
Abstract
Background
Wucai (Brassica campestris L. ssp. chinensis var. rosularis Tsen) is a variant of nonheading Chinese cabbage (Brassica campestris L.), which is one of the major vegetables in China. Cytoplasmic male sterility (CMS) has been used for Wucai breeding in recent years. However, the underlying molecular mechanism of Wucai CMS remains unclear. In this study, the phenotypic and cytological features of Wucai CMS were observed by anatomical analysis, and a comparative transcriptome analysis was carried out to identify genes related to male sterility using Illumina RNA sequencing technology (RNA-Seq).
Results
Microscopic observation demonstrated that tapetum development was abnormal in the CMS line, which failed to produce fertile pollen. Bioinformatics analysis detected 4430 differentially expressed genes (DEGs) between the fertile and sterile flower buds. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed to better understand the functions of these DEGs. Among the DEGs, 35 genes (53 DEGS) were implicated in anther and pollen development, and 11 genes were involved in pollen cell wall formation and modification; most of these showed downregulated expression in sterile buds. In addition, several genes related to tapetum development (A6, AMS, MS1, MYB39, and TSM1) and a few genes annotated to flowering (CO, AP3, VIN3, FLC, FT, and AGL) were detected and confirmed by qRT-PCR as being expressed at the meiosis, tetrad, and uninucleate microspore stages, thus implying possible roles in specifying or determining the fate and development of the tapetum, male gametophyte and stamen. Moreover, the top four largest transcription factor families (MYB, bHLH, NAC and WRKY) were analyzed, and most showed reduced expression in sterile buds. These differentially expressed transcription factors might result in abortion of pollen development in Wucai.
Conclusion
The present comparative transcriptome analysis suggested that many key genes and transcription factors involved in anther development show reduced gene expression patterns in the CMS line, which might contribute to male sterility in Wucai. This study provides valuable information for a better understanding of CMS molecular mechanisms and functional genome studies in Wucai.
Similar content being viewed by others


Background
Wucai (Brassica campestris L. ssp. chinensis var. rosularis Tsen) is a variant of nonheading Chinese cabbage (Brassica campestris L.), which is the most important species in the Brassicaceae family [1]. As an important autumn and winter vegetable crop, this crop is cultured widely in most parts of China, where it originated, especially in the Yangtze-Huaihe River Basin, and has become increasingly popular in other countries for its beautiful shape and significant levels of vitamins and minerals [2]. In recent years, cytoplasmic male sterility (CMS) has been used in some cultivated breeds [3] to generate stronger plants and higher hybrid seed yield [4, 5].
Owing to an interaction between mitochondrial and nuclear genes, the CMS phenotype fails to produce functional anthers, pollen or male gametes [6]. Thus, understanding the delicate and complex processes of anther and pollen development is a prerequisite for comprehension of this unique phenomenon in CMS plants [5]. However, anther and pollen development is a critical phase in the plant life cycle, which contains a series of correlated events involving a diverse range of genes in complex regulatory networks [7,8,9]. Dysfunction of these genes may lead to male sterility [10]. Although many of these genes have been isolated and analyzed to have vital roles in CMS, the regulatory network and the novel genes underlying CMS occurrence are still largely unknown [8, 10].
In recent decades, genetic research into CMS occurrence has included two main types, map-based cloning and sequence-based transcriptome assays [7]. Using AFLP and SSR techniques for gene mapping, Xu et al. [11] identified the restorer gene BrRfp from the pol-like CMS restorer line of heading Chinese cabbage (B. rapa). Compared with the gene mapping method, the Illumina sequencing (RNA-Seq) technique could offer several key advantages over existing technologies [12]. This form of transcriptional analysis allows for the determination of genome-wide expression levels as well as identification of new genes and SNPs, especially genes with very low abundance [13,14,15]. Furthermore, the results of RNA-Seq also show high levels of reproducibility for both technical and biological replicates [16]. Therefore, taking these advantages into account, RNA-Seq has been used successfully in the pollen and anther development of Brassica crops, such as B. napus [6, 9, 14, 17], B. rapa [7, 18], B. oleracea [19,20,21], B. campestris [5], and B. juncea [22]. However, to the best of our knowledge, the genome-wide transcriptional profiles and related genes of fertile and sterile flower buds from Wucai have not yet been reported through RNA-Seq technology.
In our previous study, a newly sterile plant of Wucai was generated by hybridization with nonheading Chinese cabbage, and a stable sterility line was developed via backcrossing for ten generations. In this present study, the objective was to further understand the differences in the transcriptome between the CMS line and its maintainer line and to find some molecular clues to this CMS system. Accordingly, mRNA was isolated from the flower buds of fertile and sterile plants, respectively, and then, genome-wide transcriptional profiling was performed using the Illumina RNA-Seq platform. Based on bioinformatics analysis, a large number of candidate genes and transcription factors involved in anther and pollen development were isolated, and various screened candidate genes related to pollen development were further analyzed by qRT-PCR. Our results may contribute to an understanding of CMS molecular mechanisms and provide useful information for further heterosis breeding in Wucai.
Results
Phenotypic and cytological characterization
After ten generations of backcrossing, there was no difference in morphological phenotype between the sterile line 12-14A and its maintainer line 12-14B (Fig. 1a and b), and the forms of the corolla and flower seemed normal (Fig. 1c-f). However, compared with those of the fertile flower, shorter filaments and undeveloped anthers were observed on the stamens of the sterile flower (Fig. 1g-h).

