cancer paired data set of 50 patients (tumor and normal adjacent tissue) were downloaded from TCGA [18] using gdc-client tool. In addition, a colorectal cancer data set of 18 pairs of samples were downloaded from SRA, PRJNA218851 BioProject [19] using the SRA toolkit [20]. The quality control of the PRJNA218851 raw dataset was checked using the FASTQC tool and low quality reads were discarded using fastx-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Later, the reads were mapped using STAR with the GRCh38 human genome as the reference one. Then the SAM files were converted to sorted BAM files using Samtools [21]. Finally, the count matrix was generated using the summarizeOverlaps function of GenomicAlignments R package [22]. At this point, we have the counts of both data sets (PRJNA218851 and TCGA), so they are included in a single matrix using SummarizedExperiment R package [23]. A detailed description can be found in the Additional file 1: Methods.
OMICfpp methodology
The major problem in statistical analysis of omics data is the small sample size with respect to the large number of variables (genes, exons, locii, …). From an statistical point of view we are dealing with counts and covariables describing the samples i.e. a count response model is the suitable approach. These models are part of the generalized linear models and should be the natural approach. However, the small sample sizes do make it more difficult to apply such kind of models. In this paper, we propose a method for RNA-Seq data in paired designs where we tackle the issue of small sample.
In our approach, a p-value for each case-control pair is obtained, using a binomial test. These p-values are aggregated using an ordered weighted average (OWA) with a given orness previously chosen by the user or using the chooseOrness function (from the package OMICfpp) for the automatic orness choice. The aggregated p-value from the original data is compared with the aggregated p-value obtained using the same method applied to random pairs. These new pairs are generated using a randomization distribution (“Randomization distributions” section). This randomization p-value is used as a raw p-value to test the differential expression of each gene (“Marginal gene analysis” section). Figure 1a displays the outline of our approach. A detailed software implementation is contained in Additional file 1: Methods.

OMICfpp method. a) Workflow used by OMICfpp paired data analysis. The 68 paired RNA-Seq data from TCGA and PRJNA218851 BioProject were analyzed by our proposed method OMICfpp and by conventional methods edgeR and DESEq2. In the OMICfpp approach, original and randomized p-values are obtained for each paired data, applying different randomization distributions. The p-values must be aggregated using the OWA to obtain a single value per gene. The user decides, by choosing an orness, the weights assigned to the genes. Finally, a marginal gene analysis is performed and a list of genes ordered by importance according to the assigned weights is obtained. These results are compared with those obtained using edgeR. b) IL11, c) HIST2H3C and d) AC012414.3 are examples of genes with area under the cumulative distribution function, respectively. Top-left, the kernel density estimator corresponding to the original p-values of the binomial test; top-right: the corresponding cumulative distribution function of these original p-values; bottom-left: the between-pair p-values corresponding to all the values of orness used in the study; bottom-right, the complete p-values corresponding to all orness. e) Optimal orness by comparing n0 extreme genes. f) Proportion of significant genes for different α values obtained using the complete distribution. g) Density function used in “Results” section to calculate the score of Eq. 3
Randomization distributions
The data are paired samples. It will be denoted as (yi1,yi2) the i-th pair of counts for a given gene. The whole expression profile would be (yi1,yi2) with i=1,…,n with 2n samples and N genes. We are going to consider different randomization distributions.
-
Between-pairs. The first element of each pair is maintained as the original one. The second element of each pair is obtained permuting the second components of all pairs between them. We have (yi,1,yγ(i),2) for i=1,…,n where γ is now a permutation of (1,…,n). The number of possible permutations is n!.
-
Complete. Let us choose I={i1,…,in} a random subset of {1,…,2n}. The indices of {1,…,2n} not in {i1,…,in} can be denoted J={j1,…,jn}. A random correspondence between I and J will produce the pairs. Cases can be considered controls and the pairs are randomly assigned too. The number of possible values is \(\frac {(2n)!}{n!}\).
From now on, they will be named between-pair and complete distributions. Let (y1,y2) be a pair of counts to be compared and (m1,m2) the corresponding library sizes. A simple approach to compare the counts by taking into account the library sizes was proposed in [15]. In fact, assuming given the total number of counts per gene and the library sizes, we can test the null hypothesis Hi:pi1=m1/(m1+m2) against Hi:pi1≠m1/(m1+m2) where pi1 is the proportion of the i-th gene in the first sample. Under the null hypothesis, the statistic Yi1 follows a binomial distribution with Yi1+Yi2 trials and the success probability m1/(m1+m2). Note that the null distribution assume that the (random) value of Yi1+Yi2 is given.
Other testing procedures for this null hypothesis could be used and incorporated in our approach. For a given statistical test and for the i-th gene we will have \(\phantom {\dot {i}\!}(t_{i1},\ldots,t_{i_{n}})\) where tij is the statistic or p-value obtained in the j-th test. It is well known that a few pairs could produce extreme values of these statistics. The simplest approach could be to aggregate the values \(\phantom {\dot {i}\!}(t_{i1},\ldots,t_{i_{n}})\) using the mean or a median. In our opinion, a more general and really interesting point of view is to use ordered weighted averages (in short, OWA) [16].
Let us remember this aggregation operators. Let a=(a1,…,an) be the column vector of values aggregated and a′ is the transpose of the column vector a. Let \(\phantom {\dot {i}\!}\boldsymbol {a}_{r} = (a_{r_{1}},\ldots,a_{r_{n}})'\) be the ordered version a i.e. \(a_{r_{1}} \geq \ldots \geq a_{r_{n}}\). An ordered weighted average (OWA) operator of dimension n is a mapping \(f: {\mathbb R}^{n} \rightarrow {\mathbb R}\) with an associated weighting vector w=(w1,…,wn) such that \({\sum \nolimits }_{j=1}^{n} w_{j} =1\) and where \(f(a_{1},\ldots,a_{n})={\sum \nolimits }_{j=1}^{n} w_{j} a_{r_{j}} = \boldsymbol {w}' \boldsymbol {a}_{r}\). The particular cases shown in Table 1 can better illustrate the idea underlying OWA operators.
In this paper we have used the weights proposed in [24]. The method uses, for an orness δ, the probability function of a binomial distribution with n−1 trials and success probability 1−δ: \(w_{i} = \binom {n-1}{i-1} (1-\delta)^{i-1} \delta ^{n-i}\) for i=1,…,n. No weight is associated with any particular input. The relative magnitude of the input decides which weight corresponds to each input. We have chosen this approach with the following problem in mind. A major problem with paired RNA-Seq counts is that just a single pair of samples is responsible for the global observed difference or global effect. The whole pair or just an element of the pair could be an outlier or a real observation. The OWA operator permit us to control the influence of a particular pair. Each pair is marginally evaluated and the obtained statistics (p-values) are aggregated by taking into account their ordered values.
The OWA operators are bounded by the maximum and minimum operator. Yager [16] introduced a measure called orness to characterize the degree to which the aggregation is like an or (max) operation:
Note that orness((1,0,…,0))=1, orness((0,0,…,1))=0 and \(\text {orness} \left ((\frac {1}{n},\frac {1}{n}, \ldots,\frac {1}{n}) \right)= 0.5\).
Up to now the OWA has been presented using the usual decreasing ordering. If the original values are increasingly ordered then the interpretation change. In our experiment we will aggregate p-values and these p-values will be increasingly ordered per gene, from the most significant pair (lowest p-value) to the less significant pair (highest p-value). An orness near 1 corresponds to the minimum of the p-values and an orness near 0 corresponds with the maximum of the p-values. Thus, an orness close to one uses the most significant pairs and an orness close to zero will use the less significant pairs. So, when the orness goes from 0 to 1, we are going from the maximum to the minimum of the p-values.
Marginal gene analysis
The original pairs for a given gene are \(\left (y_{i1}^{(0)},y_{i2}^{(0)}\right)\) for i=1,…,n. First, we choose a given orness δ and calculate the weights w. Second, we choose a test to compare both counts, between-pair or complete. Third, we choose a randomization distribution and generates B realizations using it being \(\left (y^{(b)}_{i1},y^{(b)}_{i2}\right)\) (with i=1,…,n) the b-th realization generated. The statistics observed (for the n comparisons) corresponding to the b-th realization generated will be \(\boldsymbol {t}^{(b)} = \left (t_{1}^{(b)},\ldots,t_{n}^{(b)}\right)\) where b=0 corresponds with the original data. The corresponding p-values under the null hypothesis of no association with the phenotype would be \(\boldsymbol {p}^{(b)} = \left (p_{1}^{(b)},\ldots,p_{n}^{(b)}\right)\). Fourth, we aggregate the generated p-values using an ordered weighted average. The b-th aggregated value will be \(v_{b} = {\sum \nolimits }_{j=1}^{n} w_{j} p_{r_{j}}^{(b)} = \boldsymbol {w}' \boldsymbol {p}_{r}^{(b)}\).
Under the null distribution (any of them) the value v0 is like v1,…,vB and any possible ordering of the vector (v0,v1,…,vB) has the same probability. If a one-tail test is used where low values correspond to the alternative hypothesis then the randomization p-value is given by
where |·| denotes the number of elements. This p-value measures how extreme is v0 with respect to the others vbs and depends on the δ-orness used and the randomization distribution chosen. From now on, it will be denoted pb(δ) and pc(δ) for the between and complete distributions and a δ orness.
The between-pair p-values are evaluating the pair (or sample) factor i.e. we are looking for if there is a pair effect. Different orness will permit us to focus over a certain number of pairs from the lowest to the highest significant pairs. We are going to comment some genes in order to understand the utility of these p-values. We think that their interest is not just to declare a gene as significant or non significant. They shows a wider evaluation of the differential expression of the gene with respect to the pair effect (possibly outlier pairs) when the between-pair distribution is used and the condition effect (control vs cases) when the complete distribution is evaluated.
We have chosen three genes of the data used in section “Results” corresponding to extreme cases. Figures 1b, c and d shows a simple graphical description of the different p-values used in our approach. The top-left plot shows a kernel density estimator of the raw p-values corresponding to the original pairs. The top-right plot shows the empirical cumulative distribution function of these raw p-values. The bottom-left (respectively bottom-right) plot shows the between-pair (respectively complete) p-values for the different values of orness.
The first gene, Fig. 1b is a significant one with low p-values for all pairs. No outlier pair i.e. no pair with a clearly different p-value with respect to the other pairs. This can be seen in the plot bottom-left where the pb is horizontal. The bottom-right shows the gene is considered as significant using any orness.
The second and third genes, Fig. 1c and d are non significant genes, for all orness in all samples Fig. 1c and for some orness Fig. 1d. The gene in Fig. 1d has the highest area under the cumulative distribution function of the original p-values. The mass probability is close to one. It is clear in the cumulative distribution function almost null along the whole unit interval.
