
Abstract
Background
Sequencing data has become a standard measure of diverse cellular activities. For example, gene expression is accurately measured by RNA sequencing (RNA-Seq) libraries, protein-DNA interactions are captured by chromatin immunoprecipitation sequencing (ChIP-Seq), protein-RNA interactions by crosslinking immunoprecipitation sequencing (CLIP-Seq) or RNA immunoprecipitation (RIP-Seq) sequencing, DNA accessibility by assay for transposase-accessible chromatin (ATAC-Seq), DNase or MNase sequencing libraries. The processing of these sequencing techniques involves library-specific approaches. However, in all cases, once the sequencing libraries are processed, the result is a count table specifying the estimated number of reads originating from each genomic locus. Differential analysis to determine which loci have different cellular activity under different conditions starts with the count table and iterates through a cycle of data assessment, preparation and analysis. Such complex analysis often relies on multiple programs and is therefore a challenge for those without programming skills.
Results
We developed DEBrowser as an R bioconductor project to interactively visualize every step of the differential analysis, without programming. The application provides a rich and interactive web based graphical user interface built on R’s shiny infrastructure. DEBrowser allows users to visualize data with various types of graphs that can be explored further by selecting and re-plotting any desired subset of data. Using the visualization approaches provided, users can determine and correct technical variations such as batch effects and sequencing depth that affect differential analysis. We show DEBrowser’s ease of use by reproducing the analysis of two previously published data sets.
Conclusions
DEBrowser is a flexible, intuitive, web-based analysis platform that enables an iterative and interactive analysis of count data without any requirement of programming knowledge.
Similar content being viewed by others
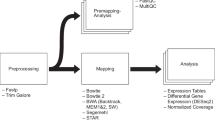

Background
Sequencing techniques have been widely used to measure the activity of genomic regions across conditions. Typical uses include differential expression [1,2,3], small RNA abundances [4, 5], epigenetic state [6, 7], protein/RNA interactions [8,9,10] and DNA/RNA interactions [11, 12]. Even though each of these sequencing libraries requires very specific processing steps to determine the genomic loci underlying the observed sequencing reads [13,14,15], the outputs are always count matrices with rows representing the genomic features of interest such as genes, exons, DNA accessible or DNA bound regions, and the columns being the samples. The values in this table are the estimated number of reads originated from each defined locus for each sample. The most common goal in the analysis of such tables is to find loci exhibiting significant differences between different groups of samples. Differential analysis of count data typically involves an iterative approach that heavily relies on visualization and unsupervised statistical analysis. A typical analysis consists of an iterative application of three main tasks; data assessment, data preparation, and differential expression analysis (Fig. 1). Here we present an application to enable interactive exploration of count matrices obtained after processing of the sequencing output.

DEBrowser overview
Data assessment evaluates the impact of latent factors that may not be related to biological differences. Such differences might come from technical factors, such as DNA/RNA fragmentation, the number of PCR cycles, or sequencing depth that may altogether confound the actual biologically relevant differences present in different samples [16, 17]. Therefore, the count matrix represents a combination of both biological and technical variation. Unsupervised methods such as clustering and principal component analysis (PCA) are powerful ways to identify technical sources of variance [18,19,20]. Data preparation builds on data assessment to determine and then apply the best approaches to reduce the impact of unwanted sources of variance. Typically this includes the elimination of low quality samples, and filtering or removal of features having low counts [21,22,23,24,25]. Data preparation may also include batch effect correction [26], which removes variability between groups of samples resulting from technical differences in the day, reagents or experimental personnel involved with sample preparation. Following these pre-processing steps often leads to more accurate differential analysis. Differential Analysis seeks to identify features that have higher or lower counts between groups of samples that represent different biological conditions.
We created DEBrowser to enable iterative analysis by non-programmers to achieve similar results to those obtained by investigators well versed in the R programming language. DEBrowser facilitates a modular iterative analysis and visualization process through an intuitive user interface, integrating multiple algorithms and visualization techniques. The goal is to allow users to iteratively inspect and apply each of the many approaches comprised of the three stages described above. DEBrowser provides an evaluation of the results at each analysis step and determines whether further improvements are necessary. DEBrowser goes further than providing static plots or heatmaps: it allows users to explore any anomaly or potential result in an interactive and dynamic manner by zooming-in on data subsets and selecting or hovering over any regions or genes of interest to plot a heatmap, bar, or box plots that updates dynamically based on the user’s selection. For example, users may select the most significantly differentially expressed genes on a volcano plot, and re-display them in a heatmap, that can be further used to inspect the expression of each individual genes across all replicates.