Morphological characteristics of flowers and microscopic observations of anthers from the sterile and fertile lines of Wucai. a and b, CMS line 12-14A and its maintainer line 12-14B. c, e, g and d, f, h, Inflorescences, flowers, and petals and stamens from the sterile and fertile lines, respectively. i-l, Transverse sections of sterile anthers; abnormal tapetum was formed, and the pollen sacs of sterile anthers did not produce pollen, in the CMS line. m-p, Transverse sections of fertile anthers; normal tapetum and mature anthers developed in the maintainer line. AT, abnormal tapetum; DPG, degenerated pollen grain; E, epidermis; En, endothecium; ML, middle layer; MSp, microspore; PG, pollen grain; T, tapetum. Scale bars in a, b, c and d represent 1 cm; scale bars in e, f, g, h represent 0.5 cm; scale bars in I-P represent 100 μm
To accurately characterize the cause of the pollen abortion, semithin sections of the buds from the two lines of Wucai were observed. As shown in Fig. 1i and IM, there was no obvious difference in the meiosis period between sterile and fertile anthers. However, at the tetrad stage, the tapetal cells expanded, and the microspore could not carry out meiotic division (Fig. 1j). After this stage of anther development, the tapetal cells vacuolated and filled in the sacs, and the microspores degraded (Fig. 1k), which caused pollen abortion (Fig. 1l). In contrast, a normal tapetum and fertile pollen grains developed in the fertile anthers (Fig. 1m-p).
De novo assembly and sequence annotation
To further understand the molecular mechanisms of CMS differences in Wucai, RNA-Seq was performed using Illumina technology. After the raw data were trimmed, a total of 52,936,673 clean reads for the fertile samples and 52,606,810 for the sterile samples were obtained, and the Q20 and Q30 were > 96.61 and > 92.53%, respectively (Table 1). In addition, the GC contents were consistently approximately 45% for both sterile and fertile samples (Table 1), suggesting that the sequencing was highly accurate. All clean reads (105,543,483) were assembled using the Trinity program [23]. As the result, 117,332 contigs were obtained with a mean length of 901 nt (Table 1). After clustering, 80,851 unigenes (> 200 bp) were generated; the average length was 1054 nt, and the N50 was 1586 nt (Table 1). The lengths of all unigenes were longer than 199 bp, and 86.95% of them ranged from 200 to 1999 bp (Additional file 1: Table S1). The assembled unigenes were subjected to search against the Nr, Swiss-Prot and COG databases, and 66,143 (81.81%), 54,857 (67.85%) and 28,129 (34.79%) unigenes were aligned against these three protein databases, respectively (Additional file 2: Table S2). The species distribution showed that that almost all of the sequences matched sequences from the Brassicaceae (Additional file 3: Figure S1).
Identification of differentially expressed genes
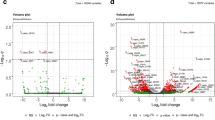
To gain better insight into the differences in gene expression patterns, we identified differentially expressed genes (DEGs) between the sterile and fertile lines. A total of 4430 genes (including 147 novel genes) were identified in the sterile and fertile comparison, including 980 genes upregulated and 3450 downregulated in sterile buds (Fig. 2; Additional file 4: Table S3). Among these DEGs, 1384 specifically expressed genes were observed that were expressed in only the fertile (1044) or sterile (340) samples. These results showed that the number of downregulated DEGs was considerably higher than that of upregulated DEGs. In addition, 147 novel genes were identified that were not annotated to any database. The biological functions of these novel genes remain to be determined (Additional file 5: Table S4).

Number of DEGs between sterile and fertile buds. Red indicates upregulated DEGs, and green indicates downregulated DEGs
Functional annotation by Gene Ontology
To investigate the function of the DEGs, the genes that showed significant differential expression were subjected to analysis by Blast2GO software. As shown in Additional file 6: Figure S2, 4430 DEGs were categorized into 53 functional groups under three main classifications. Among these groups, ‘cellular process’ (2180; 49.21%) in biological process, ‘cell’ (2618; 59.10%) and ‘cell part’ (48,388; 87.59%) in cellular component, and ‘binding’ (1450; 32.73%) in molecular function were the dominant categories. Conversely, three categories (‘cell killing’, ‘virion’ and ‘metallochaperone activity’) had only a few unigenes.
Pathway mapping by Kyoto Encyclopedia of Genes and Genomes
To understand the biological functions of DEGs that might be active in Wucai, pathway annotation was performed against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The results showed that 2217 of 4430 DEGs were assigned to 119 KEGG pathways (Additional file 7: Table S5). The 20 most significantly enriched KEGG pathways are shown in Fig. 3. The pathways with significantly more DEGs were metabolic pathways (676, 15.26%), biosynthesis of secondary metabolites (284, 6.41%), plant-pathogen interaction (162, 3.61%), and starch and sucrose metabolism (124, 2.80%). In starch and sucrose metabolism, a total of 124 DEGs were screened, and 38 of these DEGs were expressed in only fertile buds, while 71 DEGs were downregulated in sterile buds (Additional file 8: Table S6). These pathway annotations provide a basis for investigating gene functions involved in male sterility in Wucai.

Twenty most significantly enriched KEGG pathways
Genes related to anther and pollen development
Pollen development is a complex process that involves many events and plays an important role in plant propagation. In this study, all of the DEGs were annotated against the processes of anther and pollen development of A. thaliana. As shown in Table 2, 35 genes are considered to regulate male gametophyte development in Wucai. From A6 to ZAT5, 30 genes were downregulated in sterile buds. In contrast, 5 other genes, BT2, SCC12, TCMO, VAL2 and XPO1, were upregulated in sterile buds. Among these genes, A6, AMS, ENL2, MS1, MYB39, ORTH2, PLRX1 and TSM1 are also considered to be involved in tapetum development. In addition, we found several genes associated with cell wall formation and modification, such as the Pectinesterase gene (PME5), UDP-arabinose mutase gene (RGP1), and Cinnamoyl-CoA reductase gene (CCR2), which might participate in the processes leading to CMS in Wucai.
