
Abstract
Background
Bovine papillomavirus (BPV) belongs to the Papillomaviridae family and infects epithelial cells of bovines and closely related animals, causing hyperproliferative lesions known as warts or papillomas, which may regress or progress to form benign or malignant tumors. The virus enters the host cell and interacts with it by altering the regulation of genes that are responsible for controlling the cell cycle, thus triggering lesion formation. It is not yet known which host genes are regulated by viral infection. Therefore, the objective of this study was to make use of next-generation RNA sequencing methods to identify differentially expressed genes associated with BPV infection, which might elucidate possible marker genes that could be used to control the disease.
Results
Transcriptome analysis revealed that 1343 genes were differentially regulated (FDR < 0.05). A comparison of gene expression in infected and noninfected cows indicated that 655 genes were significantly upregulated, and 688 genes were significantly downregulated. Most differentially expressed genes were associated with BPV infection pathways, which supports the hypothesis that viral infection was the mechanism associated with this regulation.
Conclusions
This is the first study that focused on a large-scale evaluation of gene expression associated with BPV infection, which is important to identify possible metabolic pathways regulated by host genes for lesion development. In addition, novel targets could be identified in order to find ligands that interact with BPV, with the aim of interrupting the infection cycle.
Similar content being viewed by others
Background
With a herd of 218.23 million head of cattle and annual milk production of 33.62 billion liters [1], Brazil has the second largest cattle herd in the world and is the fifth largest producer of milk [2, 3]. US Department of Agriculture (USDA) data show that Brazil is the second largest producer of commercial beef of the world, with annual production of 2.586 million tons. These data show the strategic position of cattle in the country’s economy. However, some pathogens are known to affect the level of this production by causing severe damage to animals and great losses for producers. Among these pathogens is the bovine papillomavirus (BPV), which causes bovine papillomatosis in cattle in Brazil and several other countries. The virus mainly affects young animals, causing cutaneous and mucosal lesions that can be minimized using chemicals, surgical removal or cauterization of lesions, and autogenous vaccine usage. If left untreated, lesions may develop into malignant tumors [4, 5].
The process of viral infection is coupled to replication of the viral genome. This process depends on different host genetic factors. During infection, host genes are inhibited or activated by pathogen proteins [6]. Studies have shown that the virus regulates these genes to ensure an increase in the copy number of the viral genome and thereby trigger the infection process [6, 7]. However, it is not yet known which of these host genes are regulated by viral infection.
In the present study, we have analyzed the differential expression of genes involved in the host-pathogen relationship, thus contributing to the development of new markers associated with viral infection. Next-generation RNA sequencing technology (RNA-seq) allows detailed profiling of gene expression levels. In this type of sequencing, a large amount of RNA sequence information is generated and requires automated analysis. This approach has been used successfully to study bovine mammary tissue [8,9,10,11,12,13,14,15] and to analyze differentiated keratinocytes during human papillomavirus infection [6], but none of these studies considered the effect of gene expression during bovine papillomavirus infection on the transcriptome. In this study, we used RNA-seq to gain unprecedented insight into the regulatory mechanisms underlying the effect of gene expression during bovine papillomavirus infection.
Results
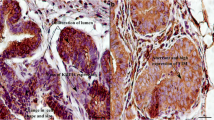
Histopathological analysis confirmed the presence of papillomatous lesions in three samples. The presence of hyperkeratosis of the epithelial layer, koilocytosis and acanthosis (Fig. 1a, b, c and d) were visualized through photomicrography. Such changes are characteristic of BPV infection [12]. In healthy animals, there was no histology compatible with papillomatous lesions. In addition, PCR detected the presence of the virus in all papillomatosis samples, and samples from healthy animals tested negative for viral DNA.

Photomicrographs of a wart stained by hematoxylin and eosin. a fragment of a papilloma. Hyperkeratosis of the epithelial layer with irregularity of the epidermis. Magnification: 40×. b visualization of koilocytosis at 40× magnification. c acanthose at 10× magnification. d noninfected animal tissue evidencing the epithelial layer without the presence of hyperkeratosis and acanthosis
Sequencing, transcript mapping, and gene expression analysis
Six libraries generated with RNA samples from cows with papillomatous infection and healthy animals were sequenced. RNA-sequencing generated a total of 121,722,238 raw paired reads with an average of 20 M reads per library. The alignment of reads showed that 87.56% (93,261,478) mapped to the bovine genome (UMD3.1.77, which has 24,617 annotated genes) (Additional file 1). Of the mapped reads, 82.38% mapped to unique positions, 14.15% mapped to multiple positions, and 3.62% were discordant alignments.