DE analysis and visualization packages
A number of graphical user interfaces address the need for user-friendly, programming-free visualizations [27,28,29,30,31,32,33] (Additional file 1: Table S1). However, all of these approaches have limited interactivity for users to carry out more sophisticated analysis. Similar to DEBrowser, existing tools accept count data as input to visualize, identify, and perform differential analysis and gene ontology. They also visualize results using scatter, MA or volcano plots, as well as heatmaps and PCA plots. DEBrowser goes one step further by enabling hands-on manipulation of the data and by enabling users to re-plot selected subsets of data. These features make DEBrowser a sophisticated tool for data exploration. DEBrowser allows users to color genes that exceed the different fold change cutoffs after differential expression analysis with only a few clicks. Furthermore, all plots are immediately redrawn immediately after changes in the plotting parameters or after any data subsetting operation. Similarly, for easy access to the underlying data, DEBrowser supports hovering to obtain detailed information of individual data points.
In contrast to existing visualization and differential analysis tools, DEBrowser places a strong focus on data assessment and preparation: it intrinsically supports normalization and batch correction methods. Once the user identifies a clear bias in data preparation, processing or sequencing, DEBrowser allows the user to minimize technical variation between samples using proven methods [26, 34,35,36,37,38]. This capability is intended to address the needs of large projects that process samples over an extended period of time, and to help users compare samples available from public repositories originating from different laboratories.
Implementation
DEBrowser is implemented in R as a shiny application using generic shiny components and layouts [39]. In particular, DEBrowser relies on R’s plotly package [40] both for interactive plots and to display multi-panel data.
Shiny’s reactive programming model is used to update plots upon detection of changes in any input control, plot, or any other bound object. Automatic re-plotting reduces the number of clicks required and therefore improves user experience. When a change in a plot parameter is detected, plots bound to that input are redrawn. As a result, with few clicks, users can, for example, change the highlighted genes from the DE results that exceed a significance of 0.05 and a 2-fold change, to those exceeding a significance of 0.01 and a 10-fold change.
In a typical DEBrowser session users upload a count table obtained by processing of sequencing reads and a table specifying sample information: Conditions and batches. No other inputs are needed (Additional file 2: Figure S1).
Design and key features
To show the general applicability of DEBrowser on “count data” from different data types; we used a large data set that we recently generated to study gene regulation in innate immune cells (human monocyte derived dendritic cells, hMDDCs) in response to Toll-like receptor signaling [41]. This study generated RNA-Seq, ChIP-Seq and ATAC-Seq [42] to track changes in transcription and regulatory element activity in the course of Toll-like receptor (TLR) signaling. We reprocessed all raw sequence reads either as described in the original publication or using more recent approaches (Additional file 3).
These data are ideal to showcase the main features of DEBrowser and how DEBrowser can be used throughout the analysis cycle. Indeed, we show how DEBrowser was used for data assessment to identify batch effects, and data preparation by filtering low count features and removing batch effects and then performing differential analysis.
Data assessment
Quality control (QC): Quality control of the count data is a fundamental step in analysis, yet it is not well supported in current applications. With DEBrowser users can easily establish whether normalization, batch correction or sample removal are necessary, or if the data is suitable for differential analysis. To this end, DEBrowser implements PCA, all2all scatter, heatmaps, interquartile range (IQR) and density plots of each sample. These plots can be drawn using a user defined subset of genes, for example by choosing the top N variable genes as defined by coefficient of variance [40, 41]. The subset of genes can be defined graphically by either an expression cutoff or by directly selecting them from another plot.
For example, the data for hMDDCs show clear donor dependent differences, which are visible in all2all plots and are captured by the second principal component (Fig. 2a). These differences may be the result of genetic heterogeneity or simply due to technical variability in library construction. Regardless of the source, the variability introduced by inter-donor differences has a direct impact on the power to detect TLR responsive genes.