Differentially expressed transcription factor genes
In the anther and pollen development processes, transcription factors are generally thought to be important regulators. To identify differentially expressed transcription factors, all of the DEGs were annotated. In this study, 131 transcription factors (182 DEGs) were found, including 128 down- and 54 upregulated DEGs (Additional file 9: Table S7). Among these transcription factors, 27 up- and 8 downregulated DEGs were specific to fertile and sterile buds, respectively. In addition, 13 DEGs were associated with 8 WRKY transcription factor genes, and WRKY19 (Unigene3849, CL2284.Contig2, CL2120.Contig3) and WRKY32 (CL4008.Contig1) were upregulated in only sterile buds. Fifteen DEGs were identified with 10 NAC transcription factor genes, and 6 of them were highly expressed in sterile buds. In the bHLH and MYB transcription factor families, a total of 43 DEGs were associated with 16 bHLH and 13 MYB transcription factors, and 10 bHLHs (15 DEGs) and 8 MYBs (16 DEGs) were downregulated in sterile buds, respectively (Fig. 4, Table 3). These differentially expressed transcription factors might result in abortion of pollen development in Wucai.

Heat map analysis of bHLH (a), MYB (b), NAC (c) and WRKY (d) transcription factors. The expression levels shown are based on RPKM data. The color key represents the value of log2(S/F). Red represents high expression, and blue represents low expression. Each row represents a DEG
Real-time qPCR validation of gene expression patterns
To validate the results of RNA-Seq, 28 DEGs, including 11 genes annotated to anther and pollen development, 8 transcription factor genes, 8 flowering genes and one gene with unknown function were subjected to verification using qRT-PCR. The results of this experiment are shown in Fig. 5. Among these genes, 19 genes were downregulated in sterile buds, including 7 tapetum-specific genes (A6, AMS, ENL2, MS1, MYB39, ORTH2 and TSM1), 3 pollen cell wall formation genes (PME5, ZAT5, RGP1), 4 transcription factor genes (WRKY9, NAC91, MY104, BH089), 4 flowering genes (FLC, AGL18, AGL104–1 and AGL104–2) and one gene with unknown function (CL11374.Contig2). All of the 28 DEGs exhibited the same tendency between the RNA-Seq analysis and qRT-PCR results, which suggested that our transcriptome analysis was accurate and reliable.

qRT-PCR verification of differentially expressed unigenes. S means sterile sample, and F means fertile sample. Relative expression levels were calculated using Actin as an internal control
To further determine the expression pattern of key genes in the anther and pollen development, 2 transcription factors and 6 tapetum and pollen cell wall development genes were selected from the above for qRT-PCR assay (Fig. 6). Among these genes, the pollen cell wall formation gene PME5 (Unigene37636) was highly and specifically expressed in the fertile buds at the tetrad stage. The five tapetum development genes and one transcription factor (BH089) were highly expressed at the meiosis or tetrad stage in the fertile buds, and all of them shoed low abundance in sterile buds. The other transcription factor, BH077, was highly expressed at meiosis in sterile buds. These results further confirmed the reliability of the RNA-Seq data.

Expression of anther and pollen development related genes at different stages using qRT-PCR. S means sterile sample, and F means fertile sample. 1–3 indicate the pollen meiosis stage (bud sizes 0.5–1.5 mm), tetrad stage (1.5–3.0 mm) and uninucleate microspore stage (3.0–4.5 mm) of anther and pollen development, respectively. Relative expression levels were calculated using Actin as an internal control
In addition, 8 flowering genes were also examined in this study (Fig. 6). Among these genes, CO, AP3 and FT were highly expressed at the tetrad stage, and VIN3 was highly expressed in the meiosis period in sterile buds. FLC, AGL18, AGL104–1 and AGL104-2 were highly expressed at the meiosis or tetrad stage in fertile buds. The abnormal expression of these genes might influence the development of male gametophytes and stamens, leading to male sterility (Fig. 7).

The mechanism of FLC affects fertility in Wucai. Black arrows indicate promotion, and inverted T bars indicate repression. Dashed lines indicate that further research is needed. Question marks indicate unknown genes. Red arrows indicate up- or downregulation. AGL18 (Unigene20308), AGAMOUS LIKE 18; AGL104 (CL3910.Contig1, CL3910.Contig2), AGAMOUS LIKE 104; AP3 (CL13421.Contig5), APETALA 3; CO (Unigene36778) CONSTANS; FLC (CL3897.Contig1), Flowering Locus C; FT (CL2127.Contig2), Flowering Locus T; VIN3 (CL8770.Contig1), VERNALIZATION 3; TFs, transcription factors
Discussion
In higher plants, male sterility is a common phenotypic trait in which the abortion of stamens occurs and plants fail to produce functional anthers, pollen or male gametes under typical natural conditions [20, 24]. As the male reproductive organ, stamens play an important role in plant inheritance [5]. In this present study, morphological comparisons were performed between fertile and sterile lines of Wucai (Fig. 1a-h), and there was no difference between them except the stamens, which had shorter filaments and aborted anthers in the sterile flowers (Fig. 1g-h). A cytological examination was further carried out to evaluate the differences in pollen development between the fertile and sterile lines, and we observed that anther abortion occurred consistently in the sterile line, in which the tapetum developed abnormally and the microspore began to degrade after the meiotic stage (Fig. 1i-l). These results were consistent with those of Liu et al. [7] and Zhou et al. [5] and suggested that the abnormal development of the tapetal cells and microspores led to pollen and anther abortion.