The distribution of transcript expression is shown in Additional file 2. A total of 14,213 genes were expressed, of which 1343 were significantly differentially regulated (FDR < 0.05) in a comparison of gene expression between infected cows and healthy animals. Of these, 655 genes were significantly upregulated and 688 were significantly downregulated in infected animals (Additional file 3).
qPCR validation of RNA-sequencing results
Our qPCR results confirm the differential expression patterns of RNA-seq obtained for infected and healthy animals. Differential gene expression identified by using qPCR was statistically significant (P < 0.0003). Differences in gene expression between the case and control groups were statistically significant when both methods were used, RNA-seq and qPCR (Table 1).
Biological function enrichment and pathway analysis of differentially expressed genes
Of the differentially expressed genes, we identified 122 genes with immune function, 489 related to the cell cycle, 112 involved in the process of ubiquitination, and 42 in the process of apoptosis. The top 24 differentially expressed genes in infected animals and their functions are listed in Table 2. We also found 26 genes involved in the process of keratinization and 162 in the process of gene expression (Additional file 4).
Figure 2 shows the genes that were upregulated and downregulated in infected animals and their molecular functions. Particularly, among these GO functional classes, the genes associated with ubiquitination and apoptosis were expressed at higher levels in infected animals, suggesting that the corresponding genes might be related to the pathogenic mechanism of BPV.

Differentially expressed genes in infected and healthy animals that are significantly enriched in several molecular and cellular functions
KEGG analysis identified several metabolic pathways associated with papillomavirus infection. The most commonly encountered pathways were cell adhesion of molecules, processing and presentation of antigen, differentiation of TH1 and TH2 cells, and the pathway of human papillomavirus infection (Additional file 4). In those metabolic pathways, protein-protein interactions were analyzed, and the associations between proteins and shared functions was possible to verify. For example, Fig. 3 shows the predicted and known interactions of ISG15 protein, which participates in the metabolic pathway of human papillomavirus infection. This gene plays a key role in the innate immune response to viral infection and ubiquitination. The ISG15 cellular partners were analyzed, and among the many functions, all had in common the functions of cellular response to viruses and ubiquitination. Two of the ISG15 partners were differentially expressed in this study, UBA7 and HERC5, which were upregulated in infected cows, suggesting that they might be related to viral pathogenicity.

Interaction between the upregulated differentially expressed gene ISG15 and its functional cellular partners. Colored nodes represent the query protein and first shell of interactors. Filled nodes are related to known or predicted 3D structures. Empty nodes correspond to proteins of unknown 3D structure. Colored lines represent known, predicted, and other interactions. The light blue line color is associated with known interactions from curated databases; the pink line represents the experimentally determined interactions; the green line represents interactions predicted from gene clusters; the red line is associated with predicted interactions according to gene fusions; the dark blue line represents predicted interactions of gene co-occurrence; yellow, black and gray lines represent other interactions
Discussion
In this study, we identified that BPV infection in the animal modified the expression profile of some of the host genes that were upregulated in infected animals, such as the keratin genes TMEM79, IVL, FOXN1, and SOSTDC1. A previous study [6] presented a change in the expression profile of keratinocytes in HPV-infected cells. The high expression of these cells is directly influenced by the pathogen, as the virus infects these cells and uses its machinery to replicate and transcribe viral genes. In another study [13], high expression of the TREM1 gene was verified on the cell surface of epithelial tissue infected by Leishmania. In this study, the overexpression of TREM-family genes was also verified in virus-damaged tissues. In this way, it is possible to infer that the gene in question is being expressed via activation of immune system cells by the host as a response to infection.
Some genes related to the immune process, specifically the response to viruses that activate the MHCI pathway, were found: 28 were upregulated genes, and only 8 were downregulated (STMN1, KYNU, CCL26, CCL14, CXCL14, HLA-A, CCL14, and IL1F10). In this study, we have identified 120 genes with a role in regulating the inflammatory process and immune response, for example, the S100A8 and S100A9 genes were upregulated in infected animals along with their partners S100A12, IL8 and IL1B genes. This expression profile was also observed in a previous study [14], when S100A8 and S100A9 protein expression in the skin of epidermodysplasia verruciformis (EV) patients was investigated. In non-lesioned skin, both proteins were occasionally detectable in the keratinocytes of the granular layer or were completely lacking. The exact mechanisms underlying the regulation of these proteins remains to be determined; however, it can be speculated that the S100A8/A9 genes might exert their effects during carcinogenesis.
