
Abstract
Background
Emerging evidence has been experimentally confirmed the tissue-specific expression of circRNAs (circRNAs). Global identification of human tissue-specific circRNAs is crucial for the functionality study, which facilitates the discovery of circRNAs for potential diagnostic biomarkers.
Results
In this study, circRNA back-splicing junctions were identified from 465 publicly available transcriptome sequencing samples. The number of reads aligned to these identified junctions was normalized with the read length and sequence depth for each sample. We generated 66 models representing enriched circRNAs among human tissue transcriptome through biclustering algorithm. The result provides thousands of newly identified human tissue-specific circRNAs.
Conclusions
This result suggests that expression of circRNAs is not prompted by random splicing error but serving molecular functional roles. We also identified circRNAs enriched within circulating system, which, along with identified tissue-specific circRNAs, can serve as potential diagnostic biomarkers.
Similar content being viewed by others

Background
Circular RNAs (circRNAs) are a type of long non-coding RNAs, whose 3′ and 5′ ends joined into a single strand circular form. Although the existence of human circRNAs has been discovered and proven with electron microscopy for more than 30 years [1], it was only until 2012 with the advance of high throughput sequencing technology the ubiquitous expression of circRNA in mammals was found and proven [2]. Emerging evidence indicates the tissue-specific circRNAs play crucial roles in post-transcriptional level. Several cases of human circRNAs were found to serve as natural microRNA sponges [3, 4]. Biogenesis of circRNAs was found competing with the mRNAs of the host gene. In recent years, cell-free circRNAs were found in salvia and blood plasma [5, 6]. CircRNAs can potentially serve as diagnostic biomarkers for the undercover correlation to the pathogenesis of diseases and human physiological functions, as well as the stable circular forms. Global identification of human tissue-specific circRNAs is crucial for the study of circRNAs functionality.
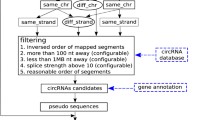
The junctions between the 3′ and 5′ ends of the circRNAs have been referred as back-splicing junctions. The existence of circRNA within transcriptome sequencing data can be detected through identification of reads spanning these junctions. In previous studies [4], threshold applied to identify certain junctions as circRNA was that at least two unique reads spanning a head-tail junction. To discover human tissue-specific circRNAs, we collected 465 human transcriptome sequencing runs and applied the established pipeline. Expression level of circRNAs was estimated using the normalized counts of reads spanning the back-splicing junctions [7]. Biclustering [8] was conducted to detect circRNA expression patterns across different types of human tissues. From the result 66 bicluster models, we found a huge portion of circRNAs express only in the specific tissue type. This result suggests that expression of circRNAs is not prompted by random splicing error but serving molecular functional roles. We also identified circRNAs enriched within circulating system, which, along with identified tissue-specific circRNAs, can serve as potential diagnostic biomarkers.
Results
A total 148,095 unique back-splicing junctions were identified from the selected transcriptome sequencing runs. Each of the junction site satisfy the threshold defined in the find_circ scripts [4, 9], as provided in the (Additional file 1). At least two reads were found spanning the identified site. More than 30000 junctions were found to have standard deviation of SRPBM among the 465 runs larger than 10. Through examining the alignment result with normalized value, we managed to find the tissue enriched circRNAs which would have been neglected. Back-splicing junctions with only one spliced read can be found in many datasets with high SRPBM values. The biclustering algorithm clustered 16,317 unique junctions into 66 coherent expression profile models (Additional file 2). The result of the biclustering reflects that the expression profiles of circRNAs are under tissue-specific regulation. A network view of the identified tissue-specific circRNAs is illustrated in Fig. 1.