Differential expression using edgeR and DESeq2
In order to compare our results with the most used methodologies for differential expression analysis, we analyzed the data using the Bioconductor packages edgeR [9] and DESeq2 [17]. The Additional file 1: Methods contains the code and further details in order to reproduce these studies.
Results
Choosing an orness
Many possible methods could be proposed for the orness choice. We have implemented the following procedure where no prior knowledge of the user is assumed. It is a non supervised method. First, an small number of simulations is performed and the randomization p-values per gene corresponding to a set of orness values are calculated. For instance, we can take ten simulations and orness from 0.01 to 0.99 with a grid of 50 points. For each orness, we evaluate the mean of the largest n0p-values and the mean of the lowest n0p-values. We choose the orness corresponding to the largest difference between them i.e. the orness where the significant and non significant genes are more clearly distinguished. This is evaluated for different n0 values and the orness with the greater difference is chosen. The evaluated n0 values have to be chosen in such a way that the two gene set are clearly contained in the significant and non significant gene sets respectively. It is implemented in the function chooseOrness of the OMICfpp package. The simulation study will use this function. Note that the idea is to choose the orness comparing clearly significant and non significant gene sets. It is convenient to have a previous estimation about the fractions of both kind of genes. It can be estimated by using the procedure proposed in [25] and implemented in the R package [26]. We have used it to choose n_0. The details are in Additional file 1: Methods.
For our data set, values for n0 from 100 to 10000 with an increment of 10 were chosen. The number of estimated non significant genes (using the method in [25]) gives us a number around 11000 genes, thus we explore up to 10000. For each n0 the optimal orness is calculated (Fig. 1e). It is clear that there are two clearly defined intervals of n0 with the same orness within the interval. This figure suggests two possible orness values: 0.37 and 0.93.
The closer the orness to 1, the more stringent is the selection of differentially expressed genes (Fig. 1f and g). Thus, only genes that are significant in most or all samples are reported. It is not always the case that a gene is differentially expressed in all patients, especially when the sample size increases. So, choosing values of orness in the range [0.8, 1] could be a excluding selection. On the other hand, choosing the range [0,0.2] is too permissive. This is illustrated in Fig. 1f, where the proportion of genes with complete p-values lesser than a α value (from 0.1 to 0.01) in each δ orness value are evaluated.
However, the orness could be chosen according to an expert judgment based on previous knowledge. First, a small set of genes with differential expression experimentally verified and a set of housekeeping genes i.e. genes with no differential expression, can be proposed. In this case, we are concerned with colorectal cancer (CRC) data set. Thus, information from the TCGA project, through the web server for cancer and normal gene expression profiling (GEPIA) [27] can be used to select a set of genes with validated differential expression in CRC and other set of housekeeping genes. For instance, the genes CDH3 [28], IL11 [29] or SLC11A1 [30] are experimentally validated as differentially expressed in CRC. Also, the genes HIST2H3C, ACTB or RPS23 do not present differential expression in TCGA, have a constitutive function and are not previously described association with CRC, thus can be used as housekeeping. We can replace the data driven procedure with a supervised selection of significant and non significant gene sets.
Finally, the user could choose the orness according with a type of strategy. For instance, a greedy choice could be to use orness close to one i.e. looking for the most significant pairs. Also, a conservative strategies can be choose an orness of 0.5 and a inclusive strategy would use values close to zero i.e. close to the maximum of the p-values using the less significant pairs.
OMICfpp results using an orness value
The between pairs distribution reports the difference between pairs allowing us to identify the influence of outlier pairs. On the other hand, the complete distribution allows us reporting the differences between the controls and cases i.e. the evaluation of the experimental condition. Our methodology allows the evaluation of both experimental factors, although the condition (colorectal cancer in our experimental study) will be evaluated using the complete distribution.
The randomization p-values have been estimated using 1000 realizations. Two thousand eight hundred ninety seven genes were differentially expressed using an orness value of 0.37 and 1564 with an orness of 0.93 (p-value < 0.001, see Additional file 1: Results). Of these, 501 genes were reported in common. We pretend to order the genes using the p-values. Obviously, if we have such a large number of null p-values, they can non ordered using only this p-value. Then, we have used a second ordering criteria using the score proposed.
The top 30 genes for both orness value are shown in Table 2 and a bibliographic search was conducted in order to determine if the genes of each list were previously reported and validated. It has been found that, using an orness value of 0.37, 46,67% and 70% of the genes were previously reported in colorectal cancer and in another type of cancer, respectively. In addition, the bold entries show that 56.6% of the first 30 genes were also reported by other methods.
Figure 2a displays the randomization p-value observed for all the orness values. Although, not all genes have a consistent low p-value pattern in a wide range of orness values, all of them show a null p-value around the 0.37 orness. For instance, genes such as ITGAL, IBSP and GUCA2B have a consistent low p-value pattern, Fig. 2a, and its differential expression in colorectal cancer were verified in previous studies (Table 2). Moreover, some genes that have less clearly defined profiles as MAGEC2, Fig. 2a, have also been experimentally validated (Table 2). However, according to our results, it is not differentially expressed in all patients, which is confirmed in the bibliography [31]. This demonstrates the utility of randomized p-value profiles for target gene selection. Thus, we can suggest, that the same result can be occur by the JADE2, ANO2 or MCF2L2 genes, that have not been previously reported.

Randomization p-values for the 30 most significant genes using an orness value of (a) 0.37, (b) 0.93 and (c) the interval score
The results obtained using an orness of 0.93, show that 43.3% and 73.3% of the genes were previously reported in CRC and in another type of cancer, respectively (Table 2). In addition, the bold entries show that only 26.67% of the first 30 genes were also reported by other methods. The genes ANOS1, CNTN1 or ARNTL2, with a well defined randomization p-values pattern and the genes MAD1L1 or ABCC2, with less clearly defined profiles (Fig. 2b), have been experimentally validated in CRC (Table 2). Also, a considerable number of the first 30 genes (33.3%) reported using an orness of 0.93, were previously experimentally validated.
In view of all the above, we suggest that the genes reported as differentially expressed using OMICfpp, which have not been previously reported in the bibliography, have a high probability of being validated experimentally. Especially those genes that present a defined profile in the randomized p-values pattern graphic and, to a lesser extent, those in which the randomized p-values pattern is less defined.
Ordering genes
We have selected in “Choosing an orness” section just two orness values according with an unsupervised method. However, it seems very interesting to explore the results using not just one or two orness values. Instead, we can use many orness values in order to sort the genes in the study. Note that for a given δ-orness the value pc(δ), randomization p-value using the complete distribution and a δ-orness, could be interpreted as the membership degree (in fuzzy set terminology) of this gene to be non significant i.e. to belong to the set of “non significant genes”. A high pc(δ) corresponds to non significant gene. The integral of this p-value with respect to the orness is a good quantification to be used to order the genes from most significant (lowest area) to lowest significant gene (highest area). This area is given by
where ψ is a density function over the unit interval [0,1]. This aggregated value is like a mean membership degree of the gene to “non significant genes”. This value is calculated for all genes and ordered in increasing order from the most significant to the lowest significant gene. The ordering obtained is consistent with the results in the next section and the whole list can be found in the file score_complete.html in Additional file 1: Results. Note that we use only the complete p-value because we are interested in the differential expression. The ordering using the between-pair distribution would order the genes according with the importance of particular pairs to the differential expression.
For our data set the density used for the orness is a beta distribution with parameters (4.310396,1.977092) shown in Fig. 1g. The automatic procedure suggested to use two possible orness values, 0.37 and 0.93. A common criteria is to use an orness close to 0.5 i.e. close to the average. Following this idea, we have chosen a beta distribution giving a probability 0.9 to the interval [.37,.93], a probability 0.05 to the interval [0,0.37] and a probability of 0.05 to the interval [0.93,1]. The probability mass is mainly concentrated in the central interval (0.9) y the two other intervals concentrate an small probability (0.05 each one).
We identify a total of 26 genes with p-value < 1.05e-12. The first 30 genes reported are shown in the Table 3. The results indicated that 83.3% of the first 30 genes reported are also reported using edgeR and DESeq2 methods. In addition, only a 30% of the genes have been reported in the CRC bibliography. The randomization p-values profiles for the genes of the Table 3, are shown in the Fig. 2c.
OMICfpp in a small sample size context
This section contains a small study about the sample size. In our case the sample size refers to the number of pairs used in the study. The original data used in the paper has 68 pairs. We have reproduced the study with two random samples from the original pairs. Firstly we have used 10 pairs and, secondly, a total number of 20 pairs. The results obtained with 10, 20 and 68 pairs will be compared (Fig. 3). We have considered two α values and evaluated the number of genes with a p-value lesser than α for each orness value. In fact, we have plotted the fraction of common significant genes with respect to the total number of genes in the study. These α values show two typical behaviour in these plots. Figure 3a corresponds with α=0.001. Shows a great overlapping between the results for 68 and 20 pairs for small values of orness. The number of these genes decreases for higher values of orness. Similar comment can be applied to the comparison of 68 with 10 pairs.

(a) Number of genes declared significant i.e. complete p-value lesser than α=0.001 using 68, 20 and 10 samples. Label n68.n20 corresponds with the proportion of genes declared significant using 68 and 20 for different orness values. Labels n68.n10 and n20.n10 corresponds with the proportion declared significant using 68 and 10 samples and 20 and 10 samples. (b) The corresponding proportions with respect to the number of significant genes using 68 pairs
As it could be expected when α is greater the number of common genes between the three studies is clearly greater. Figure 3b corresponds with α=0.001. The power of the study with 68 is much greater and only when we declare significant genes with a higher threshold the results are more similar.
Comparing OMICfpp, edgeR and DESeq2
We have compared our results with those obtained using the methods edgeR and DESeq2. We had four different methods per gene and four p-values for them. The two first will be the complete randomization p-values corresponding to the orness values 0.37 and 0.93. The third p-value corresponds to the method implemented in the package edgeR [9] and the fourth corresponds to DESeq2 [17].
In order to compare the significant genes by taking into account the four criteria an α value of 0.001 have been chose. The significant genes for a given p-value is composed by those genes with the p-value < 0.001.
Under our analysis, edgeR reported 15860 significant genes and DESeq2 reported 15563 genes. Of these, 13589 genes are reported by both methods and 86.5% of these are not reported by OMICfpp (Fig. 4a). OMICfpp method reported 2897 and 1564 genes using an orness value of 0.37 and 0.93, respectively.