Identifying and correcting batch effects. PCA plot (a) before and (b) after batch effect correction. Circles and triangles represent batch 1 and 2, respectively
Plots available for data assessment
Principal component analysis (PCA)
PCA finds an ordered coordinate transformation whose basis capture, in decreasing order, the most variance in the data. DEBrowser allows users to plot any pair of principal components in a scatter plot. Once users specify sample information (e.g. condition), DEBrowser uses colors or shapes to group samples. These plots are ideal to detect outliers or batch effects (Fig. 2).
All2all scatter plots
Gives an overview of sample similarities and variance by plotting all pairwise scatter plots (Additional file 2: Figure S2). Low correlation or high variance across replicates will negatively impact the power to detect DE.
Heatmaps
DEBrowser allows users select genes based on variance, minimum expression level, DE p-value, or after manually selecting a set of genes from any gene centric plots (e.g. scatter, volcano, and other heatmaps). Heatmaps are also useful to assess replicate variability, low quality samples, or batch effects (Fig. 3). Similar to other plots, heatmaps can be used to visualize any type of count data and are ideal to identify global patterns in the data such as dynamic changes in chromatin accessibility following TLR signaling [41] (Additional file 2: Figure S3).

Visualizing the effect of batch correction. Heatmap for normalized RNA-Seq data (log10 transformed, scaled and centered) at different time points from two donors before (a) and after (b) batch correction. Heatmaps show the top 1000 most varied genes (based on coefficient of variance) whose total minimum counts are higher than 100
IQR and density plots
Interquartile range and density plots display a sample’s quantification distribution in different ways. Using these plots, users can detect any global discrepancy across samples and evaluate the impact of normalization on the distribution of counts. DEBrowser simplifies comparisons by providing plots for both normalized and unnormalized data. Plots are re-drawn as soon as users change the normalization method.
Data preparation
Removal of low coverage features
Removing features (genes, or genomic regions) that have low coverage due to their low expression, increases the speed and accuracy of DE algorithms. It also helps to perform more accurate dispersion calculations and multiple hypothesis correction [43]. DEBrowser provides three common ways to filter these features: by specifying a minimum signal in at least one sample, by a minimum average signal across all samples or by requiring a minimum signal in at least n samples (n defined by the users). Once filtering a criterion is specified, DEBrowser reports read counts for each sample (Fig. 4a, b) and plots the feature count distributions before and after applying the filtering (Fig. 4c, d).

Setting detection levels. DEBrowser displays the total number of counts per library (a, b) and a histogram of log10 transformed unnormalized feature counts (c, d) before and after filtering features below the user defined detection level
Normalization
The count data originating from a sequencing experiment is affected by sequencing depth as well as from differences in the composition of the detected features [44, 45]. DEBrowser supports normalization methods specifically designed for count data: median ratio normalization (MRN) implemented in DESeq2 package [43, 46], Trimmed Mean of M-values (TMM), Relative Log Expression (RLE), and upper quartile methods implemented in the EdgeR package [47]. To evaluate the effect of normalization, DEBrowser immediately displays PCA, IQR and density plots after normalization.
Batch effect correction
When quality control shows a clear batch effect that can be traced back to a technical artifact (e.g. different sequencing devices, different personnel, library kits, reagent batch), DEBrowser allows the users, if the experimental design allows batch correction, to minimize the batch effect [48]. The users can specify a batch for every sample via a simple tab separated file that can be created using a text editor or spreadsheet software. Given a batch specification, DEBrowser supports two different methods: ComBat [26, 49] and Harman [50]. The resulting, batch corrected dataset can be evaluated using the same tools available for initial quality control (Figs. 2b, 3b). DEBrowser provides a platform to detect, correct and evaluate the result of batch correction.
Differential expression (DE) analysis
To demonstrate a typical usage of DEBrowser, we applied DEBrowser on a data set from a previously published study on the role of Jun terminal kinases (JNK1 and JNK2) in the liver and their role in insulin resistance [2]. For this purpose, the authors relied on four different mouse genotypes: wild type (WT), and hepatocyte specific knockouts of Jnk1 and Jnk2 independently (LΔ1, LΔ2), and a double knockout (LΔ1,2). Each genotype was fed either a regular or a high fat diet (HFD). Thereafter, hepatic expression was assayed for each genotype fed with corresponding diet in triplicate using RNA-Seq, resulting in a total of 24 libraries. This study is ideal for DE analysis as it included three replicates per condition. Therefore, we used RSEM [15] for library quantification and DEBrowser to analyze the resulting read count table.