Network summary of the tissue-specific circRNAs. The circular nodes in this graph represents amount of grouped back-splicing junctions. Red nodes represent tissue-specific circRNAs, while blue nodes represent circRNAs enriched in multiple tissue types
CircRNAs can be identified using poly A enriched RNA-Seq data
It had been assumed that since the exon originated circRNAs does not go through polyadenylation process after transcribed and spliced, they cannot be identified in the ploy-A enriched RNA-Seq data. However in several recent studies [2, 10, 11] circRNAs were identified in poly-A enriched RNA-Seq data sets, this could be due to the fact that some circular isoforms of the gene are adenine-rich. In this study, we discovered 24,589 unique back-splicing junctions from the 376 selected poly-A enriched RNA-Seq runs. One of the pivotal circRNA cdr1as [3, 4], which was proven to be nature miRNA miR-7 sponge, was found in 107 of our selected runs. Among these runs 71 are poly-A enriched.
Novel back-spicing junctions
Compared with human circRNAs reported in 22 recent studies [4, 6, 7, 12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29], we found 5680 identified circRNAs back-splicing junctions has been reported in other studies. The remaining 92,015 unique back-splicing junctions are considered as novel circRNA candidates. Isoform annotation and the expression profiling can be found in the data base CircNet [30].
Tissue-specific circRNAs
As illustrated in Fig. 1, the biclustering result provides thousands of tissue-specific expressed circRNAs. The nodes containing lower than 10 circRNAs, or connects to more than 3 types of tissues were hidden in the graph. The network demonstrates that circRNA co-expression profile following specific patterns similar to human genes [31]. Some groups of circRNAs express in multiple types of tissues with close correlated function. For example, the 332 circRNAs grouped with bowel, colon and large intestine might have potential physiological roles in the digest system, while the 243 circRNAs enriched in prostate and thyroid might correlate with male reproducing or development. The large amount of circRNA enriched in blood or blood cell samples suggests the ubiquity of circulating circRNAs, which makes circRNAs ideal diagnostic biomarkers. The tissue-specific circRNAs is available in (Additional file 3).
Brain-specific circRNA host genes are enriched with synaptic GO terms
Based on the result of the gene-set enrichment analysis, we found that host genes of the brain-specific circRNAs are specially enriched with synapse-associated GO terms [32], as listed in Table 1. This result is consistent with the recent report regarding synaptic genes hosting circRNAs [33]. Through this study we hypothesize that these brain-specific circRNAs participate in the neuron development and synaptic functions. The back-splicing junction sites were enriched into these 9 gene groups, which can be found in (Additional file 4).
Potential diagnostic biomarkers revealed from the results of the biclustering
Besides the tissue specificity of human circRNAs, putative diagnostic biomarkers can be discovered from the bicluster models. We found 607 back-splicing junctions and cancer skin/prostate samples were clustered into the same bicluster (SD10_10.txt from Additional file 2). Samples with close conditions originated from the same tissue types were biclustered with back-spicing junctions, suggesting that circRNAs originated from these back-splicing junctions express specifically to the disease condition as well as tissue type. With sufficient experiment verification as well as population studies, these circRNAs can serve as potential diagnostic biomarkers. These back-splicing junction sites and the related conditions are summarized in Table 2. A full list of these biomarkers is available in (Additional file 5).
Discussion
In this study, we identified the potential tissue specific circRNA through conducting biclustering on expression profiles of circRNA across multiple human tissue samples. Despite the promising results, several limitations are worth-mentioning.
First of all, RNA-Seq data set collected in this study are retroactive sourced. Potential Batch effect was inevitable. Expression profiles within poly-A enriched samples were also biased. On the other hand, the expression profile was based on the normalized count of back spliced junction site spanning reads. Without accurate annotation of the full-length sequence of circRNA, this kind of measurement can be limited. Finally, the gene set enrichment conducted in this study was based on the genes locus that intersect with back spliced junction sites. This analysis was based on the assumption that functions of circRNAs correlate with the functions of back spliced junction sites overlapped genes. Weather tissue specific genes correlate with the biogenesis of tissue specific circRNA Is also an ongoing research subject. Further analysis of the issue specific circRNA locus correlation with known tissue specific genes should be conducted in the near future.