Comparison between OMICfpp, edgeR and DESeq2 results. a) Venn diagram comparing the genes with raw p-values less than 0.001, using a OMICfpp and the p-value obtained by edgeR and DESeq2. b) Simulated data set using Poisson distributions. Differences between p-values using different methods, different types of genes and all orness. Rows correspond to the comparisons between methods: bc, between-pair vs complete distributions; be, between-pair distribution vs edgeR method; ce, complete distribution vs edgeR methods. c) Simulated data set using Negative binomial distributions. Differences between p-values using different methods, different types of genes and all orness. Rows correspond to the comparisons between methods: bc, between-pair vs complete distributions; be, between-pair distribution vs edgeR method; ce, complete distribution vs edgeR methods
OMICfpp reports around 85% fewer genes if a p-value < 0.001 is considered. The same applies if an adjusted p-value < 0.001 for edgeR (14332 significant genes) and DESeq2 (14606 significant genes) is considered. Thus, our method is more restrictive than edgeR or DESeq2. Thus our method is more restrictive than edgeR or DESeq2. Furthermore, 914 genes are reported in common by edgeR, DESeq2 and OMICfpp using an orness of 0.93 i.e. 54% more that when an orness of 0.37 is used. Moreover, 95.4% of the genes reported using an orness of 0.93 were also reported by the other methods, while in the case of orness of 0.37 only 35% of the genes were reported by the other methods.
The first 30 genes reported by edgeR and DESeq2 are shown in the Tables 4 and 5, respectively. The results obtained using edgeR, show that 66.6% and 60% of the genes were previously reported in CRC and in another type of cancer, respectively. In addition, the bold entries show that 76.67% of the first 30 genes were also reported by the other methods (Table 4). For the DESeq2 results, 80% and 83.3% of the genes were previously reported in CRC and in another type of cancer, respectively, and 70% of the genes are also reported in the other methods (Table 5).
Simulation study
In order to obtain a more complete evaluation of the OMICfpp method, a simulation study was performed, using Poisson counts (Fig. 4b) and negative binomial distributions approach (Fig. 4c).
In the simulation study using Poisson counts, we consider three types of features (genes for instance): significant genes, non significant genes and outliers genes. We have to simulate random pairs of counts for the three types. We consider four Poisson random variables such that the i-th variable Xi follows a Poisson distribution with mean λi for i=1,…,4. If the gene is non significant then we simulate the random vector (X1,X3)=(x1,x3). The pair of counts for the pair are (x1,x1+x3). Note that the mean of X3, λ3, is small i.e. just an small increment of the count. If the gene is significant then we simulate (X1,X2)=(x1,x2) and the counts are (x1,x1+x2). Finally, if the gene is an outlier then we have two types of pairs. The first type of pair is as a pair of a non significant gene. The second type of genes is different. We consider a realization of (X1,X4)=(x1,x4) and the counts are (x1,x1+x4). The mean λ4 is much greater than λ2. The idea is to simulate genes with no differential expression for the most of the pairs except for a few ones. We call them outlier genes. This model is implemented in the function rPairedPoisson of the package OMICfpp. We have used 1000 genes with 50 significant, 50 outliers and 900 non significant genes. For each outlier gene we have used 1,2,…,10 outlier pairs for an outlier gene. The orness values used goes from 0.01 to 0.99 with a step of 0.02. The mean of the random variables will be λ1=10 and λ3=2. The mean λ2∈{6,10,14,18} and λ4 goes from 30 to 60 with an step of 2.
Figure 4b display a simple graphical description. We have three types of genes: outliers, significant and non significant genes. We pretend to compare three methods, between-pair and complete randomization p-values and edgeR. These p-values have been estimated for each simulated data set. The mean of the differences between each pair of methods has been calculated and displayed in this figure by taking into account the value of λ4 i.e. more extreme outliers and for different values of λ2 i.e. more clearly differentiated significant genes. Our p-values are sensible to the outliers (first column) and are similar to the results of edgeR when for λ2 equal to 10, 14 and 18. However, edgeR can detect a difference of λ2=6 and our methods can not detect it. The non-signficant genes are equally non-detected by all methods. Again, edgeR is more powerful but very sensitive to the outliers. Our methods are not so powerful but they detect the outliers and are not so sensible to them.
A similar model has been performed by replacing the Poisson distribution with the negative binomial distribution (Fig. 4c). Now, the means of the four negative distribution used are μ1=10, μ3=2 and μ2 takes the values 20, 30, 40 and 50. The values for μ4 goes from 60 to 100 with a step of 5. The dispersion parameter used for all negative distributions has been 1/10. We think the comments given using Poisson distributions can be applied to the study using negative binomial distributions.
It is important to note that the method DESeq2 can not be applied to this simulated data because it probably needs a greater over dispersion in the data. We have had problems with the estimation of the prior distributions. For this reason, we compare our methodology just with the method edgeR.
Gene expression signatures for colorectal cancer
A total of 491 genes were reported in common for all methods (Fig. 4a), of these 65 genes are within the top 30 previously described (see Tables 2, 3, 4 and 5). These genes are studied in more detail, in order to propose a gene expression signatures for colorectal cancer. A total of 36 genes in common have been previously reported in CRC: ANOS1, CNTN1, SLC11A1, IBSP, MUSK, USP2, GUCA2B, TNFRSF17, COL11A1, IL11, C2orf40, PRPH, TMEFF2, KRT80, UGP2, SLC30A10, GSTM5, ESM1, CA7, WNT2, FEZF1-AS1, AJUBA, CEMIP, CLDN1, SPERT, FOXQ1, INHBA, EPHX4, KLK6, TRIB3, CST1, SPIB, GRIN2D, GCNT2, COL10A1 and MMP7. Furthermore, a total of 29 genes in common have not been previously reported in CRC: LAS1L, ICA1, TNFRSF12A, GTF2IRD1, ZNF200, MYOC, NEXMIF, POLR3E, LGI1, PCSK2, PRELID3A, BEST4, CTNND2, SLC6A19, OTOP2, IGLV2-14, AC016027.1, LINC02577, PCAT2, AC055717.2, DAO, TMEM82, SPATA12, LINC02163, OTOP3, LINC02414, AC07099.1, CPNE7 and LINC02418 (see Tables 2, 3, 4 and 5). The randomization p-value profiles are shown in Fig. 2. In the case of validated genes that are shown in the Fig. 2, all have a defined randomization p-value profiles. The same happens with the profiles of the genes not reported in the literature, with the exception of SPATA12. Probably SPATA12 corresponds to a false positive gene, despite being reported by all methods, since this gene is expressed primarily in testis and may play a role in testicular development and spermatogenesis. On the other hand, little or nothing is known about the AC016027.1, AC055717.2 and AC07099.1 genes, which makes their description difficult. This is the same situation with the coding genes of ncRNA, LINC02577, LINC02163, LINC02414, LINC02418 and the prostate cancer associated transcript 2 (PCAT2) gene. The neurite extension and migration factor (NEXMIF) gene has not been previously reported in cancer, thus its function is unknown. The same goes for the RNA polymerase III subunit E (POLR3E) gene, the PRELI domain containing 3A (PRELID3A) gene, the otopetrin 2 (OTOP2) and 3 (OTOP3) genes, the immunoglobulin lambda variable 2-14 (IGLV2-14) gene and the bestrophin 4 (BEST4) gene that encodes a transmembrane proteins. The LAS1L gene is essential for cell proliferation and also for biogenesis of the 60S ribosomal subunit [32] and has been previously related with pancreatic cancer [33]. The ICA1 gene encodes a protein with an arfaptin homology domain that is found both in the cytosol and as membrane-bound form on the Golgi complex and immature secretory granules. This protein binds to the small GTPase Rab2, thus it can be related to cancer [34]. The TNF receptor superfamily member 12A (TNFRSF12A) gene is well known in cancer, for example, it is linked to poor prognosis in breast cancer [35]. The GTF2IRD1 gene encodes a transcription factor protein, that are related to tumor-promotion [36]. The zinc finger protein 200 (ZNF200) is a little known gene and in cancer, only variants associated to ovarian cancer have been previously reported [37]. The MYOC gene encodes the protein myocilin, which is believed to have a role in cytoskeletal function and it has been previously reported in glaucoma [38]. Thus, this gene has less likely to be validated experimentally in CRC. However, the rest of the genes (LGI1, PCSK2, CTNND2, SLC6A19, DAO, TMEM82 and CPNE7) could be related to CRC and have been previously identified in other types of cancer. Thus, we propose these 20 genes as new candidate genes.
Discussion
We develop OMICfpp as a method for statistical analysis of RNA-Seq data with a paired design and small sample size context. OMICfpp, through the orness election allows to the user assign weight to the results, based on each biological context. However, we also provide the alternative of automatic orness selection. Here we use colorectal cancer data, but OMICfpp can be applied to all kinds of biological problems that involve RNA-Seq analysis.
We use the chooseOrness function to select an orness value of 0.37 and 0.93. We also tested the possibility of using a probability distribution over the orness and use the score for CRC data analysis. The results suggest that the use of the score is a more robust method for gene selection, whereas a single orness selection is a reasonable method. Besides, a large number of genes reported in the top position using the score, are also reported within the results obtained by a single orness value.
On the other hand, we tested OMICfpp results using different sample sizes (“OMICfpp in a small sample size context” section). It is clear that a smaller sample size will affect more the highest values of orness. For low orness there is a great overlapping between significant genes using lower sample sizes (Fig. 3). These results confirm that the sample size is very important in obtaining results. We suggest to use p-values < 0.001 as the cut line for the results obtained using OMICfpp in smaller sample sizes.
The results obtained by OMICfpp method were validated through bibliographic review, and also by a simulation study. An important part of the results are in agreement with the cancer bibliography, validating the OMICfpp method. Also, we compare the results of OMICfpp with those obtained by edgeR [9] and DESeq2 [17]. We obtain a considerably smaller number of candidate genes than edgeR and DESeq2 (Fig. 4), indicating that our method is more accurate. In turn, the results using an orness of 0.93 were also supported by edgeR or DESeq2 by more than 90%. In addition, there is an important coincidence between the top 30 genes reported by OMICfpp, edgeR and DESeq2 methods.
Besides, the simulation study shows that edgeR is more powerful than our procedure. However, the outliers affects more the results of edgeR than ours. If there is a suspect than the differential expression is due to just one or two outlier pairs, then our approach could complement the study.
Moreover, we identify candidate genes not reported by edgeR and DESeq2 methods, which we suggest must be validated. Furthermore, 491 genes are reported by all compared methods (Fig. 4a). Of these, 65 genes are in the top result in all methods and 36 genes have been previously reported in the bibliography as differentially expressed in colorectal cancer (Tables 2, 3, 4 and 4. All of these with a well defined randomization p-value profile. Thus, we deepened in the study of the remaining 29 genes, using the biological data obtained in the bibliography and biological data bases, and our randomization p-value profiles (Fig. 2). Therefore, we propose the use of randomization p-values profiles as an accurate method to select the candidate genes for experimental validation.