Of the genes that participate in the immune response, 61 were found to be downregulated in infected animals, such as the CCL20 gene, which is responsible for chemotaxis in dendritic cells (DC), effector/memory T-cells, and B-cells and plays an important role on skin and mucosal surfaces under homeostatic and inflammatory conditions, as well as in pathogenesis, including that of cancer and autoimmune diseases.
Sperling et al. [15] proposed a novel molecular mechanism for how HPV8 might disrupt innate immunity in the skin. They found reduced numbers of antigen-presenting Langerhans cells in epidermodysplasia verruciformis lesions and showed the reduction to be a consequence of HPV8-mediated suppression of CCL20. They showed that CCL20 is expressed in the uppermost differentiated layers of normal skin epidermis but is almost absent in HPV8-positive lesioned skin of EV patients. The virus might use this form to escape the host immune response.
Genes involved in cell proliferation and growth were also found to be differentially expressed. These genes include members of the SOX family of transcription factors mainly involved in the regulation of the malignant process through their ability to regulate numerous cancer markers, including those of cell proliferation, apoptosis, survival, invasion, migration, and differentiation [16]. Studies showed that SOX14 overexpression decreases viability and promotes apoptosis by altering the expression of apoptosis genes, promoting accumulation of p53 [16, 17]. In this study, the QSOX gene was found to be a member of the SOX family and possibly involved in the regulation of apoptosis. The presented results indicate that QSOX, as well as the SOX14 gene, may be associated with the process of apoptosis. These viruses have developed numerous strategies in order to block host-mediated apoptosis, contributing to cancer development. Programmed cell death, known as apoptosis, is a pivotal mechanism of cell death that plays an important role during diverse biological processes, including development, cell differentiation, and proliferation [18].
Several mechanisms have been found to cause bcl-2 deregulation and cancer, including activation of chromosomal translocation and upregulation upon viral infection, for instance, upregulated bcl-2 expression has been described in different tumors, including premalignant and malignant lesions of the uterine cervix induced by human papillomaviruses [19]. p53 has a significant role in cell cycle control; therefore, the loss of its suppressive function has been reported in different types of human neoplasia and animal tumors, such as BPV-induced tumors [20]. However, studies concerning the expression and impact of the anti-apoptotic protein bcl-2 and the tumor suppressor p53 in BPV-induced cutaneous tumors are lacking. In the study of Bocaneti [21], altered bcl-2 and p53 immunoreactivity in bovine cutaneous fibropapillomas was observed, suggesting the involvement of these two proteins in cutaneous neoplastic transformation through an impaired apoptotic process.
The E5, E6, E7, and E2 proteins of PVs are known to control the expression of host genes. E6 and E7 control the keratinocyte cell cycle, cell differentiation, and apoptosis programs. Thus, the alterations in gene expression that we observed in this study might be attributed to the functions of BPV oncoproteins. These changes are important for the infection cycle of the virus and persistent infection [9, 10]. These oncogenes interact with several different host proteins and thus could affect their expression. Therefore, in this study, we have identified several genes that possibly participate in papillomavirus infection pathways, which makes an important contribution to existing knowledge on the papillomavirus infection cycle, providing novel possible markers that could be used for the development of new prognostic and treatment methods.
Conclusions
In conclusion, we report RNA-Seq analysis of changes in the cattle transcriptome caused by BPV infection. Infection caused massive changes in the expression profile of keratinocyte, immune system, cell proliferation, and apoptosis genes. These changes mainly resulted in an expression profile characteristic of viral infection rather than tumor progression. The large dataset we have developed opens up the possibility of a deeper understanding of the cycle of BPV infection in the host. Therefore, this study serves as the basis for the realization of new studies aimed at understanding the functions of genes that could act in specific pathways during viral infection, increasing our understanding regarding the infectious cycle in the animal. In addition, this study provides information that could be used to find molecular targets for possible drugs that interrupt the cycle of infection of BPV, thus preventing papillomatosis.