To better identify the genes associated with pollen abortion in this CMS line of Wucai, a comprehensive analysis of transcript profiles between fertile buds and sterile buds was performed using RNA sequencing technology, which could detect low abundance transcripts and provide new insights into male sterility through global investigation of gene expression changes [5, 25]. A total of 105,543,483 clean reads and 117,332 contigs were obtained based on the RNA-Seq data, and 980 upregulated (1.21%) and 3450 downregulated (4.27%) DEGs out of 80,851 unigenes were identified based on their gene expression levels (Fig. 2; Table 1; Additional file 4: Table S3). These results indicated that changes in the expression of a large number of related genes could cause male sterility in Wucai, though the development of anther and pollen is a complicated process and involves numerous genes.
In the KEGG enrichment results, 4430 DEGs were classified into 119 metabolic pathways (Fig. 3, Additional file 7: Table S5), and these pathways might encompass all the biological pathways in anther development [26]. Among these pathways, starch and sucrose metabolism provides energy and carbon for anther development, and starch and sucrose are accumulated as energy reserves for pollen maturation [4, 27]. In our research, out of 124 DEGs involved in this pathway, 38 DEGs were expressed in only fertile buds and 2 DEGs (Unigene23056 and Unigene11909) were expressed in only sterile buds (Additional file 8: Table S6). The specific expression of these genes might lead to disturbances in the metabolism of starch and sucrose and the processing of energy reserves, which could suppress pollen development and ultimately lead to male sterility [7, 28]. This finding was consistent with those of previous works [4, 19, 29, 30].
In addition to the metabolic pathways, many key genes have been identified for pollen and anther development in Arabidopsis [31] and Brassica [9, 20, 32]. It is important to note that we identified 35 anther and pollen development related genes (53 unigenes) that have homologs in Arabidopsis and Brassica, and most of them were downregulated and associated with the development of the tapetum and pollen cell wall (Table 2). Among these 53 unigenes, 9 DEGs (6 genes: ACA2, AGD10, AGL18, PME5, TMK1 and ZAT5) were expressed in only fertile buds (log2 Ratio(S/F) > 17), which might offer new insights into the mechanisms of CMS regulation in Wucai. ACA2 encodes a Calcium-transporting ATPase 2 (plasma membrane-type), which regulates the Ca2+-mediated signaling pathway during pollen development [33, 34]. The nonexpression of this gene in sterile buds might disrupt the Ca2+ balance in the pollen mother cell. However, interestingly, AGD10 might be involved in root development as an ARF-GAP protein [35,36,37], and AGL18, encoding a MADS-box protein, has been reported as a flowering-inhibiting factor [38]. The functions of these genes in pollen development have not yet been reported and need further investigation. The other three genes (PME5, ZAT5, TMK1) and ACOX1, CCR2, GUN2, PGIP2, RBG7, RGP1, TCMO, VAL2 were involved in pollen cell wall formation, modification and organization (Table 2), in which critical chemical changes could lead to pollen abortion [5].
It has been reported that some constituents of the pollen wall are secreted from tapetal cells [39, 40]. Abnormal (early or delayed) tapetal cell degeneration can result in male sterility [5]. In conjunction with our cytological observations of Wucai buds (Fig. 1i-p), several genes related to tapetum development were revealed (Table 2). As a basic helix-loop-helix (bHLH) protein, AMS is required for tapetal cell biosynthesis, postmeiotic microspore and pollen wall formation, and tapetum programmed cell death (PCD) by directly regulating target genes involved in these biological pathways [41,42,43]. MS1 encodes a transcription factor of the PHD finger family and is specifically expressed in microsporocytes [44]. A6, a tapetum-specific protein secreted by the tapetal cells, displays similarity to β-1,3-glucanases, which degrade callose during pollen development [45]. TSM1 encodes a cation-dependent CCoAOMT-like protein involved in phenylpropanoid polyamine conjugate biosynthesis and has a function in stamen/pollen development [46, 47]. Downregulated expression of these genes could result in degeneration of the tapetum, eventually leading to abortion.
The regulation of transcription is a fundamental process in all living organisms [48]. Transcription factors can regulate multiple related downstream genes, which are essential components of the cellular machinery and play key roles in plant growth and development [49]. In the present study, 131 transcription factors (182 DEGs) were found (Additional file 9: Table S7). Among these transcription factors, the top four largest families were bHLH (16), MYB (13), NAC (10), and WRKY (8) (Table 3). The bHLH proteins, which bind as dimers to specific DNA target sites, are a superfamily of transcription factors, and several of them are critical for tapetal PCD and pollen development [41]. MYB transcription factors are also known to be required for anther and aleurone layer development, callose dissolution, and exine formation [19, 50, 51]. NAC and WRKY transcription factors consist of a large gene family involved in a wide range of biological processes [48, 50], and some of them participate in pollen development (WRKY2, WRKY27; GPC, NST1) [48, 52,53,54]. Research over the past several years has demonstrated that changes in the expression of these transcription factors often cause male sterility [5, 19].