Furthermore, in the last 20 years, it has been searched to identify “cancer signature” in terms of diagnosis, prognosis or prediction of therapeutic response [39]. Although the term refers to one or more genes, a biomarker panel with a growing number of genes is currently used. In this sense, we recommend the experimental validation of LAS1L, ICA1, TNFRSF12A, GTF2IRD1, ZNF200, NEXMIF, POLR3E, LGI1, PCSK2, PRELID3A, BEST4, CTNND2, SLC6A19, OTOP2, IGLV2-14, PCAT2, DAO, TMEM82, OTOP3 and CPNE7 genes as new targets for gene expression signature in colorectal cancer.
Conclusions
RNA-Seq is a powerful method to study the complexity of the transcriptome, however there are many challenges to solve. On the one hand, the inclusion of the experimental design in the analysis of the results can contribute to the obtaining of more precise results. In this regard, OMICfpp is an accurate method for differential expression analysis in RNA-Seq data with paired design. On the other hand, a large number of genes identified as differentially expressed in silico are not experimentally validated. In this sense, we propose the use of randomized p-values profile graphic as a powerful and robust method to select the target genes for experimental validation.
Abbreviations
- CRC:
-
Colorectal cancer
- OWA:
-
Ordered Weighted Average
- RNA-Seq:
-
RNA sequencing TCGA: The Cancer Genome Atlas
References
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009; 10(1):57–63. https://doi.org/10.1038/nrg2484.RNA-Seq.
Zhong W, Mark G, Michael S. RNA-Seq: a revolutionary tool for transcriptomics Zhong. Nat Rev Genet. 2009; 10(1):57–63. https://doi.org/10.1038/nrg2484.
Casamassimi A, Federico A, Rienzo M, Esposito S, Ciccodicola A. Transcriptome profiling in human diseases: New advances and perspectives. Int J Mol Sci. 2017;18(8). https://doi.org/10.3390/ijms18081652.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29(1):15–21. https://doi.org/10.1093/bioinformatics/bts635.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012; 7:562–78. https://doi.org/10.1038/nprot.2012.016.
Langmead B. Aligning short sequencing reads with Bowtie. Current protocols in bioinformatics / editoral board, Andreas D. Baxevanis... [et al.] 2010; CHAPTER:11–7. https://doi.org/10.1002/0471250953.bi1107s32.
Anders S, Pyl PT, Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31(2):166–9. https://doi.org/10.1093/bioinformatics/btu638.
Liao Y, Smyth GK, Shi W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013; 41(10):108. https://doi.org/10.1093/nar/gkt214.
Lun ATL, Chen Y, Smyth GK. In: Mathé E, Davis S, (eds).It’s DE-licious: A Recipe for Differential Expression Analyses of RNA-seq Experiments Using Quasi-Likelihood Methods in edgeR, vol. 1418; 2016. Chap. 19. https://doi.org/10.1007/978-1-4939-3578-9_19.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010; 11(10):106. https://doi.org/10.1186/gb-2010-11-10-r106.
Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016; 17:13. https://doi.org/10.1186/s13059-016-0881-8.
Robert C, Watson M. Errors in RNA-Seq quantification affect genes of relevance to human disease. Genome Biol. 2015; 16(1):1–16. https://doi.org/10.1186/s13059-015-0734-x.
Al Seesi S, Tiagueu Y, Zelikovsky A, Mǎndoiu II. Bootstrap-based differential gene expression analysis for RNA-Seq data with and without replicates. BMC Genomics. 2014; 15(Suppl 8):2. https://doi.org/10.1186/1471-2164-15-S8-S2.
Zhou Q, Su X, Jing G, Chen S, Ning K. RNA-QC-chain: comprehensive and fast quality control for RNA-Seq data. BMC Genomics. 2018; 19(1):144. https://doi.org/10.1186/s12864-018-4503-6.
Kal AJ, van Zonneveld AJ, Benes V, van den Berg M, Koerkamp MG, Albermann K, Strack N, Ruijter JM, Richter A, Dujon B, Ansorge W, Tabak HF. Dynamics of Gene Expression Revealed by Comparison of Serial Analysis of Gene Expression Transcript Profiles from Yeast Grown on Two Different Carbon Sources. Mol Biol Cell. 1999; 10(6):1859–72. https://doi.org/10.1091/mbc.10.6.1859.
Yager RR. On ordered weighted averaging aggregation operators in multicriteria decisionmaking. IEEE Trans Syst Man Cybern. 1988; 18(1):183–90. https://doi.org/10.1109/21.87068.
Love M, Huber W, Anders S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014; 15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. 2015; 19(1A):68–77. https://doi.org/10.5114/wo.2014.47136.
Kim S-K, Kim S-Y, Kim J-H, Roh SA, Cho D-H, Kim YS, Kim JC. A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients. Mol Oncol. 2014; 8(8):1653–66. https://doi.org/10.1016/j.molonc.2014.06.016.
Leinonen R, Sugawara H, Shumway M, on behalf of the International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011; 39(suppl_1):19–21. https://doi.org/10.1093/nar/gkq1019.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25(16):2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. Software for computing and annotating genomic ranges. PLOS Comput Biol. 2013; 9(8):1–10. https://doi.org/10.1371/journal.pcbi.1003118.
Morgan M, Obenchain V, Pagès H. Summarizedexperiment: Summarizedexperiment container. 2018. r package version 1.12.0. Bioconductor.org. https://bioconductor.org/packages/release/bioc/html/SummarizedExperiment.html.
Ayala G, Leon T, Zapater V. Different averages of a fuzzy set with an application to vessel segmentation. IEEE Trans Fuzzy Syst. 2005; 13(3):384–93. https://doi.org/10.1109/TFUZZ.2004.839667.
Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci. 2003; 100(16):9440–5. https://doi.org/10.1073/pnas.1530509100.
with contributions from, Bass AJ, Storey JD, Dabney A, Robinson D. Qvalue: Q-value Estimation for False Discovery Rate Control. 2015. R package version 2.10.0. http://github.com/jdstorey/qvalue.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017; 45(W1):98–102. https://doi.org/10.1093/nar/gkx247.
Bellini GA, Caballero OL, Sonali AC, Su T, Ahmed A, Njoh L, Cekic V, Whelan RL. P-Cadherin (CDH3) is overexpressed in colorectal tumors and has potential as a serum marker for colorectal cancer monitoring. Oncoscience. 2017;4(September). https://doi.org/10.18632/oncoscience.370.
Xu DH, Zhu Z, Wakefield MR, Xiao H, Bai Q, Fang Y. The role of il-11 in immunity and cancer. Cancer Lett. 2016; 373(2):156–63. https://doi.org/10.1016/j.canlet.2016.01.004.
Yu L, Yin B, Qu K, Li J, Jin Q, Liu L, Liu C, Zhu Y, Wang Q, Peng X, Zhou J, Cao P, Cao K. Screening for susceptibility genes in hereditary non-polyposis colorectal cancer. Oncol Lett. 2018; 15(6):9413–9. https://doi.org/10.3892/ol.2018.8504.
Chen Y-T, Panarelli NC, Piotti KC, Yantiss RK. Cancer–testis antigen expression in digestive tract carcinomas: Frequent expression in esophageal squamous cell carcinoma and its precursor lesions. Cancer Immunol Res. 2014; 2(5):480–6. https://doi.org/10.1158/2326-6066.CIR-13-0124.
Castle CD, Cassimere EK, Lee J, Denicourt C. Las1L is a nucleolar protein required for cell proliferation and ribosome biogenesis. Mol Cell Biol. 2010; 30(18):4404–14. https://doi.org/10.1128/MCB.00358-10.
Kashiwaya K, Nakagawa H, Hosokawa M, Mochizuki Y, Ueda K, Piao L, Chung S, Hamamoto R, Eguchi H, Ohigashi H, Ishikawa O, Janke C, Shinomura Y, Nakamura Y. Involvement of the tubulin tyrosine ligase-like family member 4 polyglutamylase in pelp1 polyglutamylation and chromatin remodeling in pancreatic cancer cells. Cancer Res. 2010; 70(10):4024–33. https://doi.org/10.1158/0008-5472.CAN-09-4444.
Buffa L, Fuchs E, Pietropaolo M, Barr F, Solimena M. Ica69 is a novel rab2 effector regulating er–golgi trafficking in insulinoma cells. Eur J Cell Biol. 2008; 87(4):197–209. https://doi.org/10.1016/j.ejcb.2007.11.003.
Yang J, Min KW, Kim DH, Son BK, Moon KM, Wi YC, Bang SS, Oh YH, Do SI, Chae SW, Oh S, Kim YH, Kwon MJ. High TNFRSF12A level associated with MMP-9 overexpression is linked to poor prognosis in breast cancer: Gene set enrichment analysis and validation in large-scale cohorts. PLoS ONE. 2018; 13(8):1–13. https://doi.org/10.1371/journal.pone.0202113.
Huo Y, Su T, Cai Q, Macara IG. An In Vivo Gain-of-Function Screen Identifies the Williams-Beuren Syndrome Gene GTF2IRD1 as a Mammary Tumor Promoter. Cell Rep. 2016; 15(10):2089–96. https://doi.org/10.1016/j.celrep.2016.05.011. 15334406.
Peedicayil A, Vierkant RA, Hartmann LC, Fridley BL, Fredericksen ZS, White KL, Elliott EA, Phelan CM, Tsai YY, Berchuck A, Iversen ES, Couch FJ, Peethamabaran P, Larson MC, Kalli KR, Kosel ML, Shridhar V, Rider DN, Liebow M, Cunningham JM, Schildkraut JM, Sellers TA, Goode EL. Risk of ovarian cancer and inherited variants in relapse-associated genes. PLoS ONE. 2010;5(1). https://doi.org/10.1371/journal.pone.0008884.
Kennedy KD, AnithaChristy SA, Buie LK, Borrás T. Cystatin a, a potential common link for mutant myocilin causative glaucoma. PloS ONE. 2012;7(5). https://doi.org/10.1371/journal.pone.0036301.
Chibon F. Cancer gene expression signatures – The rise and fall?,. Eur J Cancer. 2013; 49(8):2000–9. https://doi.org/10.1016/j.ejca.2013.02.021.
Vendrell E, Ribas M, Valls J, Solé X, Grau M, Moreno V, Capellà G, Peinado MA. Genomic and transcriptomic prognostic factors in R0 Dukes B and C colorectal cancer patients. Int J Oncol. 2007; 30(5):1099–107.