Methods
Animals, management, and sampling
The animals used in this study were owned by the São José farm in the Central Agreste region of the state of Sergipe, northeastern Brazil. Sample collection was performed by qualified veterinary medical professionals and registered in the class council, according to the procedures established by the corresponding ethics committee. Briefly, six female Girolando dairy breeders in the Central Agreste region of the state of Sergipe, northeastern Brazil, were used. Animal care, management, and usage procedures were approved by the ethics committee on production animals of the Federal University of Sergipe (protocol number: 05/14). Three animals with papillomatosis and three healthy animals were used. We collected samples of skin from the neck region of each animal. We used different animals because recent studies have detected the DNA of HPV in various parts of the host, such as peripheral blood mononuclear cells (PBMC), plasma, urine, semen, spermatozoa, trophoblasts, and the bladder. These studies have shown that PVs are not strictly epitheliotropic [22,23,24,25,26,27,28]. In addition, each lesion was analyzed in triplicate. All animals were females approximately 2 years old. Each sample was divided into three parts: one was used to confirm the presence of BPV in the lesion and was then wrapped in foil and stored at − 20 °C; another, for confirmation of the injured tissue, was placed in 10% formalin; and another, for the evaluation of gene expression, was placed in RNAlater and then stored at − 80 °C.
Histopathological analysis
Formalin-fixed tissue samples were processed by standard paraffin wax techniques. Samples were then cut into 3–5-μm sections and stained by the hematoxylin and eosin method (HE). Slides were evaluated microscopically at increasing magnifications (5×, 10×, 20×, 40×).
Viral detection
Each sample was individually macerated for total DNA extraction with the aid of the DNeasy Blood and Tissue kit (Qiagen), following the supplier’s instructions. About 100 ng of DNA were used for polymerase chain reaction (PCR) using the general detection primers FAP59/64 [29]. All amplification reactions were prepared to a final volume of 25 μL using the Master Mix PCR kit (Promega), according to the supplier’s protocol. All primers used were diluted to 10 μM.
RNA isolation and sequencing
Total RNA (~ 15 mg/sample) were purified using the Realipred™ RNA Tissue Miniprep System Kit (Promega) according to the manufacturer’s instructions. The concentration of RNA was measured with Nanodrop ND-2000 instrument (NanoDrop Thermo Scientific, Wilmington, DE, USA), and the quality was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Libraries were generated from 1000 ng of total RNA using the TruSeq stranded mRNA Sample Preparation LS Protocol Kit (Illumina Inc. San Diego, CA, USA) as per the manufacturer’s recommendations. Libraries were quantified using the Kapa Library Quantification Kits (Illumina Inc. San Diego, CA, USA). The average fragment size was determined using the 2100 Bioanalyzer instrument (Agilent Technologies). Cluster formation on the flow cell was performed using the cBot instrument (Illumina Inc.,). All libraries were sequenced on a HiSeq 2500 Version 4 (Illumina Inc.) running HCS software v2.2.6.
After sequencing, the Galaxy Project [30] interface to cloud computing resources was used to bring command-line-driven tools. The Illumina output files were converted to FASTQ file format and sequence quality trimming was performed using Trimmomatic [31], with a phred quality score > 30 over the length of the reads. The reads were aligned to the UMD3.1 Bos taurus 8 reference genome using Tophat v2.0.11 [32] and then assembled into transcripts using Cufflinks v2.2.1 [33] with genome annotation using UCSC Main Cow RefSeq genome. Reads per kilobase per million mapped reads (RPKM) were used to establish the transcript expression rate. Identification of the differentially expressed genes was performed using Cuffdiff v 2.2.1 software, and visualization was generated by CummeRbund [33]. Because a small number of genes may be highly expressed in the epithelial tissue and could hamper accurate quantification of genes with low expression, differential gene expression was defined as significant according to a false discovery rate (FDR) < 0.05, adjusted with the procedure of Benjamini and Hochberg [34].