In addition, FLOWERING LOCUS C (FLC), which encodes a MADS-box transcription factor and functions as a repressor of flowering [55], was noted in our comparative analysis (CL3897.Contig1; Additional file 9: Table S7). It has been reported that overexpression of this gene from B. campestris could affect fertility by the GA pathway in Arabidopsis [56]. However, in our study, we found that the FLC gene was downregulated in sterile buds, and several identified genes involved in stamens (AP3) [57] and the male gametophyte (AGL18 and AGL104) [58, 59] (Fig. 6; Additional file 10: Table S8) were downstream targets of FLC (Fig. 7). Among these genes, AGL18 and AGL104 showed low expression in fertile buds (Fig. 6; Additional file 10: Table S8). We speculated that the downregulation of FLC, which is associated with male infertility, might influence the expression of key genes in anther and pollen development, along with other fertility related genes (Fig. 7). This hypothesis must be further verified.
Taken together, the present investigation of the transcriptome could increase our knowledge and understanding of the molecular mechanisms of male sterile in Wucai and provide numerous candidate genes that can be verified through transgenic technology in future.
Conclusions
In this study, a comparative transcriptome analysis of sterile and fertile buds from Wucai was performed through an Illumina sequencing approach, and the different biological processes and genes that regulated anther and pollen development were analyzed using comparative analysis. As a result, a total of 4430 DEGs, 174 novel genes, 35 anther and pollen development related genes, and 47 transcription factors (the top four largest families) were revealed. The RNA-Seq analysis was further confirmed through qRT-PCR. Based on the functional annotation and expression patterns, it was concluded that the occurrence of male sterility is probably related to the functional and metabolic abnormalities of these candidate DEGs in Wucai. These transcriptome data will be important to serve as a reference and provide insights for future elucidation of male sterility in Wucai.
Materials and Methods
Plant materials
Buds from near-isogenic lines of Wucai, CMS line 12-14A and its maintainer line 12-14B (Fig. 1a and b), were used as the plant materials in this study. Backcrossed continuously for over ten generations, the male sterile line 12-14A of Wucai was generated from a CMS line of nonheading Chinese cabbage. The sterile line 12-14A and its maintainer line, 12-14B, were planted in the vegetable breeding fields of Anhui Agricultural University (Hefei, Anhui Province, China; longitude 117°14′E, latitude 31°52’N) from October until April of the following year.
Morphological and cytological observations
At the full-bloom stage, the flower structures of the CMS and fertile lines were observed using a Canon EOS550D digital camera (Canon, Japan), and the images from petals and stamens were captured with an Olympus SZX10 stereomicroscope (Olympus, Japan). The sections of flower buds from the CMS and fertile plants were obtained following the method described by Peng et al. [60]. The semithin sections were observed and photographed using an Olympus BX61 light microscope (Olympus, Japan) equipped with a Mshot MD30 camera (Olympus, Japan).
RNA extraction and Illumina sequencing
In this experiment, the buds of the sterile or fertile lines were collected from three different plants, respectively. According to the methods of Huang et al. [25], total RNA was isolated from the mixed bud samples using the TRIzol Reagent Kit (Invitrogen, USA) and purified using the Dynabeads® mRNA Purification Kit (Ambion, USA). The isolated RNA samples were sent to 1GENE Technology Co., Ltd. (Hangzhou, China; http://www.1gene.com.cn/) for Illumina sequencing (Illumina MiSeq platform) and unigene annotation. The raw transcriptome data of six samples from three biological replicates of sterile or fertile lines were deposited in the NCBI Short Read Archive (SRA, accession number: SRP145484).
De novo assembly and functional annotation analysis
The paired-end clean reads of each sample were de novo assembled into contigs using Trinity (http://trinityrnaseq.sourseforge.net), and the nonredundant unigenes were further obtained with the TGI Clustering tools [61]. Among these unigenes, there were several unigenes with a high degree of similarity (more than 70%) in the same cluster (starting with CL, followed by the gene family’s number); the rest were singletons (starting with Unigene), which had low similarity (less than 70%) or no similarity and could not be clustered with each other. Then, functional annotation of the unigenes was performed using BLASTX alignment (E-value<1e-5) in the nonredundant (nr), Swiss-Prot, and COG databases. With the nr annotations, the Gene Ontology (GO) annotations of the unigenes were obtained through the Blast2GO program [62], and GO functional classification was carried out with the WEGO software [63]. The KEGG pathway annotation was performed using a BLAST search against the KEGG database (KEGG, http://www.genome.jp/kegg/).
Differentially expressed gene (DEG) identification
Reads per kilobase per million reads (RPKM) was adopted to compare the differences in unigene expression between the sterile and fertile lines. The DEGs were identified by a false discovery rate (FDR) ≤0.001 and an absolute value of log2 ratio ≥ 1 (ratio = the fold change of differential expression) [13]. The DEGs were used for GO and KEGG enrichment analyses according to the method described by An et al. [14] and Liu et al. [13].
Quantitative real-time PCR verification
RNA was isolated from different samples as described above. The DNase-treated RNA (1 mg) was reverse transcribed to cDNA using the PrimeScript™ RT Reagent Kit (TaKaRa, Japan). Quantitative real-time PCR was then performed with the SYBR® Premix Ex Taq™ II Kit (TaKaRa, Japan). The specific primers designed based on the selected DEG sequences are listed in Additional file 11: Table S9. PCR amplification was performed in the Bio-Rad CFX96 instrument according to the manufacturer’s instructions. Data normalization was carried out using the expression levels of Actin as the internal control. Three biological repeats for each sample and three technical replicates for each gene were performed, and the relative expression level was calculated as 2-ΔΔCt.
Abbreviations
- CMS:
-
Cytoplasmic male sterility
- DEGs:
-
Differentially expressed genes
- FDR:
-
False discovery rate
- FLC:
-
FLOWERING LOCUS C
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PCD:
-
Programmed cell death
- RNA-Seq:
-
RNA sequencing technology
- RPKM:
-
Reads per kilobase per million reads
- SRA:
-
Short Read Archive
References
Zou M, Yuan L, Zhu S, Liu S, Ge J, Wang C. Response of osmotic adjustment and ascorbate-glutathione cycle to heat stress in a heat-sensitive and a heat-tolerant genotype of wucai (Brassica campestris L.). Sci Horic. 2016;211:87–94.