Hutterer E, Asslaber D, Caldana C, Krenn PW, Zucchetto A, Gattei V, Greil R, Hartmann TN. CD18 (ITGB2) expression in chronic lymphocytic leukaemia is regulated by DNA methylation-dependent and -independent mechanisms. Br J Haematol. 2015; 169(2):286–9. https://doi.org/10.1111/bjh.13188.
Hlavata I, Mohelnikova-Duchonova B, Vaclavikova R, Liska V, Pitule P, Novak P, Bruha J, Vycital O, Holubec L, Treska V, Vodicka P, Soucek P. The role of abc transporters in progression and clinical outcome of colorectal cancer. Mutagenesis. 2012; 27(2):187–96. https://doi.org/10.1093/mutage/ger075.
Dvorak P, Pesta M, Soucek P. Abc gene expression profiles have clinical importance and possibly form a new hallmark of cancer. Tumor Biol. 2017; 39(5):1010428317699800. https://doi.org/10.1177/1010428317699800. PMID:28468577.
Qi L, Zhang W, Cheng Z, Tang N, Ding Y. Study on molecular mechanism of ANOS1 promoting development of colorectal cancer. PLoS ONE. 2017; 12(8):1–10. https://doi.org/10.1371/journal.pone.0182964.
Kanda M, Shimizu D, Fujii T, Sueoka S, Tanaka Y, Ezaka K, Takami H, Tanaka H, Hashimoto R, Iwata N, Kobayashi D, Tanaka C, Yamada S, Nakayama G, Sugimoto H, Koike M, Fujiwara M, Kodera Y. Function and diagnostic value of Anosmin-1 in gastric cancer progression. Int J Cancer. 2016; 138(3):721–30. https://doi.org/10.1002/ijc.29803.
Staub E, Groene J, Heinze M, Mennerich D, Roepcke S, Klaman I, Hinzmann B, Castanos-Velez E, Pilarsky C, Mann B, Brümmendorf T, Weber B, Buhr HJ, Rosenthal A. An expression module of WIPF1-coexpressed genes identifies patients with favorable prognosis in three tumor types. J Mol Med. 2009; 87(6):633–44. https://doi.org/10.1007/s00109-009-0467-y.
Frugtniet BA, Martin TA, Zhang L, Jiang WG. Neural Wiskott-Aldrich syndrome protein (nWASP) is implicated in human lung cancer invasion. BMC Cancer. 2017; 17(1):1–11. https://doi.org/10.1186/s12885-017-3219-3.
Oh BY, Cho J, Hong HK, Bae JS, Park WY, Joung JG, Cho YB. Exome and transcriptome sequencing identifies loss of PDLIM2 in metastatic colorectal cancers. Cancer Manag Res. 2017; 9:581–9. https://doi.org/10.2147/CMAR.S149002.
Sung P-J, Boulos N, Tilby MJ, Andrews WD, Newbold RF, Tweddle DA, Lunec J. Identification and characterisation of stmn4 and robo2 gene involvement in neuroblastoma cell differentiation. Cancer Lett. 2013; 328(1):168–75. https://doi.org/10.1016/j.canlet.2012.08.015.
Kok-Sin T, Mokhtar NM, Hassan NZA, Sagap I, Rose IM, Harun R, Jamal R. Identification of diagnostic markers in colorectal cancer via integrative epigenomics and genomics data. Oncol Rep. 2015; 34(1):22–32. https://doi.org/10.3892/or.2015.3993.
Chen DH, Yu JW, Wu JG, Wang SL, Jiang BJ. Significances of contactin-1 expression in human gastric cancer and knockdown of contactin-1 expression inhibits invasion and metastasis of MKN45 gastric cancer cells. J Cancer Res Clin Oncol. 2015; 141(12):2109–20. https://doi.org/10.1007/s00432-015-1973-7.
Zhang Z, Pal S, Bi Y, Tchou J, Davuluri RV. Isoform level expression profiles provide better cancer signatures than gene level expression profiles. Genome Med. 2013; 5(4):33. https://doi.org/10.1186/gm437.
Fedarko NS, Jain A, Karadag A, Van Eman MR, Fisher LW. Elevated serum bone sialoprotein and osteopontin in colon, breast, prostate, and lung cancer. Clin Cancer Res. 2001; 7(12):4060–6. http://clincancerres.aacrjournals.org/content/7/12/4060.full.pdf.
Xu G, Zhang M, Zhu H, Xu J. A 15-gene signature for prediction of colon cancer recurrence and prognosis based on svm. Gene. 2017; 604:33–40. https://doi.org/10.1016/j.gene.2016.12.016.
Chakraborty S, Lakshmanan M, Swa HLF, Chen J, Zhang X, Ong YS, Loo LS, Akıncılar SC, Gunaratne J, Tergaonkar V, Hui KM, Hong W. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat Commun. 2015; 6(1):6184. https://doi.org/10.1038/ncomms7184.
Davis MI, Pragani R, Fox JT, Shen M, Parmar K, Gaudiano EF, Liu L, Tanega C, McGee L, Hall MD, McKnight C, Shinn P, Nelson H, Chattopadhyay D, D’Andrea AD, Auld DS, DeLucas LJ, Li Z, Boxer MB, Simeonov A. Small molecule inhibition of the ubiquitin-specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. J Biol Chem. 2016; 291(47):24628–40. https://doi.org/10.1074/jbc.M116.738567.
Qu Q, Mao Y, Xiao G, Fei X, Wang J, Zhang Y, Liu J, Cheng G, Chen X, Wang J, Shen K. Usp2 promotes cell migration and invasion in triple negative breast cancer cell lines. Tumor Biol. 2015; 36(7):5415–23. https://doi.org/10.1007/s13277-015-3207-7.
El-Saghire H, Vandevoorde C, Ost P, Monsieurs P, Michaux A, De Meerleer G, Baatout S, Thierens H. Intensity modulated radiotherapy induces pro-inflammatory and pro-survival responses in prostate cancer patients. Int J Oncol. 2014; 44(4):1073–83. https://doi.org/10.3892/ijo.2014.2260.
Jinawath N, Shiao MS, Norris A, Murphy K, Klein AP, Yonescu R, Iacobuzio-Donahue C, Meeker A, Jinawath A, Yeo CJ, Eshleman JR, Hruban RH, Brody JR, Griffin CA, Harada S. Alterations of type II classical cadherin, cadherin-10 (CDH10), is associated with pancreatic ductal adenocarcinomas. Genes Chromosome Cancer. 2017; 56(5):427–35. https://doi.org/10.1002/gcc.22447.
Nagaraj SH, Reverter A. A Boolean-based systems biology approach to predict novel genes associated with cancer: Application to colorectal cancer. BMC Syst Biol. 2011; 5(1):35. https://doi.org/10.1186/1752-0509-5-35.
Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, Waldner MJ, Bindea G, Mlecnik B, Galon J, Trajanoski Z. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015; 16(1):1–17. https://doi.org/10.1186/s13059-015-0620-6. arXiv:1011.1669v3.
Pelekanou V, Notas G, Athanasouli P, Alexakis K, Kiagiadaki F, Peroulis N, Kalyvianaki K, Kampouri E, Polioudaki H, Theodoropoulos P, Tsapis A, Castanas E, Kampa M. BCMA (TNFRSF17) Induces APRIL and BAFF Mediated Breast Cancer Cell Stemness. Front Oncol. 2018; 8(August):301. https://doi.org/10.3389/fonc.2018.00301.
Zhang D, Zhu H, Harpaz N. Overexpression of a1 chain of type xi collagen (col11a1) aids in the diagnosis of invasive carcinoma in endoscopically removed malignant colorectal polyps,. Pathol Res Pract. 2016; 212(6):545–8. https://doi.org/10.1016/j.prp.2016.03.005.
Jia D, Liu Z, Deng N, Tan TZ, Huang RY-J, Taylor-Harding B, Cheon D-J, Lawrenson K, Wiedemeyer WR, Walts AE, Karlan BY, Orsulic S. A COL11A1-correlated pan-cancer gene signature of activated fibroblasts for the prioritization of therapeutic targets. Cancer Lett. 2016; 382(2):203–14. https://doi.org/10.1016/j.canlet.2016.09.001. 15334406.
Yan M, Song M, Bai R, Cheng S, Yan W. Identification of potential therapeutic targets for colorectal cancer by bioinformatics analysis. Oncol Lett. 2016; 12(6):5092–8. https://doi.org/10.3892/ol.2016.5328.
Sun Q, Zhang X, Liu T, Liu X, Geng J, He X, Liu Y, Pang D. Increased expression of mitotic arrest deficient-like 1 (MAD1L1) is associated with poor prognosis and insensitive to taxol treatment in breast cancer. Breast Cancer Res Treat. 2013; 140(2):323–30. https://doi.org/10.1007/s10549-013-2633-8.
Huang Z, Li S, Song W, Li X, Li Q, Zhang Z, Han Y, Zhang X, Miao S, Du R, Wang L. Lysine-Specific Demethylase 1 (LSD1/KDM1A) Contributes to Colorectal Tumorigenesis via Activation of the Wnt/B-Catenin Pathway by Down-Regulating Dickkopf-1 (DKK1). PLoS ONE. 2013; 8(7):1–12. https://doi.org/10.1371/journal.pone.0070077.
Ambrosio S, Saccà CD, Majello B. Epigenetic regulation of epithelial to mesenchymal transition by the Lysine-specific demethylase LSD1/KDM1A. Biochim Biophys Acta - Gene Regul Mech. 2017; 1860(9):905–10. https://doi.org/10.1016/j.bbagrm.2017.07.001.
Knific T, Grazio SF, Rižner TL. Detection of aristaless-related homeobox protein in ovarian sex cord-stromal tumors. Exp Mol Pathol. 2018; 104(1):38–44. https://doi.org/10.1016/j.yexmp.2017.12.005.
Solmi R, Ugolini G, Rosati G, Zanotti S, Lauriola M, Montroni I, del Governatore M, Caira A, Taffurelli M, Santini D, Coppola D, Guidotti L, Carinci P, Strippoli P. Microarray-based identification and RT-PCR test screening for epithelial-specific mRNAs in peripheral blood of patients with colon cancer. BMC Cancer. 2006; 6:1–9. https://doi.org/10.1186/1471-2407-6-250.
Chen CY, Chung IH, Tsai MM, Tseng YH, Chi HC, Tsai CY, Lin YH, Wang YC, Chen CP, Wu TI, Yeh CT, Tai DI, Lin KH. Thyroid hormone enhanced human hepatoma cell motility involves brain-specific serine protease 4 activation via ERK signaling. Mol Cancer. 2014; 13(1):1–17. https://doi.org/10.1186/1476-4598-13-162.