DEBrowser supports differential analysis using DESeq2 [43], EdgeR [47], and Limma [51]. We used all three methods and present the analysis done with DESeq2 in the main figures and the comparisons between all three methods in the supplementary figures (Additional file 2: Figures S6 and S7). Users can perform differential analysis after defining the groups of samples to compare. DE results can be visualized through the same scatter, volcano and MA plots used for data assessment (Fig. 5a-c). Users can highlight results by specifying desired significance and fold change cut-offs. All plots allow interactive access: Users may select a point within the plot to zoom-in and re-display only selected data. Plots are redrawn as soon as the users change any parameter or select points to zoom-in on any data point or set of points that can be investigated by graphically selecting them.

DE visualization. All detected genes are plotted as a (a) scatter plot, (b) volcano plot, and (c) MA plot. Genes that pass a threshold of padj < 0.01 and |log2foldChange| > 1 in differential expression analysis are colored in blue when they are down regulated in control (WT) and red when they are upregulated in the double KO
As reported by the authors, high fat diet has a stronger effect on LΔ2 animals compared LΔ1 animals (Additional file 2: Figure S8). To examine genes that are dysregulated in the liver under high fat diet fed mice, we performed DE analysis between WT mice fed with a normal or high fat diet. In all, 493 genes are significantly down regulated and 350 are up regulated in livers of mice fed with a high-fat diet (p < 0.01, |log2foldChange| > 1). Disease ontology analysis of up regulated genes shows, not surprisingly, a clear enrichment of diseases resulting from poor diet (urinary, kidney and other obesity related ailments). Here to show an example, we overlaid enriched insulin signaling pathway genes on a scatter plot (Fig. 6a) and easily created a heatmap (Fig. 6b) by using selected genes on this scatter plot similar to that in Additional file 2: Figure S3-B in the original report [2].

Heat map visualization. Heatmaps of scaled and centered Log10 transformed normalized read counts (a). As in 5A but with genes annotated in the insulin signaling pathway highlighted in green and (b). Only differentially expressed genes involved insulin signaling. c All genes differentially expressed in at least one pairwise comparison across all genotypes fed HFD and regular diet (|log2foldChange| > 1 and padj< 0.01). d Heatmap showing only genes in the PPARα pathway, averaged across all replicates of a given condition
We then compared the effect of both normal and HFD on all four genotypes. To do so we performed pairwise comparisons between all conditions and selected all genes with a |log2foldChange| > 1 and padj< 0.01 in at least one comparison to display in a heatmap similar to Additional file 2: Figure S3-A in the original report (Fig. 6c). Finally, we used DEBrowser to reproduce Additional file 2: Figure S3-B in original report. To do so we performed gene ontology analysis of the DE genes between WT and LΔ1,2 on HFD, selected genes that are annotated as part of the PPARα pathway and visualized them as a heatmap (Fig. 6d).
It is important to note that the original publication used much older DE methods [52]. When we applied DESeq2 to replicate the analysis we found that it had much greater power to detect differentially expressed genes and indeed at a similar threshold many more genes are called DE. Nevertheless, there is a very good agreement (73%) in the calls made by both methods (Additional file 2: Figure S4), and most importantly, there are no differences with the Gene Ontology enrichments reported in the original publication (Additional file 2: Figure S5).
Further, users can explore individual regions by hovering over points. The gene or region id is shown and a bar plot displaying the values of the gene or region across all samples is drawn. This is especially useful to investigate, for example, why certain genes may have large differences in between samples but fail to achieve statistical significance. In Fig. 7a, the, Fabp3 gene fails to achieve a significance of 0.05 despite of its mean expression between the conditions exceeding a 50-fold change. Hovering over Fabp3 shows the high variance of this gene across samples, which explains why statistical tests that account for both inter and intra condition variability fail to achieve significance in cases of high intra condition variability (Fig. 7b).

