
Abstract
Background
This study used next generation sequencing to generate the mitogenomes of four African pangolin species; Temminck’s ground pangolin (Smutsia temminckii), giant ground pangolin (S. gigantea), white-bellied pangolin (Phataginus tricuspis) and black-bellied pangolin (P. tetradactyla).
Results
The results indicate that the mitogenomes of the African pangolins are 16,558 bp for S. temminckii, 16,540 bp for S. gigantea, 16,649 bp for P. tetradactyla and 16,565 bp for P. tricuspis. Phylogenetic comparisons of the African pangolins indicated two lineages with high posterior probabilities providing evidence to support the classification of two genera; Smutsia and Phataginus. The total GC content between African pangolins was observed to be similar between species (36.5% – 37.3%). The most frequent codon was found to be A or C at the 3rd codon position. Significant variations in GC-content and codon usage were observed for several regions between African and Asian pangolin species which may be attributed to mutation pressure and/or natural selection. Lastly, a total of two insertions of 80 bp and 28 bp in size respectively was observed in the control region of the black-bellied pangolin which were absent in the other African pangolin species.
Conclusions
The current study presents reference mitogenomes of all four African pangolin species and thus expands on the current set of reference genomes available for six of the eight extant pangolin species globally and represents the first phylogenetic analysis with six pangolin species using full mitochondrial genomes. Knowledge of full mitochondrial DNA genomes will assist in providing a better understanding on the evolution of pangolins which will be essential for conservation genetic studies.
Similar content being viewed by others

Background
Worldwide, the eight extant pangolin species are classified in the order Pholidota which consists of one family, Manidae (Class: Mammalia). The four African species includes Temminck’s ground pangolin (Smutsia temminckii), giant ground pangolin (S. gigantea), white-bellied pangolin (Phataginus tricuspis) and black-bellied pangolin (P. tetradactyla) [1,2,3]. The four Asian species include Philippine pangolin (Manis culionensis), Indian pangolin (M. crassicaudata), Chinese pangolin (M. pentadactyla) and Malayan pangolin (M. javanica). All African pangolin species are listed as Vulnerable on the International Union for Conservation of Nature (IUCN) Red List of Threatened Species [4,5,6,7]. Of the Asian species, two are listed as Critically Endangered (Chinese and Malayan pangolin) [8, 9] and two are listed as Endangered (Philippine and Indian pangolin) [10, 11]. Pangolins face numerous threats, including habitat destruction [12,13,14], electrocution [15,16,17] as well as poaching and illegal trade [18,19,20,21,22,23]. In 2016, the IUCN voted in support of transferring all eight pangolin species from Appendix II to Appendix I at the Convention on International Trade in Endangered Species of Fauna and Flora (CITES), which was approved at the 17th Conference of Parties (COP17). The listing has resulted in worldwide commercial trade in pangolins being banned as from January 2017 [24, 25]. The taxonomy of pangolins is still under debate, with disagreement regarding the number of genera due to lack of molecular phylogenetic analysis [1, 3, 26,27,28,29]. These species have been placed into six genera by Pocock [26]. Other authors have classified all eight extant species of pangolins into a single genus; Manis [3, 29, 30]. Corbet and Hill [31] suggested two genera; Manis (Asian pangolins) and Phataginus (African pangolins) while Koeningswald [32] and Gaudin and Wible [1] proposed three genera; Manis (Asian pangolins), Phataginus (African tree pangolins) and Smutsia (African ground pangolins). Based on osteological characteristics from the entire skeleton [33], three genera were supported, with the first two genera (Phataginus and Smutsia) forming a monophyletic African clade in the subfamily Smutsiinae [33]. Lastly, four genera have been proposed by McKenna and Bell [28] and Kingdon [34] namely Manis, Smutsia, Phataginus and Uromanis. Several authors follow the single genus classification [35,36,37,38], however an in-depth taxonomic study of pangolin genera is required in order to clarify this issue.