Shang HS, Liu JY, Lu HF, Chiang HS, Lin CH, Chen A, Lin YF, Chung JG. Casticin induced apoptotic cell death and altered associated gene expression in human colon cancer colo 205 cells. Environ Toxicol. 2017; 32(8):2041–52. https://doi.org/10.1002/tox.22381. 0307025v1.
Sha J, Xue W, Dong B, Pan J, Wu X, Li D, Liu D, Huang Y. PRKAR2B plays an oncogenic role in the castration-resistant prostate cancer. Oncotarget. 2017; 8(4):6114–29. https://doi.org/10.18632/oncotarget.14044.
Yuan Y, Qi G, Shen H, Guo A, Cao F, Zhu Y, Xiao C, Chang W, Zheng S. Clinical significance and biological function of WD repeat domain 54 as an oncogene in colorectal cancer. 2018:1–37. https://doi.org/10.1002/ijc.31736.
Crawford NPS, Walker RC, Lukes L, Officewala JS, Williams RW, Hunter KW. The Diasporin Pathway: a tumor progression-related transcriptional network that predicts breast cancer survival. Clin Exp Metastasis. 2008; 25(4):357–69. https://doi.org/10.1007/s10585-008-9146-6. NIHMS150003.
Maiga A, Lemieux S, Pabst C, Lavallée VP, Bouvier M, Sauvageau G, Hébert J. Transcriptome analysis of G protein-coupled receptors in distinct genetic subgroups of acute myeloid leukemia: Identification of potential disease-specific targets. Blood Cancer J. 2016; 6(6):1–9. https://doi.org/10.1038/bcj.2016.36.
Sun X, Liu S, Wang D, Zhang Y, Li W, Guo Y, Zhang H, Suo J, Sun X, Liu S, Wang D, Zhang Y, Li W, Guo Y, Zhang H, Suo J. Colorectal cancer cells suppress CD4<sup>+</sup> T cells immunity through canonical Wnt signaling. Oncotarget. 2017; 8(9):15168–81. https://doi.org/10.18632/oncotarget.14834.
Wang C, Chen J, Zhang Q, Li W, Zhang S, Xu Y, Wang F, Zhang B, Zhang Y. Elimination of CD4 low HLA-G + T cells overcomes castration- resistance in prostate cancer therapy. 2018;August. https://doi.org/10.1038/s41422-018-0089-4.
Gao JL, Lv GY, He BC, Zhang BQ, Zhang H, Wang N, Wang CZ, Du W, Yuan CS, He TC. Ginseng saponin metabolite 20(S)-protopanaxadiol inhibits tumor growth by targeting multiple cancer signaling pathways. Oncol Rep. 2013; 30(1):292–8. https://doi.org/10.3892/or.2013.2438.
Yang Y, Dong X, Xie B, Ding N, Chen J, Li Y, Zhang Q, Qu H, Fang X. Databases and Web Tools for Cancer Genomics Study. Genomics Proteomics Bioinforma. 2015; 13(1):46–50. https://doi.org/10.1016/j.gpb.2015.01.005.
Bogdanov A, Moiseenko F, Dubina M. Abnormal expression of ATP1A1 and ATP1A2 in breast cancer. F1000Research. 2017; 6(May):10. https://doi.org/10.12688/f1000research.10481.1.
Bigagli E, De Filippo C, Castagnini C, Toti S, Acquadro F, Giudici F, Fazi M, Dolara P, Messerini L, Tonelli F, Luceri C. DNA copy number alterations, gene expression changes and disease-free survival in patients with colorectal cancer: a 10 year follow-up. Cell Oncol. 2016; 39(6):545–58. https://doi.org/10.1007/s13402-016-0299-z.
MacIejczyk A, Jagoda E, Wysocka T, Matkowski R, Györffy B, Lage H, Surowiak P. ABCC2 (MRP2, cMOAT) localized in the nuclear envelope of breast carcinoma cells correlates with poor clinical outcome. Pathol Oncol Res. 2012; 18(2):331–42. https://doi.org/10.1007/s12253-011-9449-9.
Matsushita N, Yamamoto S, Inoue Y, Aruga A, Yamamoto M. RT-qPCR analysis of the tumor antigens TOMM34 and RNF43 in samples extracted from paraffin-embedded specimens of colorectal cancer. Oncol Lett. 2017; 14(2):2281–7. https://doi.org/10.3892/ol.2017.6412.
Aleskandarany MA, Negm OHN, Rakha EA, Ahmed MAH, Nolan CC, Ball GR, Caldas C, Green AR, Tighe PJ, Ellis IO. TOMM34 expression in early invasive breast cancer: A biomarker associated with poor outcome. Breast Cancer Res Treat. 2012; 136(2):419–27. https://doi.org/10.1007/s10549-012-2249-4.
Mazzoccoli G, Pazienza V, Panza A, Valvano MR, Benegiamo G, Vinciguerra M, Andriulli A, Piepoli A. ARNTL2 and SERPINE1: Potential biomarkers for tumor aggressiveness in colorectal cancer. J Cancer Res Clin Oncol. 2012; 138(3):501–11. https://doi.org/10.1007/s00432-011-1126-6.
Ha NH, Long J, Cai Q, Shu XO, Hunter KW. The Circadian Rhythm Gene Arntl2 Is a Metastasis Susceptibility Gene for Estrogen Receptor-Negative Breast Cancer. PLoS Genet. 2016; 12(9):1–20. https://doi.org/10.1371/journal.pgen.1006267.
Kunapuli P, Kasyapa CS, Hawthorn L, Cowell JK. LGI1, a putative tumor metastasis suppressor gene, controls in Vitro invasiveness and expression of matrix metalloproteinases in glioma cells through the ERK1/2 pathway. J Biol Chem. 2004; 279(22):23151–7. https://doi.org/10.1074/jbc.M314192200.
Cai Z, Liang P, Xuan J, Wan J, Guo H. Ecrg4 as a novel tumor suppressor gene inhibits colorectal cancer cell growth in vitro and in vivo. Tumor Biol. 2016; 37(7):9111–20. https://doi.org/10.1007/s13277-015-4775-2.
You Y, Li H, Qin X, Ran Y, Wang F. Down-regulated ecrg4 expression in breast cancer and its correlation with tumor progression and poor prognosis - a short report. Cell Oncol. 2016; 39(1):89–95. https://doi.org/10.1007/s13402-015-0260-6.
Kozar I, Cesi G, Margue C, Philippidou D, Kreis S. Impact of braf kinase inhibitors on the mirnomes and transcriptomes of melanoma cells. Biochim Biophys Acta (BBA) Gen Subj. 2017; 1861(11, Part B):2980–92. https://doi.org/10.1016/j.bbagen.2017.04.005. Biochemistry of Synthetic Biology - Recent Developments.
Ishida M, Kushima R, Chano T, Okabe H. Immunohistochemical demonstration of the type III intermediate filament peripherin in human rectal mucosae and well-differentiated endocrine neoplasms. Oncol Rep. 2007; 18(3):633–7.
Puertas MC, Carrillo J, Pastor X, Ampudia RM, Planas R, Alba A, Bruno R, Pujol-Borrell R, Estanyol JM, Vives-Pi M, Verdaguer J. Peripherin is a relevant neuroendocrine autoantigen recognized by islet-infiltrating B lymphocytes. J Immunol (Baltimore, Md. : 1950). 2007; 178(10):6533–9. https://doi.org/10.4049/jimmunol.178.10.6533.
Liu HY, Zhang CJ. Identification of differentially expressed genes and their upstream regulators in colorectal cancer. Cancer Gene Ther. 2017; 24(6):244–50. https://doi.org/10.1038/cgt.2017.8.
Sun T, Du W, Xiong H, Yu Y, Weng Y, Ren L, Zhao H, Wang Y, Chen Y, Xu J, Xiang Y, Qin W, Cao W, Zou W, Chen H, Hong J, Fang J-Y. Tmeff2 deregulation contributes to gastric carcinogenesis and indicates poor survival outcome. Clin Cancer Res. 2014; 20(17):4689–704. https://doi.org/10.1158/1078-0432.CCR-14-0315.
Li C, Liu X, Liu Y, Liu X, Wang R, Liao J, Wu S, Fan J, Peng Z, Li B, Wang Z. Keratin 80 promotes migration and invasion of colorectal carcinoma by interacting with PRKDC via activating the AKT pathway. Cell Death Dis. 2018;9(10). https://doi.org/10.1038/s41419-018-1030-y.
Ulbrich C, Pietsch J, Grosse J, Wehland M, Schulz H, Saar K, Hubner N, Hauslage J, Hemmersbach R, Braun M, van Loon J, Vagt N, Egli M, Richter P, Einspanier R, Sharbati S, Baltz T, Infanger M, Ma X, Grimm D. Differential Gene Regulation under Altered Gravity Conditions in Follicular Thyroid Cancer Cells: Relationship between the Extracellular Matrix and the Cytoskeleton. Cell Physiol Biochem. 2011; 28(2):185–98. https://doi.org/10.1159/000331730.
Thorsen K, Schepeler T, Øster B, Rasmussen MH, Vang S, Wang K, Hansen KQ, Lamy P, Pedersen JS, Eller A, Mansilla F, Laurila K, Wiuf C, Laurberg S, Dyrskjøt L, Ørntoft TF, Andersen CL. Tumor-specific usage of alternative transcription start sites in colorectal cancer identified by genome-wide exon array analysis. BMC Genomics. 2011; 12(1):505. https://doi.org/10.1186/1471-2164-12-505.
Li S, Hu Z, Zhao Y, Huang S, He X. Transcriptome-Wide Analysis Reveals the Landscape of Aberrant Alternative Splicing Events in Liver Cancer. Hepatology. 2018:0–1. https://doi.org/10.1002/hep.30158.
Lu Q. δ-Catenin dysregulation in cancer: interactions with E-cadherin and beyond. J Pathol. 2010; 222(2):119–23. https://doi.org/10.1002/path.2755.
Tian W, Fu H, Xu T, Xu SL, Guo Z, Tian J, Tao W, Xie HQ, Zhao B. Slc6a19 is a novel putative gene, induced by dioxins via ahr in human hepatoma hepg2 cells. Environ Pollut. 2018; 237:508–14. https://doi.org/10.1016/j.envpol.2018.02.079.
Shangkuan W-C, Lin H-C, Chang Y-T, Jian C-E, Fan H-C, Chen K-H, Liu Y-F, Hsu H-M, Chou H-L, Yao C-T, Chu C-M, Su S-L, Chang C-W. Risk analysis of colorectal cancer incidence by gene expression analysis. PeerJ. 2017; 5:3003. https://doi.org/10.7717/peerj.3003.