Functional annotation and pathway analyses of differentially expressed genes
For the functional annotation of differentially expressed genes, the Gene Ontology (GO) and UNIPROT databases were used to obtain functional information. The genes were searched for functions associated with the progression of papillomatous lesions in cattle, and the metabolic pathways in which the products of these genes participate were mapped with the KEGG database (Additional file 4). Protein-protein interactions was analyzed using the STRING database.
qPCR verification of RNA sequencing results
The expression levels of nine differentially expressed genes (BOLA-DYB, DAPL1, KRT78, KRTAP3–1, UBA7, BNIP3, IVL, FABP4, and CST6) were analyzed using real-time quantitative PCR (qPCR) to validate RNA sequencing results. These genes were chosen because they present different functions important for the virus infection cycle. Two reference genes (RPS15 and GAPDH) were used following a published protocol [16]. Reverse transcription was performed with the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific), using aliquots (2 μg) of the same total RNA used in RNA sequencing analysis. The cDNA samples were diluted to 20 ng/μl based on the starting concentration of RNA. The PCR reaction mix was composed of 12.5 μL Power SYBR® Green PCR Master Mix (Life Technologies Inc., Burlington, ON, Canada), 2.5 μL cDNA, 600 nM of each forward and reverse primers (Additional file 5). The thermal cycling conditions were 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. A melting curve was generated by the end of each reaction to verify the formation of a single peak and to exclude the possibility of non-specific product formation. The experiments were carried out in triplicate for each data point. Relative quantification of gene expression was determined using the 2-ΔΔCt method [35]. A Student’s t-test was used to identify significant differences in gene expression (p < 0.05 was considered significant).
Primer validation was performed by PCR using the Master Mix PCR kit (Promega). For each reaction, 12 μL of PCR Master Mix, 1 μL of each primer, 9 μL of ultrapure water, and 2 μL of DNA was used. The PCR cycling conditions were as follows: initial denaturation at 94 °C for 10 min, followed by 35 cycles of 94 °C for 1 min, 60 °C for 1 min, and 72 °C for 1 min, and a final extension at 72 °C for 10 min. PCR products were visualized using 1.5% agarose gel electrophoresis.
References
Instituto Brasileiro de Geografia e Estatística (IBGE). Produção da pecuária mundial. 2016. https://www.ibge.gov.br/estatisticas-novoportal/economicas/agricultura-e-pecuaria/. Accessed 22 Feb 2018.
Livestock and Poultry: world markets and trade. Washington, DC: United States Departament of Agriculture - USDA, Foreign Agricultural Service, abr. 2017. http://usda.mannlib.cornell.edu/MannUsda/homepage.do. Accessed 17 May 2018.
Landes M, Melton A, Edwards S. United States. Department of Agriculture. Psd: from where the buffalo roam: india’s beef exports. A report from the economic research service. Washington, D.C.: United States Department of Agriculture – USDA; 2016. https://www.ers.usda.gov/webdocs/publications/37672/59707_ldpm-264-01.pdf?v=42543. Accessed 1 May 2018
Thomas I, Juliane H-J, Shwu-Yuan W, Cheng-Ming C. Involvement of Brd4 in different steps of the papillomavirus life cycle. Virus Res. 2017. https://doi.org/10.1016/j.virusres.2016.12.006.
Borzacchiello G, Roperto F. Bovine papillomaviruses, papillomas and cancer in cattle. Vet Res. 2008. https://doi.org/10.1051/vetres.2008022.
Klymenko T, Gu Q, Herbert I, Stevenson A, Iliev V, Watkins G, Pollock C, Bhatia R, Cuschieri K, Herzyk P, Colector D, Graham SV. RNA-Seq analysis of differentiated keratinocytes reveals a massive response to late events during human papillomavirus 16 infection, including loss of epithelial barrier function. J Virol. 2017. https://doi.org/10.1128/JVI.01001-17.
Culp TD, Budgeon LR, Christensen ND. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology. 2005. https://doi.org/10.1016/j.virol.2005.11.025.
Wickramasinghe S, Rincon G, Islas-Trejo A, Medrano JF. Transcriptional profiling of bovine milk using RNA sequencing. BMC Genomics. 2012. https://doi.org/10.1186/1471-2164-13-45.
Cui X, Hou Y, Yang S, Xie Y, Zhang S, Zhang Y, Zhang Q, Lu X, Liu GE, Sun D. Transcriptional profiling of mammary gland in Holstein cows with extremely different milk protein and fat percentage using RNA sequencing. BMC Genomics. 2014. https://doi.org/10.1186/1471-2164/15/226.