Why are genes with high fold change not significantly DE? a FABP3 gene is hovered over on a scatter plot. b The read counts for FABP3 gene is shown as a bar plot, showing high intra condition variability which explains the lack of significance in DE
Volcano, Scatter and MA plots work on summary statistics: significance averages or fold change of averages. To explore the underlying data for any set of regions in a plot, DEBrowser can draw heatmaps for any selected region from any main plot. Selection can be made in a rectangular form or as a free-form using plotly’s lasso select (Fig. 8a), which then dynamically generates a heatmap of the selection. (Fig. 8b). Conversely, in any heatmap the users can select a subset of regions (such as based on similar expression pattern) for downstream analysis such as gene ontology, disease and pathway analysis.

Gene selection using free form drawing. a Scatter plot of up and down regulated genes. b Heatmap of lasso selected of two up (Apob, C3) and two down regulated genes (Apoa1, Ftl1)
Gene ontology, disease and pathway discovery
For gene expression analysis in particular, DEBrowser supports Gene Ontology (GO) [53], KEGG pathway [53] and disease ontology analysis [54]. Users can perform GO or Pathway analysis directly on the results of differential expression analysis or on a subset of selected genes from any of the plots described above. For KEGG pathway analysis, in particular, DEBrowser provides pathway diagram for each enriched category (Fig. 9).

Pathway visualization. Insulin signaling pathway. Detected DE genes shown with colors according to their fold changes
To further assist users in differential analysis, DEBrowser provides k-means clustering of differential regions, and when these regions are associated with genes, a gene ontology enrichment analysis is performed using enrichGO function in ClusterProfiler package [53] (Fig. 10).

Ontology comparisons in subclusters. An example of a cluster profiler plot
Comparison to related applications
There are several applications, with varying functionalities, available for the exploration and analysis of DE. Most notable ones are, OASIS [27], VisRseq [28], DEGUST [29], DEIVA [30], WebMeV [31], Chipster [32], and DEapp [33]. A comparison of DEBrowser features to those applications is shown in Additional file 1: Table S1.
DEBrowser modular design
To reduce the code complexity and manage the program easier the components were designed in a modular fashion, so that while DEBrowser grows larger, it is easy to build on top of the simple modules. To this end, bar, box and scatter plots, heatmaps modules could be reused multiple times in DEBrowser. We also shared example shiny applications that use individual modules. This modularity increased our development, test speed, and code reusability. For example, the size and the margins of the plots are controlled within the same module in all the plots in DEBrowser. This modular design allows other users to repurpose any of the tools built into DEBrowser for their own packages.
Conclusion
Existing tools do not fully support the full process of differential expression analysis and visualization. Additionally, the plots are usually static and do not allow interactivity to understand the different parts of the data using different parameters reducing the efficiency of data exploration.
In contrast, the DEBrowser application provides users, who do not have any programming experience, the ability to perform their own analysis in an iterative and interactive process that responds dynamically to user inputs. DEBrowser leverages open source components that are in active development in bioconductor [55, 56], thus it benefits from a large community of developers. Its modular design makes it easy to swap components shall new paradigms or projects emerge that provide more ideal functionality than currently available. Therefore, it fills a much-needed void in graphical user interfaces for the analysis of count data that is typical of sequencing assays.
Abbreviations
- ATAC-Seq:
-
Assay for transposase-accessible chromatin
- ChIP-Seq:
-
Chromatin immunoprecipitation sequencing
- CLIP-Seq:
-
Cross-linking immunoprecipitation sequencing
- GO:
-
Gene Ontology
- hMDDCs:
-
Human monocyte derived mouse dendritic cells
- IQR:
-
Interquartile range
- JNK1 and JNK1:
-
Jun terminal kinases
- LΔ1 :
-
JNK1 knockout
- LΔ1,2 :
-
JNK1 and JNK2 double knockout
- LΔ2 :
-
JNK2 knockout
- MRN:
-
Median ratio normalization
- PCA:
-
Principal component analysis
- QC:
-
Quality control
- RIP-Seq:
-
RNA immunoprecipitation sequencing
- RLE:
-
Relative Log Expression
- RNA-Seq:
-
RNA sequencing
- TLR:
-
Toll-like receptor
- TMM:
-
Trimmed Mean of M-values
- WT:
-
Wild type
References
Blodgett DM, Nowosielska A, Afik S, Pechhold S, Cura AJ, Kennedy NJ, et al. Novel observations from next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes. 2015;64:3172–81.
Vernia S, Cavanagh-Kyros J, Garcia-Haro L, Sabio G, Barrett T, Jung DY, et al. The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014;20:512–25.