Singh CK, Malas KM, Tydrick C, Siddiqui IA, Iczkowski KA, Ahmad N. Analysis of Zinc-Exporters Expression in Prostate Cancer. Sci Rep. 2016; 6(1):36772. https://doi.org/10.1038/srep36772.
El Sayed SM, Abou El-Magd RM, Shishido Y, Chung SP, Sakai T, Watanabe H, Kagami S, Fukui K. D-amino acid oxidase gene therapy sensitizes glioma cells to the antiglycolytic effect of 3-bromopyruvate. Cancer Gene Ther. 2012; 19(1):1–18. https://doi.org/10.1038/cgt.2011.59.
Zhu D-J, Chen X-W, Wang J-Z, Ju Y-L, Ou Yang M-Z, Zhang W-J. Proteomic analysis identifies proteins associated with curcumin-enhancing efficacy of irinotecan-induced apoptosis of colorectal cancer LOVO cell. Int J Clin Exp Pathol. 2014; 7(1):1–15. https://doi.org/24427321.
Schulten H-J, Hussein D, Al-Adwani F, Karim S, Al-Maghrabi J, Al-Sharif M, Jamal A, Al-Ghamdi F, Baeesa SS, Bangash M, Chaudhary A, Al-Qahtani M. Microarray Expression Data Identify DCC as a Candidate Gene for Early Meningioma Progression. PloS ONE. 2016; 11(4):0153681. https://doi.org/10.1371/journal.pone.0153681.
Lin K-T, Shann Y-J, Chau G-Y, Hsu C-N, Huang C-YF. Identification of latent biomarkers in hepatocellular carcinoma by ultra-deep whole-transcriptome sequencing. Oncogene. 2014; 33(39):4786–94. https://doi.org/10.1038/onc.2013.424.
Liu Z, Lin Y, Liu X, Yu W, Zhang Y, Li D. [Experimental study of inhibition of tumor cell proliferation by a novel gene SPATA12]. Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences. 2012; 37(3):222–7. https://doi.org/10.3969/j.issn.1672-7347.2012.03.002.
Dong L, Hong H, Chen X, Huang Z, Wu W, Wu F. LINC02163 regulates growth and epithelial-to-mesenchymal transition phenotype via miR-593-3p/FOXK1 axis in gastric cancer cells. Artif Cells Nanomedicine Biotechnol. 2018; 0(0):1–9. https://doi.org/10.1080/21691401.2018.1464462.
Sagara A, Igarashi K, Otsuka M, Kodama A, Yamashita M, Sugiura R, Karasawa T, Arakawa K, Narita M, Kuzumaki N, Narita M, Kato Y. Endocan as a prognostic biomarker of triple-negative breast cancer. Breast Cancer Res Treat. 2017; 161(2):269–78. https://doi.org/10.1007/s10549-016-4057-8.
Feodorova Y, Tashkova D, Koev I, Todorov A, Kostov G, Simitchiev K, Belovejdov V, Dimov R, Sarafian V. Novel insights into transcriptional dysregulation in colorectal cancer. Neoplasma. 2018; 65(3):415–24. https://doi.org/10.4149/neo_2018_170707N467.
Jung Y-S, Jun S, Lee SH, Sharma A, Park J-I. Wnt2 complements Wnt/ β-catenin signaling in colorectal cancer. Oncotarget. 2015; 6(35):37257–68. https://doi.org/10.18632/oncotarget.6133.
Mercer KE, Hennings L, Ronis MJJ. Alcohol Consumption, Wnt/ β-Catenin Signaling, and Hepatocarcinogenesis. In: Journal of Arboriculture vol. 27: 2015. p. 185–95. https://doi.org/10.1007/978-3-319-09614-8_11.
Eskandari E, Mahjoubi F, Motalebzadeh J. An integrated study on TFs and miRNAs in colorectal cancer metastasis and evaluation of three co-regulated candidate genes as prognostic markers. Gene. 2018; 679(3):150–9. https://doi.org/10.1016/j.gene.2018.09.003.
Mesquita D, Barros-Silva JD, Santos J, Skotheim RI, Lothe RA, Paulo P, Teixeira MR. Specific and redundant activities of <i>ETV1</i> and <i>ETV4</i> in prostate cancer aggressiveness revealed by co-overexpression cellular contexts. Oncotarget. 2015; 6(7):5217–36. https://doi.org/10.18632/oncotarget.2847.
Taflin H, Odin E, Derwinger K, Carlsson G, Gustavsson B, Wettergren Y. Relationship between folate concentration and expression of folate-associated genes in tissue and plasma after intraoperative administration of leucovorin in patients with colorectal cancer. Cancer Chemother Pharmacol. 2018. https://doi.org/10.1007/s00280-018-3690-9.
Selcuklu SD, Donoghue MTA, Rehmet K, De Gomes MS, Fort A, Kovvuru P, Muniyappa MK, Kerin MJ, Enright AJ, Spillane C. MicroRNA-9 inhibition of cell proliferation and identification of novel miR-9 targets by transcriptome profiling in breast cancer cells. J Biol Chem. 2012; 287(35):29516–28. https://doi.org/10.1074/jbc.M111.335943.
Chen N, Guo D, Xu Q, Yang M, Wang D, Peng M, Ding Y, Wang S, Zhou J. Long non-coding RNA <i>FEZF1-AS1</i> facilitates cell proliferation and migration in colorectal carcinoma. Oncotarget. 2016; 7(10):8630–8. https://doi.org/10.18632/oncotarget.7168.
Zhang Z, Sun L, Zhang Y, Lu G, Li Y, Wei Z. Long non-coding RNA FEZF1-AS1 promotes breast cancer stemness and tumorigenesis via targeting miR-30a/Nanog axis. J Cell Physiol. 2018; 233(11):8630–8. https://doi.org/10.1002/jcp.26611.
Jia H, Song L, Cong Q, Wang J, Xu H, Chu Y, Li Q, Zhang Y, Zou X, Zhang C, Chin YE, Zhang X, Li Z, Zhu K, Wang B, Peng H, Hou Z. The LIM protein AJUBA promotes colorectal cancer cell survival through suppression of JAK1/STAT1/IFIT2 network. Oncogene. 2017; 36(19):2655–66. https://doi.org/10.1038/onc.2016.418.
Jia L, Gui B, Zheng D, Decker KF, Tinay I, Tan M, Wang X, Kibel AS. Androgen receptor-regulated miRNA-193a-3p targets AJUBA to promote prostate cancer cell migration. Prostate. 2017; 77(9):1000–11. https://doi.org/10.1002/pros.23356.
Zhang Y, Jia S, Jiang WG. KIAA1199 and its biological role in human cancer and cancer cells (Review). Oncol Rep. 2014; 31(4):1503–8. https://doi.org/10.3892/or.2014.3038.
Nakagawa S, Miyoshi N, Ishii H, Mimori K, Tanaka F, Sekimoto M, Doki Y, Mori M. Expression of CLDN1 in colorectal cancer: a novel marker for prognosis. Int J Oncol. 2011; 39(4):791–6. https://doi.org/10.3892/ijo.2011.1102.
Zhang Y, Zhang Y, Geng L, Yi H, Huo W, Talmon G, Kim YC, Wang SM, Wang J. Transforming growth factor β mediates drug resistance by regulating the expression of pyruvate dehydrogenase kinase 4 in colorectal cancer. J Biol Chem. 2016; 291(33):17405–16. https://doi.org/10.1074/jbc.M116.713735.
Royo F, Zuñiga-Garcia P, Torrano V, Loizaga A, Sanchez-Mosquera P, Ugalde-Olano A, González E, Cortazar AR, Palomo L, Fernández-Ruiz S, Lacasa-Viscasillas I, Berdasco M, Sutherland JD, Barrio R, Zabala-Letona A, Martín-Martín N, Arruabarrena-Aristorena A, Valcarcel-Jimenez L, Caro-Maldonado A, Gonzalez-Tampan J, Cachi-Fuentes G, Esteller M, Aransay AM, Unda M, Falcón-Pérez JM, Carracedo A. Transcriptomic profiling of urine extracellular vesicles reveals alterations of CDH3 in prostate cancer. Oncotarget. 2016; 7(6):1000–11. https://doi.org/10.18632/oncotarget.6899.
Zheng L-Z, Chen S-Z. shRNAinduced knockdown of the SPERT gene inhibits proliferation and promotes apoptosis of human colorectal cancer RKO cells,. Oncol Rep. 2018; 40(2):813–22. https://doi.org/10.3892/or.2018.6455.
Galamb O, Kalmár A, Barták BK, Patai Á,V, Leiszter K, Péterfia B, Wichmann B, Valcz G, Veres G, Tulassay Z, Molnár B. Aging related methylation influences the gene expression of key control genes in colorectal cancer and adenoma. World J Gastroenterol. 2016; 22(47):10325. https://doi.org/10.3748/wjg.v22.i47.10325.
Liu J, Liu Z, Liu Q, Li L, Fan X, Wen T, An G. CLEC3B is downregulated and inhibits proliferation in clear cell renal cell carcinoma. Oncol Rep. 2018; 7(6):1000–11. https://doi.org/10.3892/or.2018.6590.
Vishnubalaji R, Hamam R, Yue S, Al-Obeed O, Kassem M, Liu F-F, Aldahmash A, Alajez NM. MicroRNA-320 suppresses colorectal cancer by targeting SOX4, FOXM1, and FOXQ1. Oncotarget. 2016; 7(24):2752–60. https://doi.org/10.18632/oncotarget.8937.
Feng A, Yuan X, Li X. MicroRNA-345 inhibits metastasis and epithelial-mesenchymal transition of gastric cancer by targeting FOXQ1. Oncol Rep. 2017; 38(5):2752–60. https://doi.org/10.3892/or.2017.6001.
Okano M, Yamamoto H, Ohkuma H, Kano Y, Kim H, Nishikawa S, Konno M, Kawamoto K, Haraguchi N, Takemasa I, Mizushima T, Ikeda M, Yokobori T, Mimori K, Sekimoto M, Doki Y, Mori M, Ishii H. Significance of INHBA expression in human colorectal cancer. Oncol Rep. 2013; 30(6):2903–8. https://doi.org/10.3892/or.2013.2761.
Katayama Y, Oshima T, Sakamaki K, Aoyama T, Sato T, Masudo K, Shiozawa M, Yoshikawa T, Rino Y, Imada T, Masuda M. Clinical Significance of INHBA Gene Expression in Patients with Gastric Cancer who Receive Curative Resection Followed by Adjuvant S-1 Chemotherapy. In vivo (Athens, Greece). 2017; 31(4):565–71. https://doi.org/10.21873/invivo.11095.