Canovas A, Rincon G, Bevilacqua C, Islas-Trejo A, Brenaut P, Hovey RC, Boutinaud M, Morgenthaler C, Vanklompenberg MK, Martin P, Medrano JF. Comparison of five different RNA sources to examine the lactating bovine mammary gland transcriptome using RNA-sequencing. Sci Rep. 2014. https://doi.org/10.1038/srep05297.
Eveline IAM, Ran L, Adolf AA, Pier-Luc D, Nathalie B, Chaouki B, Xin Z. Transcriptome adaptation of the bovine mammary gland to diets rich in unsaturated fatty acids shows greater impact of linseed oil over safflower oil on gene expression and metabolic pathways. BMC Genomics. 2016. https://doi.org/10.1186/s12864-016-2423-x.
Araldi RP, Melo TC, Neves AC, Spadacci-Morena DD, Magnelli RF, Modolo DG, de-Sá-Júnior PL, Mazucchelli-de-Souza J, Carvalho RF, Beçak W, Stocco RC. Hyperproliferative action of bovine papillomavirus: genetic and histopathological aspects. Genet Mol Res. 2015. https://doi.org/10.4238/2015.
Bomfim LGS, Magalhães LS, Santos-Filho MAA, Peres NTA, Corrêa CB, Tanajura DM, Silva AM, Lipscomb MW, Borges VM, Jesus AR, Almeida RP, de Moura TR. Leishmania infantum induces the release of sTREM-1 in visceral Leishmaniasis. Front Microbiol. 2017. https://doi.org/10.3389/fmicb.2017.02265.
Podgórska M, Oldak M, Marthaler A, Fingerle A, Walch-rückheim B, Lohse S, Müller CSL, Vogt T, Ustav M, Wnorowski A, Malejczyk M, Majewski S, Smola S. Chronic inflammatory microenvironment in epidermodysplasia Verruciformis skin lesions: role of the synergism between HPV8 E2 and C/EBPb to induce pro-inflammatory S100A8/A9 proteins. Front Microbiol. 2018. https://doi.org/10.3389/fmicb.2018.00392.
Sperling T, Ołdak M, Walch-ru B, Wickenhauser C, Doorbar J, Pfister H, Malejczyk M, Majewski S, Keates AC, Smola S. Human papillomavirus type 8 interferes with a novel C/ EBPb-mediated mechanism of keratinocyte CCL20 chemokine expression and Langerhans cell migration. PLoS Pathog. 2012. https://doi.org/10.1371/journal.ppat.1002833.
Stanisavljevic D, Petrovic I, Vukovic V, Schwirtlich M, Gredic M, Stevanovic M, Popovic J. SOX14 activates the p53 signaling pathway and induces apoptosis in a cervical carcinoma cell line. PLoS One. 2017. https://doi.org/10.1371/journal.pone.0184686.
Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R, Zhang AW, Parcy F, Lenhard B, Sandelin A, Wasserman WW. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2015. https://doi.org/10.1093/nar/gkv1176.
Ulukaya E, Acilan C, Yilmaz Y. Apoptosis: why and how does it occur in biology? Cell Biochem Funct. 2011;29:468–80.
Singh M, Srivastava S, Singh U, Mathur N, Shukla Y. Co-expression of p53 and Bcl-2 proteins in human papillomavirus-induced premalignant lesions of the uterine cervix: correlation with progression to malignancy. Tumor Biol. 2009;30:276–85.
Finlay M, Yuan Z, Morgan IM, Campo MS, Nasir L. Equine sarcoids: bovine papillomavirus type 1 transformed fibroblasts are sensitive to cisplatin and UVB induced apoptosis and show aberrant expression of p53. Vet Res. 2012;43:81.
Bocaneti F, Altamura G, Corteggio A, Velescu E, Borzacchiello G. Expression of bcl-2 and p53 in bovine cutaneous fibropapillomas. Infect Agent Cancer. 2015. https://doi.org/10.1186/1750-9378-10-2.
Bodaghi S, Wood LV, Roby G, Ryder C, Steinberg SM, Zheng Z. Could human papillomaviruses be spread through blood? J Clin Microbiol. 2005. https://doi.org/10.1128/JCM.43.11.5428-5434.2005.
Lindsey CJ, Almeida ME, Vicari CF, Carvalho C, Yaguiu A, Freitas AC, Beçak W, Stocco RC. Bovine papillomavirus DNA in milk, blood, urine, semen, and spermatozoa of bovine papillomavirus-infected animals. Genet Mol Res. 2009;8:310–8.