Chen G, Zhang S, Yuan L, Zhu S, Liu S, Zhang H, Wang C. Establishment of effective regeneration system of Wucai in vitro. Mol Plant Breed. 2017;15(4):1466–72 (in Chinese).
Yuan L, Zhu S, Liu S, Chen G, Zou M, Ge J, Wang C. A new male sterility savoy hybrid 'Huiwu 11′. Acta Hortic Sin. 2016;43(7):1423–4 (in Chinese).
Wei M, Song M, Fan S, Yu S. Transcriptomic analysis of differentially expressed genes during anther development in genetic male sterile and wild type cotton by digital gene-expression profiling. BMC Genomics. 2013;14(1):1–16.
Zhou X, Liu Z, Ji R, Feng H. Comparative transcript profiling of fertile and sterile flower buds from multiple-allele-inherited male sterility in Chinese cabbage (Brassica campestris L. ssp. pekinensis). Mol Gen Genomics. 2017;292:967–90.
Xu HM, Kong XD, Chen F, Huang JX, Lou XY, Zhao JY. Transcriptome analysis of Brassica napus pod using RNA-Seq and identification of lipid-related candidate genes. BMC Genomics. 2015;16(1):1–10.
Liu C, Liu Z, Li C, Zhang Y, Feng H. Comparative transcriptome analysis of fertile and sterile buds from a genetically male sterile line of Chinese cabbage. In Vitro Cell Dev Biol Plant. 2016;52(2):130–9.
Chen C, Chen G, Cao B, Lei J. Transcriptional profiling analysis of genic male sterile–fertile Capsicum annuum reveal candidate genes for pollen development and maturation by RNA-Seq technology. Plant Cell Tissue Organ Cult. 2015;122(2):465–76.
Liu XQ, Yu CY, Dong JG, Xu AX, Hu SW. De novo transcriptome reconstruction of a thermo-sensitive male sterility mutant in rapeseed (Brassica napus; Brassicaceae). App Plant Sci. 2017;5(12):1700077.
Yang J, Liu X, Xu B, Zhao N, Yang X, Zhang M. Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of Brassica juncea. BMC Genomics. 2013;14(1):9.
Xu X, Sun X, Zhang Y, Zhang L, Fang Z. Identification of AFLP and SSR markers linked with the male fertility restorer gene of CMS 06J45 in heading Chinese cabbage (Brassica rapa L. ssp. pekinensis). Plant Breed. 2014;133:615–9.
Wang Z, Gerstein MM. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63.
Liu CH, Fan C. De novo transcriptome assembly of floral buds of pineapple and identification of differentially expressed genes in response to ethephon induction. Front Plant Sci. 2016;7(168):203.
An H, Yang Z, Yi B, Wen J, Shen J, Tu J, Ma C, Fu T. Comparative transcript profiling of the fertile and sterile flower buds of pol CMS in B. napus. BMC Genomics. 2014;15(2):258.
Harper AL, Trick M, Higgins J, Fraser F, Clissold L, Wells R, Hattori C, Werner P, Bancroft I. Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat Biotechnol. 2012;30(8):798–802.
Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320(5881):1344–9.
Yan X, Dong C, Yu J, Liu W, Jiang C, Liu J, Hu Q, Fang X, Wei W. Transcriptome profile analysis of young floral buds of fertile and sterile plants from the self-pollinated offspring of the hybrid between novel restorer line NR1 and Nsa CMS line in Brassica napus. BMC Biochem. 2013;14(3):1–16.
Huang Q, Xin X, Guo Y, Hu S. Cytological observation of anther structure and genetic investigation of a new type of cytoplasmic male sterile 0A193-CMS in Brassica rapa L. Plant Breed. 2016. 135(6). https://doi.org/10.1111/pbr.12416.
Wang S, Wang C, Zhang XX, Chen X, Liu JJ, Jia XF, Jia SQ. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in cabbage. Plant Physiol Bioch. 2016;105:224–32.
Pei X, Jing Z, Tang Z, Zhu Y. Comparative transcriptome analysis provides insight into differentially expressed genes related to cytoplasmic male sterility in broccoli (Brassica oleracea var. italica ). Sci Horic. 2017;217:234–42.
Lyu M, Liang Y, Yu Y, Ma Z, Song L, Yue X, Cao J. Identification and expression analysis of BoMF25, a novel polygalacturonase gene involved in pollen development of Brassica oleracea. Plant Reprod. 2015;28(2):121–32.
Heng S, Liu S, Xia C, Tang HY, Xie F, Fu T, Wan Z. Morphological and genetic characterization of a new cytoplasmic male sterility system (oxa CMS) in stem mustard (Brassica juncea). Theor Appl Genetics. 2017;131(1):1–8.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644.
Liu J, Pang C, Wei H, Song M, Meng Y, Ma J, Fan S, Yu S. iTRAQ-facilitated proteomic profiling of anthers from a photosensitive male sterile mutant and wild-type cotton (Gossypium hirsutum L.). J Proteomics. 2015;126:68–81.
Huang S, Liu Z, Yao R, Li D, Feng H. Comparative transcriptome analysis of the petal degeneration mutant pdm in Chinese cabbage (Brassica campestris ssp. pekinensis) using RNA-Seq. Mol Gen Genet. 2015;290(5):1–15.