Roberts DL, O’Dwyer ST, Stern PL, Renehan AG. Global gene expression in pseudomyxoma peritonei, with parallel development of two immortalized cell lines. Oncotarget. 2015; 6(13):10786–800. https://doi.org/10.18632/oncotarget.3198.
Vakrakou A, Devetzi M, Papachristopoulou G, Malachias A, Scorilas A, Xynopoulos D, Talieri M. Kallikrein-related peptidase 6 (KLK6) expression in the progression of colon adenoma to carcinoma. Biol Chem. 2014; 395(9):1105–17. https://doi.org/10.1515/hsz-2014-0166.
Khoury N, Zingkou E, Pampalakis G, Sofopoulos M, Zoumpourlis V, Sotiropoulou G. KLK6 protease accelerates skin tumor formation and progression. Carcinogenesis. 2018; 395(9):1105–17. https://doi.org/10.1093/carcin/bgy110.
Miyoshi N, Ishii H, Mimori K, Takatsuno Y, Kim H, Hirose H, Sekimoto M, Doki Y, Mori M. Abnormal expression of TRIB3 in colorectal cancer: a novel marker for prognosis. Br J Cancer. 2009; 101(10):1664–70. https://doi.org/10.1038/sj.bjc.6605361.
Li K, Zhang T-T, Hua F, Hu Z-W. Metformin reduces TRIB3 expression and restores autophagy flux: an alternative antitumor action. Autophagy. 2018; 14(7):1278–9. https://doi.org/10.1080/15548627.2018.1460022.
Liu Y, Wang G, Yang Y, Mei Z, Liang Z, Cui A, Wu T, Liu C-Y, Cui L. Increased TEAD4 expression and nuclear localization in colorectal cancer promote epithelial–mesenchymal transition and metastasis in a YAP-independent manner. Oncogene. 2016; 35(21):2789–800. https://doi.org/10.1038/onc.2015.342.
Lim B, Kim H-J, Heo H, Huh N, Baek S-J, Kim J-H, Bae D-H, Seo E-H, Lee S-I, Song K-S, Kim S-Y, Kim YS, Kim M. Epigenetic silencing of miR-1271 enhances MEK1 and TEAD4 expression in gastric cancer. Cancer Med. 2018; 35(21):2789–800. https://doi.org/10.1002/cam4.1605.
Jiang J, Liu H-L, Tao L, Lin X-Y, Yang Y-D, Tan S-W, Wu B. Let-7d inhibits colorectal cancer cell proliferation through the CST1/p65 pathway. Int J Oncol. 2018; 53(2):781–90. https://doi.org/10.3892/ijo.2018.4419.
Dai D-N, Li Y, Chen B, Du Y, Li S-B, Lu S-X, Zhao Z-P, Zhou A-J, Xue N, Xia T-L, Zeng M-S, Zhong Q, Wei W-D. Elevated expression of CST1 promotes breast cancer progression and predicts a poor prognosis. J Mol Med (Berlin, Germany). 2017; 95(8):873–86. https://doi.org/10.1007/s00109-017-1537-1.
Wang N, Chen Y, Han Y, Zhao Y, Liu Y, Guo K, Jiang Y. Proteomic analysis shows down-regulations of cytoplasmic carbonic anhydrases, CAI and CAII, are early events of colorectal carcinogenesis but are not correlated with lymph node metastasis. Tumori. 2012; 98(6):783–91. https://doi.org/10.1700/1217.13504.
Zheng Y, Xu B, Zhao Y, Gu H, Li C, Wang Y, Chang X. CA1 contributes to microcalcification and tumourigenesis in breast cancer. BMC Cancer. 2015; 15(6):679. https://doi.org/10.1186/s12885-015-1707-x.
Takagi Y, Shimada K, Shimada S, Sakamoto A, Naoe T, Nakamura S, Hayakawa F, Tomita A, Kiyoi H. SPIB is a novel prognostic factor in diffuse large B-cell lymphoma that mediates apoptosis via the PI3K-AKT pathway. Cancer Sci. 2016; 107(9):1270–80. https://doi.org/10.1111/cas.13001.
Ferguson HJM, Wragg JW, Ward S, Heath VL, Ismail T, Bicknell R. Glutamate dependent NMDA receptor 2D is a novel angiogenic tumour endothelial marker in colorectal cancer. Oncotarget. 2016; 7(15):20440–54. https://doi.org/10.18632/oncotarget.7812.
Sharma A, Jiang C, De S. Dissecting the sources of gene expression variation in a pan-cancer analysis identifies novel regulatory mutations. Nucleic Acids Res. 2018; 46(9):4370–81. https://doi.org/10.1093/nar/gky271.
Meyer RD, Zou X, Ali M, Ersoy E, Bondzie PA, Lavaei M, Alexandrov I, Henderson J, Rahimi N. TMIGD1 acts as a tumor suppressor through regulation of p21Cip1/p27Kip1 in renal cancer. Oncotarget. 2018; 9(11):9672–84. https://doi.org/10.18632/oncotarget.23822.
Wang W, Li Q, Yang T, Bai G, Li D, Li Q, Sun H. Expression of AQP5 and AQP8 in human colorectal carcinoma and their clinical significance. World J Surg Oncol. 2012; 10(11):242. https://doi.org/10.1186/1477-7819-10-242.
Ma J, Zhou C, Yang J, Ding X, Zhu Y, Chen X. Expression of AQP6 and AQP8 in epithelial ovarian tumor. J Mol Histol. 2016; 47(2):129–34. https://doi.org/10.1007/s10735-016-9657-4.
Chao C-C, Wu P-H, Huang H-C, Chung H-Y, Chou Y-C, Cai B-H, Kannagi R. Downregulation of miR-199a/b-5p is associated with GCNT2 induction upon epithelial-mesenchymal transition in colon cancer. FEBS Lett. 2017; 591(13):1902–17. https://doi.org/10.1002/1873-3468.12685.
Murakami M, Yoshimoto T, Nakabayashi K, Tsuchiya K, Minami I, Bouchi R, Izumiyama H, Fujii Y, Abe K, Tayama C, Hashimoto K, Suganami T, Hata K-i, Kihara K, Ogawa Y. Integration of transcriptome and methylome analysis of aldosterone-producing adenomas. Eur J Endocrinol. 2015; 173(2):185–95. https://doi.org/10.1530/EJE-15-0148.
Yang B, Cao L, Liu B, McCaig CD, Pu J. The Transition from Proliferation to Differentiation in Colorectal Cancer Is Regulated by the Calcium Activated Chloride Channel A1. PLoS ONE. 2013;8(4). https://doi.org/10.1371/journal.pone.0060861.
Yu Y, Walia V, Elble RC. Loss of CLCA4 promotes epithelial-to-mesenchymal transition in breast cancer cells. PLoS ONE. 2013; 8(12):4–12. https://doi.org/10.1371/journal.pone.0083943.
Atsumi T, Singh R, Sabharwal L, Bando H, Meng J, Arima Y, Yamada M, Harada M, Jiang J-J, Kamimura D, Ogura H, Hirano T, Murakami M. Inflammation amplifier, a new paradigm in cancer biology. Cancer Res. 2014; 74(1):8–14. https://doi.org/10.1158/0008-5472.CAN-13-2322.
Solé X, Crous-Bou M, Cordero D, Olivares D, Guinó E, Sanz-Pamplona R, Rodriguez-Moranta F, Sanjuan X, de Oca J, Salazar R, Moreno V. Discovery and validation of new potential biomarkers for early detection of colon cancer. PloS ONE. 2014; 9(9):106748. https://doi.org/10.1371/journal.pone.0106748.
Li T, Huang H, Shi G, Zhao L, Li T, Zhang Z, Liu R, Hu Y, Liu H, Yu J, Li G. TGF- β1-SOX9 axis-inducible COL10A1 promotes invasion and metastasis in gastric cancer via epithelial-to-mesenchymal transition. Cell Death Dis. 2018; 9(9):849. https://doi.org/10.1038/s41419-018-0877-2.
Klupp F, Neumann L, Kahlert C, Diers J, Halama N, Franz C, Schmidt T, Koch M, Weitz J, Schneider M, Ulrich A. Serum MMP7, MMP10 and MMP12 level as negative prognostic markers in colon cancer patients. BMC Cancer. 2016; 16(5):494. https://doi.org/10.1186/s12885-016-2515-7.
Zhang Q, Liu S, Parajuli KR, Zhang W, Zhang K, Mo Z, Liu J, Chen Z, Yang S, Wang AR, Myers L, You Z. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene. 2017; 36(5):687–99. https://doi.org/10.1038/onc.2016.240.
Chung S, Furihata M, Tamura K, Uemura M, Daigo Y, Nasu Y, Miki T, Shuin T, Fujioka T, Nakamura Y, Nakagawa H. Overexpressing PKIB in prostate cancer promotes its aggressiveness by linking between PKA and Akt pathways. Oncogene. 2009; 28(32):2849–59. https://doi.org/10.1038/onc.2009.144.
Acknowledgements
Not Applicable.
Funding
This work has been supported by Project DPI2017-87333-R (G.A.) with FEDER funds from the Spanish Ministry of Economy and Competitiveness; and by Chilean CONICYT/FONDECYT-POSTDOCTORADO N o3180486 (A.L.R.-C).
Availability of data and materials
All the data used in this manuscript are public and available in the repositories of The Cancer Genome Atlas(TCGA, on https://gdc.cancer.gov/, projects TCGA-COAD and TCGA-READ) and NCBI Sequence Read Archive (SRA, on https://www.ncbi.nlm.nih.gov/sra, accession number PRJNA218851), as we indicated in the Materials and Methods section. The OMICfpp is available at http://www.uv.es/ayala/software/OMICfpp_0.2.tar.gz. All methodology and results have been uploaded as part of the electronic supplementary material.
Author information
Authors and Affiliations
Contributions
GA and ALR-C conceived and designed the study. AB-G and ALR-C performed downloaded and pre-processing of data. GA developed the R package OMICfpp. AB-G, ALR-C and GA analyzed and interpreted the data. ALR-C and GA carried out the figures, tables and drafting the manuscript. AB-G, ALR-C and GA participated in the critical reading and manuscript edition. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1
All procedures and data needed to reproduce the whole study have been included in the file SupplementaryMaterial.tar.gz. Once decompressed the file SupplementaryMaterialMethods.pdf contains a detailed description of the methods used and the results obtained. The whole paper can be reproduced reading this file. Other data files generated during the analysis are included in the folder Methods. The detailed html reports with the results can be found in the folder Results. (GZ 118,244 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Berral-Gonzalez, A., Riffo-Campos, A. & Ayala, G. OMICfpp: a fuzzy approach for paired RNA-Seq counts. BMC Genomics 20, 259 (2019). https://doi.org/10.1186/s12864-019-5496-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5496-5