Sarkola M, Rintala M, Grenman S, Syrjänen S. Human papillomavirus DNA detected in breast milk. Pediatr Infect Dis J. 2008. https://doi.org/10.1097/INF.0b013e318169ef47.
Shimada T, Yamaguchi N, Nishida N, Yamasaki K, Miura K, Katamine S, Masuzaki H. Human papillomavirus DNA in plasma of patients with HPV16 DNA-positive uterine cervical Cancer. Jpn J Clin Oncol. 2010. https://doi.org/10.1093/jjco/hyp193.
Santos EUD, Silva MAR, Pontes NE, Coutinho LCA, Paiva SSL, Castro RS, Freitas AC. Detection of different bovine papillomavirus types and co-infection in bloodstream of cattle. Transbound Emerg Dis. 2013. https://doi.org/10.1111/tbed.12237.
Roperto S, Brun R, Paolini F, Urraro C, Russo V, Borzacchiello G, Pagnini U, Raso C, Rizzo C, Roperto F, Venuti A. Detection of bovine papillomavirus type 2 in the peripheral blood of cattle with urinary bladder tumours: possible biological role. J Gen Virol. 2008. https://doi.org/10.1099/vir.0.2008/004457-0.
Daudt C, Da Silva FRC, Lunardi M, Alves CBDT, Weber MN, Cibulski SP, Alfieri AF, Alfieri AA, Ca CW. Papillomaviruses in ruminants: An update. Transbound Emerg Dis. 2017. https://doi.org/10.1111/tbed.12868.
Forslund O, Antonsson A, Nordin P, Stenquist B, Hansson BG. A broad range of human papillomavirus types detected with a general PCR method suitable for analysis of cutaneous tumours and normal skin. J Gen Virol. 1999. https://doi.org/10.1099/0022-1317-80-9-2437.
Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014. https://doi.org/10.1093/bioinformatics/btu170.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013. https://doi.org/10.1186/gb-2013-14-4-r36.
Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression. Analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012. https://doi.org/10.1038/nprot.2012.016.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol. 1995;57:289–300.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 − ΔΔCT method. Methods. 2001. https://doi.org/10.1006/meth.2001.1262.
Acknowledgements
The authors would like to thank Luana Batista and Osmario Marques for the animal care during the collection. The authors also thank the Center for Functional Genomics of the Department of Animal Science of ESALQ-USP for the RNA sequencing.
Funding
This study was conducted with funding from Brazilian Institutes CNPq (Grant: 446425/2014-1), CAPES (Grant: 23038.010050/2013-04), and FAPITEC/SE (Grant: 8444/2013-02), which provided financial support for the study. DB and GB were granted with CAPES fellowships.
Availability of data and materials
The datasets generated and analyzed during the current study are available in the GEO repository, https://www.ncbi.nlm.nih.gov/geo/, under accession number GSE122853.
The datasets supporting the conclusions of this article are included within the article and its additional files.
Author information
Authors and Affiliations
Contributions
DB e GB performed the whole experiment from the animal phase to the bioinformatics analysis, and drafted the manuscript. LS participated in the functional annotation. RS participated in the histological experiments. MB drafted the manuscript, performed data analysis, and was the coordinator of the all study. All authors revised and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Ethical approval was obtained by the Ethics Committee of Production of Animals of the Federal University of Sergipe (protocol number: 05/14). In addition, written informed consent from the owner of the animals used in this study was obtained.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Reads and mapping statistics. (XLSX 11 kb)
Additional file 2:
RPKM density histogram of transcripts from RNA-seq of BPV infected and non-infected groups. The diagram shows the distribution of the density of expressed genes at different RPKM levels. 2a) distribution of the density of expressed genes in infected animals. 2b) distribution of the density of expressed genes in non-infected animals. (PDF 96 kb)
Additional file 3:
Differentially expressed genes between infected and healthy cows. (XLSX 80 kb)
Additional file 4:
Functional annotation of differentially expressed genes. (XLSX 293 kb)
Additional file 5:
Genes and primer sequences used in qPCR validation of RNA-sequencing data. (XLSX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Barreto, D.M., Barros, G.S., Santos, L.A.B.O. et al. Comparative transcriptomic analysis of bovine papillomatosis. BMC Genomics 19, 949 (2018). https://doi.org/10.1186/s12864-018-5361-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5361-y