Mezan S, Ashwal-Fluss R, Shenhav R, Garber M, Kadener S. Genome-wide assessment of post-transcriptional control in the fly brain. Front Mol Neurosci. 2013;6:49.
Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391–6.
Beaulieu LM, Vitseva O, Tanriverdi K, Kucukural A, Mick E, Hamburg N, et al. Platelet functional and transcriptional changes induced by intralipid infusion. Thromb Haemost. 2016;115:1147–56.
Shea JM, Serra RW, Carone BR, Shulha HP, Kucukural A, Ziller MJ, et al. Genetic and epigenetic variation, but not diet, Shape the Sperm Methylome. Dev Cell. 2015;35:750–8.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–54.
Ricci EP, Kucukural A, Cenik C, Mercier BC, Singh G, Heyer EE, et al. Staufen1 senses overall transcript secondary structure to regulate translation. Nat Struct Mol Biol. 2014;21:26–35.
Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, et al. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 2012;151:750–64.
Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem. 2011;286:1204–15.
Engreitz J, Lander ES, Guttman M. RNA antisense purification (RAP) for mapping RNA interactions with chromatin. Methods Mol Biol. 2015;1262:183–97.
Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667–78.
Kucukural A, Özadam H, Singh G, Moore MJ, Cenik C. ASPeak: an abundance sensitive peak detection algorithm for RIP-Seq. Bioinformatics. 2013;29:2485–6.
Liu T. Use model-based analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein–DNA interactions in embryonic stem cells. In: Methods in Molecular Biology; 2014. p. 81–95.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323.
Head SR, Komori HK, LaMere SA, Whisenant T, Van Nieuwerburgh F, Salomon DR, et al. Library construction for next-generation sequencing: overviews and challenges. Biotechniques. 2014;56:61–4 66, 68, passim.
Tyler AD, Christianson S, Knox NC, Mabon P, Wolfe J, Van Domselaar G, et al. Comparison of sample preparation methods used for the next-generation sequencing of Mycobacterium tuberculosis. PLoS One. 2016;11:e0148676.
Alter O, Brown PO, Botstein D. Singular value decomposition for genome-wide expression data processing and modeling. Proc Natl Acad Sci U S A. 2000;97:10101–6.
Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–8.
Bono H, Nakao M, Kanehisa M. Cluster analysis of genome-wide expression profiles to predict gene functions with KEGG. Nat Genet. 1999;23:33–4.
Lin Y, Golovnina K, Chen Z-X, Lee HN, Negron YLS, Sultana H, et al. Comparison of normalization and differential expression analyses using RNA-Seq data from 726 individual Drosophila melanogaster. BMC Genomics. 2016;17:28.
Bourgon R, Gentleman R, Huber W. Independent filtering increases detection power for high-throughput experiments. Proc Natl Acad Sci U S A. 2010;107:9546–51.
Rau A, Gallopin M, Celeux G, Jaffrézic F. Data-based filtering for replicated high-throughput transcriptome sequencing experiments. Bioinformatics. 2013;29:2146–52.
Maza E, Frasse P, Senin P, Bouzayen M, Zouine M. Comparison of normalization methods for differential gene expression analysis in RNA-Seq experiments: a matter of relative size of studied transcriptomes. Commun Integr Biol. 2013;6:e25849.
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25.
Leek JT, Evan Johnson W, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–3.
Fernandez-Banet J, Esposito A, Coffin S, Schefzick S, Ding Y, Ching K, et al. Abstract 4874: OASIS: a centralized portal for cancer omics data analysis. Cancer Res. 2015;75(15 Supplement):4874.
Younesy H, Möller T, Lorincz MC, Karimi MM, Jones SJM. VisRseq: R-based visual framework for analysis of sequencing data. BMC Bioinf. 2015;16(Suppl 11):S2.
Powell DR. Degust: Visualize, explore and appreciate RNA-seq differential gene-expression data; 2015.
Harshbarger J, Kratz A, Carninci P. DEIVA: a web application for interactive visual analysis of differential gene expression profiles. BMC Genomics. 2017;18:47.