Yuan Q, Song C, Gao L, Zhang H, Yang C, Sheng J, Ren J, Chen D, Wang Y. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in onion. Plant Physiol Bioch. 2018;125:35.
Wu Z, Cheng J, Qin C, Hu Z, Yin C, Hu K. Differential proteomic analysis of anthers between cytoplasmic male sterile and maintainer lines in Capsicum annuum L. Int J Mol Sci. 2013;14(11):22982–296.
Mamun EA, Alfred S, Cantrill LC, Overall RL, Sutton BG. Effects of chilling on male gametophyte development in rice. Cell Biol Int. 2006;30(7):583–91.
Zhang Y, Chen J, Liu J, Xia M, Wang W, Shen F. Transcriptome analysis of early anther development of cotton revealed male sterility genes for major metabolic pathways. J Plant Growth Regul. 2015;34(2):223–32.
Li J, Yang S, Gai J. Transcriptome comparative analysis between the cytoplasmic male sterile line and fertile line in soybean (Glycine max (L.) Merr.). Genes Genomics. 2017;39(10):1117–27.
Li DD, Xue JS, Zhu J, Yang ZN. Gene regulatory network for tapetum development in Arabidopsis thaliana. Front Plant Sci. 2017;8:1559.
Xing M, Sun C, Li H, Hu S, Lei L, Kang J. Integrated analysis of transcriptome and proteome changes related to the Ogura cytoplasmic male sterility in cabbage. PLoS One. 2018;13(3):e0193462.
Xiang L, Qasim SM, Wu J, Lan W, Liu X, Lu Y. Comparative small RNA analysis of pollen development in autotetraploid and diploid rice. Int J Mol Sci. 2016;17(4):499.
Yan J, Zhang H, Zheng Y, Ding Y. Comparative expression profiling of miRNAs between the cytoplasmic male sterile line MeixiangA and its maintainer line MeixiangB during rice anther development. Planta. 2015;241(1):109–23.
Vernoud V, Horton AC, Yang Z, Nielsen E. Analysis of the small GTPase gene superfamily of Arabidopsis. Plant Physiol. 2003;131(3):1191–208.
Dümmer M, Michalski C, Essen LO, Rath M, Galland P, Forreiter C. EHB1 and AGD12, two calcium-dependent proteins affect gravitropism antagonistically in Arabidopsis thaliana. J Plant Physiol. 2016;206:114–24.
Yoo CM, Naramoto S, Sparks JA, Khan BR, Nakashima J, Fukuda H, Blancaflor EB. Deletion analysis of AGD1 reveals domains crucial for its plasma membrane recruitment and function in root hair polarity. J Cell Sci. 2017;131(2):jcs.203828.
Li CC, Ma GP, Xie T, Chen J, Wang ZM, Song M, Tang QL. SOC1 and AGL24 interact with AGL18-1, not the other family members AGL18-2 and AGL18-3 in Brassica juncea. Acta Physiol Plant. 2018;40(1):3.
Zou T, Liu M, Xiao Q, Wang T, Chen D, Luo T, Yuan G, Li Q, Zhu J, Liang Y. OsPKS2 is required for rice male fertility by participating in pollen wall formation. Plant Cell Rep. 2018;37:1–15.
Xiong SX, Lu JY, Lou Y, Teng XD, Gu JN, Zhang C, Shi QS, Yang ZN, Zhu J. The transcription factors MS188 and AMS form a complex to activate the expression of CYP703A2 for sporopollenin biosynthesis in Arabidopsis thaliana. Plant J. 2016;88(6):936–46.
Sheng Y, Wang Y, Jiao S, Jin Y, Ji P, Luan F. Mapping and preliminary analysis of ABORTED MICROSPORES (AMS) as the candidate gene underlying the male sterility (MS-5) mutant in melon (Cucumis melo L.). Front Plant Sci. 2017. 8. https://doi.org/10.3389/fpls.2017.00902.
Xu J, Ding Z, Vizcaybarrena G, Shi J, Liang W, Yuan Z, Werckreichhart D, Schreiber L, Wilson ZA, Zhang D. ABORTED MICROSPORES acts as a master regulator of pollen wall formation in Arabidopsis. Plant Cell. 2014;26(4):1544–56.
Xu J, Yang C, Yuan Z, Zhang D, Gondwe MY, Ding Z, Liang W, Zhang D, Wilson ZA. The ABORTED MICROSPORES regulatory network is required for postmeiotic male reproductive development in Arabidopsis thaliana. Plant Cell. 2010;22(1):91–107.
Wang Z, Li J, Chen S, Heng Y, Chen Z, Yang J, Zhou K, Pei J, He H, Deng XW. Poaceae-specific MS1 encodes a phospholipid-binding protein for male fertility in bread wheat. Proc Natl Acad Sci U S A. 2017;114(47):12614–9.
Hird DL, Worrall D, Hodge R, Smartt S, Paul W, Scott R. The anther-specific protein encoded by the Brassica napus and Arabidopsis thaliana A6 gene displays similarity to β-1,3-glucanases. Plant J. 1993;4(6):1023–33.
Fellenberg C, Milkowski C, Hause B, Lange PR, Böttcher C, Schmidt J, Vogt T. Tapetum-specific location of a cation-dependent O-methyltransferase in Arabidopsis thaliana. Plant J. 2008;56(1):132–45.
Liu H, Tan M, Yu H, Li L, Zhou F, Yang M, Zhou T, Zhao Y. Comparative transcriptome profiling of the fertile and sterile flower buds of a dominant genic male sterile line in sesame (Sesamum indicum L.). BMC Plant Biol. 2016;16(1):250.