Conclusions
From the result 66 bicluster models, we found a huge portion of circRNAs express only in the specific tissue type. This result suggests that expression of circRNAs is not prompted by random splicing error but serving molecular functional roles. We also identified circRNAs enriched within circulating system, which, along with identified tissue-specific circRNAs, can serve as potential diagnostic biomarkers after sufficient experiment verification as well as population studies.
Methods
As illustrated in Fig. 2, 465 RNA-Seq runs were collected from a wide range of independent experiments across 26 human tissues and 104 disease conditions from the NCBI Sequence Read Archive [34], in which 89 runs are non-polyA enriched RNA-Seq data. Selection of the RNA-Seq runs was made on purpose of covering as many different conditions as possible. Quality control of the sequence reads was conducted through the NGS QC toolkit [35] with default setting. The algorithm referred as find_circ [4, 9] was applied to detect back-splicing junctions. To normalize the amount of the normalized sequence reads spanning the junctions, a concept of spliced reads per billion mapping (SRPBM) was applied [7]. Amount of reads mapped onto hg19 human genome was acquired through the tool STAR [36]. The equation applied to calculate SRPBM is:

Summary of the data analysis process. The data analysis process conducted in this study is summarized in this flowchart
The junction sites with standard deviation of SRPBM among the 465 runs larger than 10 were further selected for biclustering analysis. For searching the coherent expression profiles. The R package ‘isa2’ [37] was used for Iterative Signature Algorithm analysis. Coherent expression pro-files of selected 30000 junctions among the 465 runs was acquired from the bicluster models generated from Iterative Signature Algorithm [38]. A network analysis was conducted on the grouped junctions, and the network was illustrated through Cytoscape [39]. Gene sets enrichment of the circRNA host genes was conducted through DAVID [40]. Back spliced junction sites clustered into models containing only one types of tissue were considered as tissue-specific circRNAs originated.
References
Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976;73(11):3852–6.
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript Isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7(2):e30733.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–8.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–8.
Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J, Chen D, Gu J, He X, Huang S. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25(8):981.
Bahn JH, Zhang Q, Li F, Chan T-M, Lin X, Kim Y, Wong DT, Xiao X. The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin Chem. 2015;61(1):221–30.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–57.
Tanay A, Sharan R, Shamir R. Discovering statistically significant biclusters in gene expression data. Bioinformatics. 2002;18(suppl 1):S136–44.
Glažar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20(11):1666–70.
Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15(7):409.
Salzman J. Circular RNA expression: its potential regulation and function. Trends Genet. 2016;32(5):309–16.
Boeckel J-N, Jaé N, Heumüller AW, Chen W, Boon RA, Stellos K, Zeiher AM, John D, Uchida S, Dimmeler S. Identification and characterization of hypoxia-regulated endothelial circular RNA. Circ Res. 2015;117(10):884–90.
Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, Mesteri I, Grunt TW, Zeillinger R, Pils D. Correlation of circular RNA abundance with proliferation-exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep. 2015;5:8057.
Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160(6):1125–34.
Memczak S, Papavasileiou P, Peters O, Rajewsky N. Identification and characterization of circular RNAs as a new class of putative biomarkers in human blood. PLoS One. 2015;10(10):e0141214.
Alhasan AA, Izuogu OG, Al-Balool HH, Steyn JS, Evans A, Colzani M, Ghevaert C, Mountford JC, Marenah L, Elliott DJ. Circular RNA enrichment in platelets is a signature of transcriptome degradation. Blood. 2016;127(9):e1–e11.
Cheng J, Metge F, Dieterich C. Specific identification and quantification of circular RNAs from sequencing data. Bioinf. 2016;32(7):1094-6.
Zhang X-O, Dong R, Zhang Y, Zhang J-L, Luo Z, Zhang J, Chen L-L, Yang L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016;26:1277–87.
Song X, Zhang N, Han P, Moon B-S, Lai RK, Wang K, Lu W. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res. 2016;44:e87.
Dang Y, Yan L, Hu B, Fan X, Ren Y, Li R, Lian Y, Yan J, Li Q, Zhang Y. Tracing the expression of circular RNAs in human pre-implantation embryos. Genome Biol. 2016;17(1):1.