Wang YE, Kuznetsov L, Partensky A, Farid J, Quackenbush J. WebMeV: A Cloud Platform for Analyzing and Visualizing Cancer Genomic Data. 2017. doi: https://doi.org/10.1101/147884
Kallio MA, Aleksi Kallio M, Tuimala JT, Hupponen T, Klemelä P, Gentile M, et al. Chipster: user-friendly analysis software for microarray and other high-throughput data. BMC Genomics 2011;12. doi:https://doi.org/10.1186/1471-2164-12-507
Li Y, Andrade J. DEApp: an interactive web interface for differential expression analysis of next generation sequence data. Source Code Biol Med. 2017;12. https://doi.org/10.1186/s13029-017-0063-4.
Smyth GK, Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–73.
Giordan M. A two-stage procedure for the removal of batch effects in microarray studies. Stat Biosci. 2014;6:73–84.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Nyamundanda G, Poudel P, Patil Y, Sadanandam A. A novel statistical method to diagnose, Quantify and Correct Batch Effects in Genomic Studies. Sci Rep. 2017;7:10849.
Nygaard V, Rødland EA, Hovig E. Methods that remove batch effects while retaining group differences may lead to exaggerated confidence in downstream analyses. Biostatistics. 2016;17:29–39.
Chang W, Cheng J, Allaire JJ, Xie Y, McPherson J. shiny: Web Application Framework for R. 2016. https://CRAN.R-project.org/package=shiny.
Sievert C, Parmer C, Hocking T, Chamberlain S, Ram K, Corvellec M, et al. plotly: Create Interactive Web Graphics via “plotly. js.” R package version, vol. 3; 2016.
Donnard E, Vangala P, Afik S, McCauley S, Nowosielska A, Kucukural A, et al. Comparative analysis of immune cells reveals a conserved regulatory lexicon. Cell Syst. 2018. https://doi.org/10.1016/j.cels.2018.01.002.
Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109:21–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Lee S, Seo CH, Lim B, Yang JO, Oh J, Kim M, et al. Accurate quantification of transcriptome from RNA-Seq data by effective length normalization. Nucleic Acids Res. 2011;39:e9.
Li P, Piao Y, Shon HS, Ryu KH. Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data. BMC Bioinf. 2015;16:347.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Bass B, Hundley H, Li JB, Peng Z, Pickrell J, Xiao XG, et al. The difficult calls in RNA editing. Nat Biotechnol. 2012;30:1207.
Johnson WE, Evan Johnson W, Li C. Adjusting batch effects in microarray experiments with small sample size using empirical bayes methods. In: Batch effects and noise in microarray experiments, Biostatistics. 2007;8(1):118–27.
Oytam Y, Sobhanmanesh F, Duesing K, Bowden JC, Osmond-McLeod M, Ross J. Risk-conscious correction of batch effects: maximising information extraction from high-throughput genomic datasets. BMC Bioinformatics. 2016;17:332.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7:562–78.
Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Yu G, Wang L-G, Yan G-R, He Q-Y. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 2015;31:608–9.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80.
Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015;12:115–21.
Acknowledgements
We would like to thank Kyle Gellatly, Pranitha Vangala, Elisa Donnard, Rachel Murphy, Alan Derr, Laney Zuerlein, Stephen McGregor, Michael P. Czech, Leonardo Collado-Torres, and all members of the Garber Lab for their suggestions and comments.
Funding
This work was supported by fund from the National Human Genome Research Institute NHGRI grant #U01 HG007910–01 and the National Center for Advancing Translational Sciences grant #UL1 TR001453–01 (M.G).
Availability of data and materials
Project name: DEBrowser
Project home page: https://bioconductor.org/packages/release/bioc/html/debrowser.html, https://github.com/UMMS-Biocore/debrowser
Operation systems: Platform independent
Programming language: R (> = 3.5.0)
License: GPL-v3
Restrictions to use by non-academics: None
Author information
Authors and Affiliations
Contributions
AK and MJM AK implemented the package and wrote the manuscript. OY and DO interpreted data. MG supervised the project and wrote the manuscript. AK, MJM and MG conceived the project. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Application feature comparison table. (DOCX 24 kb)
Additional file 2:
Supplementary figures. (DOCX 4395 kb)
Additional file 3:
Data processing methods and installation instructions. (DOCX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kucukural, A., Yukselen, O., Ozata, D.M. et al. DEBrowser: interactive differential expression analysis and visualization tool for count data. BMC Genomics 20, 6 (2019). https://doi.org/10.1186/s12864-018-5362-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5362-x