Mukhtar MS, Liu X, Somssich IE. Elucidating the role of WRKY27 in male sterility in Arabidopsis. Plant Signal Behav. 2017. 12(9). https://doi.org/10.1080/15592324.15592017.11363945.
Li C, Zhao Z, Liu Y, Liang B, Guan S, Lan H, Wang J, Lu Y, Cao M. Comparative transcriptome analysis of isonuclear-alloplasmic lines unmask key transcription factor genes and metabolic pathways involved in sterility of maize CMS-C. Peerj. 2017;5:e3408.
Hong Y, Zhang H, Huang L, Li D, Song F. Overexpression of a stress-responsive NAC transcription factor gene ONAC022 improves drought and salt tolerance in rice. Front Plant Sci. 2016;7:4.
Ko S-S, Li M-J, Lin Y-J, Hsing H-X, Yang T-T, Chen T-K, Jhong C-M, Ku MS-B. Molecular basis underlying male sterility in bHLH142 overexpressing rice. bioRxiv. 2017; https://doi.org/10.1101/116996.
Mei S, Liu T, Wang Z. Comparative transcriptome profile of the cytoplasmic male sterile and fertile floral buds of radish (Raphanus sativus L.). Int J Mol Sci. 2016;17(1):42.
Distelfeld A, Pearce SP, Avni R, Scherer B, Uauy C, Piston F, Slade A, Zhao R, Dubcovsky J. Divergent functions of orthologous NAC transcription factors in wheat and rice. Plant Mol Biol. 2012;78(4–5):515–24.
Nakano Y, Yamaguchi M, Endo H, Rejab NA, Ohtani M. NAC-MYB-based transcriptional regulation of secondary cell wall biosynthesis in land plants. Front Plant Sci. 2015;6:288.
Zhou JX, Liu ZW, Li YQ, Li L, Wang B, Chen S, He XJ. Arabidopsis PWWP domain proteins mediate H3K27 trimethylation on FLC and regulate flowering time. J Integr Plant Biol. 2018. https://doi.org/10.1111/jipb.12630.
Liu T, Li Y, Zhang C, Qian Y, Wang Z, Hou X. Overexpression of FLOWERING LOCUS C, isolated from non-heading Chinese cabbage (Brassica campestris ssp. chinensis Makino), influences fertility in Arabidopsis. Plant Mol Biol Rep. 2012;30(6):1444–9.
Zhao YH, Larson-Rabin Z, Li DZ, Wang GY, Peng S, Li CY. The expression and phylogenetic analysis of four AP3 -like paralogs in the stamens, carpels, and single-whorl perianth of the paleoherb Asarum caudigerum. Mol Biol Rep. 2013;40(8):4691–9.
Alvarezbuylla ER, Liljegren SJ, Pelaz S, Gold SE, Burgeff C, Ditta GS, Vergarasilva F, Yanofsky MF. MADS-box gene evolution beyond flowers: expression in pollen, endosperm, guard cells, roots and trichomes. Plant J. 2000;24(4):457–66.
Adamczyk BJ, Fernandez DE. MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol. 2009;149(4):1713–23.
Peng Y, Shi D, Zhang T, Li X, Fu T, Xu Y, Wan Z. Development and utilization of an efficient cytoplasmic male sterile system for Cai-xin (Brassica rapa L.). Sci Horic. 2015;190:36–42.
Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19(5):651–2.
Conesa A, Götz S, Garcíagómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(Web Server issue):W293–7.
Acknowledgements
Heat map analysis was performed using the OmicShare tools (http://www.omicshare.com/tools) and the R software (R i386 3.4.2).
Funding
National Key R & D Program of China (2017YFD0101803).
Provincial Science and Technology Major Project of Anhui (17030701013).
National Natural Science Foundation of China (31801853, 31701910).
Major (KJ2017ZD15) and Key (KJ2017A153) Projects of Natural Science Research Funds in Support of Colleges.
Availability of data and materials
The raw RNA-Seq data used in this study have been deposited in the Nation Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database under the accession number SRP145484 (https://www.ncbi.nlm.nih.gov/sra/SRP145484).
Author information
Authors and Affiliations
Contributions
CW is the project leader. GC designed the experiments, and wrote the manuscript. GC and CW designed the experiment. GC and XY carried out the experiments. SZ, LY and JH supervised the study and SZ helped perform the experiments. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Summary of de novo transcriptome assembly. (XLS 18 kb)
Additional file 2:
Table S2. Statistics of annotation results. (XLS 18 kb)
Additional file 3:
Figure S1. Characteristics of homology search of Illumina sequences against the NR database. (DOC 505 kb)
Additional file 4:
Table S3. All DEGs between sterile and fertile buds. (XLS 5575 kb)
Additional file 5:
Table S4. Novel genes. (XLS 50 kb)
Additional file 6:
Figure S2. Gene Ontology (GO) assignment. of DEGs. (DOC 190 kb)
Additional file 7:
Table S5. Statistics of DEGs matched in KEGG pathways. (XLS 41 kb)
Additional file 8:
Table S6. DEGs associated with starch and sucrose metabolism. (XLS 46 kb)
Additional file 9:
Table S7. Annotation and relative expression of differentially expressed transcription factors in Wucai. (XLS 60 kb)
Additional file 10:
Table S8. Flowering genes identified from the DEGs. (XLS 21 kb)
Additional file 11:
Table S9. Primers used in this study. (XLS 26 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, G., Ye, X., Zhang, S. et al. Comparative Transcriptome Analysis between Fertile and CMS Flower Buds in Wucai (Brassica campestris L.). BMC Genomics 19, 908 (2018). https://doi.org/10.1186/s12864-018-5331-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5331-4