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215.
Zhang Y, Zhang X-O, Chen T, Xiang J-F, Yin Q-F, Xing Y-H, Zhu S, Yang L, Chen L-L. Circular intronic long noncoding RNAs. Mol Cell. 2013;51(6):792–806.
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9(9):e1003777.
Rybak-Wolf A, Stottmeister C, Glažar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58(5):870–85.
Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15(7):1.
Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015;16(1):1.
Zhang X-O, Wang H-B, Zhang Y, Lu X, Chen L-L, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159(1):134–47.
Caiment F, Gaj S, Claessen S, Kleinjans J. High-throughput data integration of RNA–miRNA–circRNA reveals novel insights into mechanisms of benzo [a] pyrene-induced carcinogenicity. Nucleic Acids Res. 2015;43(5):2525–34.
Kelly S, Greenman C, Cook PR, Papantonis A. Exon skipping is correlated with exon circularization. J Mol Biol. 2015;427(15):2414–7.
Liu YC, Li JR, Sun CH, Andrews E, Chao RF, Lin FM, Weng SL, Hsu SD, Huang CC, Cheng C. CircNet: a database of circular RNAs derived from transcriptome sequencing data. Nucleic Acids Res. 2016;44(D1):D209-15.
Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397–406.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9.
You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci. 2015;18:603–10.
Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2011;39(Database issue):D19-21.
Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2):e30619.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Csárdi G, Kutalik Z, Bergmann S. Modular analysis of gene expression data with R. Bioinformatics. 2010;26(10):1376–7.
Bergmann S, Ihmels J, Barkai N. Iterative signature algorithm for the analysis of large-scale gene expression data. Phys Rev E. 2003;67(3):031902.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35(suppl_2):W169–75.
Acknowledgements
The authors would like to thank the Ministry of Science and Technology, the National Chiao Tung University and Ministry of Education, Taiwan, and University System of Taiwan (VGHUST), for financially supporting this research.
Funding
Publication charges for this work were supported by the Ministry of Science and Technology, Taiwan [MOST103-2628-B-009-001-MY3, MOST105-2627-M-009-007-, MOST105-2319-B-400-002-, MOST104-2911-I-009-509 and MOST 106-2633-B-009-001]. The research reported in this paper was mainly supported by “Aiming for the Top University Program” of the National Chiao Tung University and Ministry of Education, Taiwan, R.O.C.
Availability of data and materials
The data from this study are available as Additional files 1, 2, 3, 4 and 5.
About this supplement
This article has been published as part of BMC Genomics Volume 19 Supplement 1, 2018: 16th International Conference on Bioinformatics (InCoB 2017): Genomics. The full contents of the supplement are available online at https://bmcgenomics.biomedcentral.com/articles/supplements/volume-19-supplement-1.
Author information
Authors and Affiliations
Contributions
This study was proposed by YCL, under the supervision of HDH and CCL. YCL performed the circRNA detection and data collection. The biclustering was conducted by YJC, JRL and CHS. All authors participated in the audition and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
One hundred forty-eight thousand ninety-five unique back-splicing junctions were identified from the selected transcriptome sequencing runs. Each of the junction site satisfy the threshold defined in the find_circ scripts. (XLSX 10182 kb)
Additional file 2:
The biclustering algorithm clustered 16,317 unique junctions into 66 coherent expression profile models. (RAR 1442 kb)
Additional file 3:
The tissue-specific circRNAs. (XLSX 426 kb)
Additional file 4:
The back-splicing junction sites were enriched into these 9 gene groups. (TXT 133 kb)
Additional file 5:
Collection of circRNAs can serve as potential diagnostic biomarkers. (XLSX 110 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, YC., Chiu, YJ., Li, JR. et al. Biclustering of transcriptome sequencing data reveals human tissue-specific circular RNAs. BMC Genomics 19 (Suppl 1), 958 (2018). https://doi.org/10.1186/s12864-017-4335-9
Published:
DOI: https://doi.org/10.1186/s12864-017-4335-9