Mitochondrial DNA (mtDNA) accounts for 1-2% of total DNA content found in mammalian species [39] and is circular, double-stranded and between 14 and 19 kb in length [40]. The vertebrate mitochondrial genome generally consists of 37 genes, specifying 13 proteins, two ribosomal RNAs, 22 tRNAs, and a control region [41]. The control region is non-coding and contains elements that may regulate replication and transcription [42]. Mitochondrial DNA is generally suitable for evolutionary studies due to its high mutation rate, well-structured genome with restricted non-coding DNA sequences and lack of recombination. Several studies have used portions of the mitochondrial genome including the control region (D-loop) [43, 44], cytochrome c oxidase I (CoxI) [44, 45], cytochrome B (Cob) [44, 46, 47] and 16S ribosomal RNA (16S rRNA) [47] for traceability of confiscated pangolin scales. Whole mitochondrial DNA genomes will however be more informative for phylogenetic analysis [48,49,50,51,52,53]. To date, full mitochondrial genomes of five pangolin species have been determined including M. pentadactyla, M. javanica, S. temminckii, P. tetradactyla and P. tricuspis [54,55,56,57,58,59]. However, two mitogenomes include misidentified Genbank records incorrectly accessioned as M. pentadactyla and P. tetradactyla that were noted in subsequent studies [58, 60]. Several techniques have been reported to generate whole mitochondrial genomes, however modern techniques such as next generation sequencing (NGS) using 454, Illumina and Ion Torrent technology have simplified and made sequencing mitogenomes from any eukaryotic DNA easier, quicker and more affordable compared to Sanger-based methods [61,62,63]. The vast suite of Bioinformatics software currently available facilitates the annotation and aids in analyses of large datasets [64].
In general, a quarter of the reads generated by RNA/DNA sequence experiments are from mitochondrial genomes [61, 64,65,66] which may be attributed to their high copy numbers as well as their high expression levels. Due to the AT richness of mtDNA, as well as it being polyadenylated it can contribute to an increase in poly-A RNA selection [65]. Assembling mitochondrial genomes are significantly less complex than their nuclear genome counterparts as they are smaller in size, and harbour fewer genes [64]. The mitogenomes of two Asian pangolin species (M. pentadactyla and M. javanica) have been assembled using Illumina HiSeq technology, whereby the authors extracted mitochondrial sequences from nuclear data obtained from NGS techniques [57, 59].
Current phylogenetic assessments of pangolins have been conducted using only two of the four African pangolin species namely; Temminck’s ground pangolin and white-bellied pangolin [56, 58]. In addition, the current genus-level classification of pangolins is still under debate. Thus, in this study we performed next generation sequencing for all four African pangolins using the Illumina HiSeq 2500 in order to reconstruct complete mitochondrial genomes. Here we present the first whole mitochondrial DNA genomes of two of the African pangolin species; the black-bellied pangolin (P. tetradactyla) and the giant ground pangolin (S. gigantea). In addition, we describe the mitochondrial genome features in order to understand the evolutionary forces shaping the mitochondrial genomes of African pangolins. Lastly, we conduct a phylogenetic assessment in order to provide a genus-level classification of African pangolins.
Methods
Sample collection and DNA isolation
This study used six deceased individuals, sampled by the African Pangolin Working Group (APWG) and representing the four African pangolin species. Tissue samples were placed in absolute ethanol and were stored at the National Biobank, National Zoological Gardens of South Africa (NZG), at −80 °C until analysis. The samples were from one black-bellied pangolin (P. tetradactyla; MF509825), one white-bellied pangolin (P. tricuspis; MF536683), both from Ghana [67]; and three Temminck’s ground pangolins (S. temminckii; MF536685–MF536687) from South Africa. In addition, a giant ground pangolin (S. gigantea; MF536684) scale sample was included from an illegal seizure. The species identity of samples used in this study was confirmed with Sanger sequencing of the CoxI and Cob loci which were compared to chain-of-custody voucher specimens available from the NZG species reference database [68] (see http://www.barcodeofwildlife.org). All voucher specimens were verified and identified by an acknowledged authority (Raymond Jansen; African Pangolin Working Group). DNA was isolated using the QIAamp Micro Kit (QIAGEN, Novato, CA, USA) and the respective manufacturers’ protocol for tissue was followed. DNA was quantified on the Qubit 3.0 Fluorometer (Thermo Scientific, Massachusetts, USA). Polymerase Chain Reaction (PCR) amplification and sequencing, to verify species identity, were performed as outlined in Mwale [44].
Next-generation sequencing and assembly
The products were run on an Illumina HiSeq 2500 (Illumina Incorporated, San Diego, CA, USA) using a rapid run and the TruSeq DNA LT Sample Prep Kit (Illumina Incorporated, San Diego, CA, USA). Data quality was evaluated using FastQC v0.11.2 [69] software, and trimmed and edited through Trimmomatic v0.36 [70] to remove the adapters and poor quality sections. Mitogenomes were assembled in CLC Genomics Workbench v6 (https://www.qiagenbioinformatics.com; CLC Bio, Aarhus, Denmark) using De Novo alignment, with paired reads. Sequence identity of contigs was validated by performing a BLAST search on the National Centre for Biotechnology Information (NCBI) website (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Mitogenome annotation and phylogenetic analysis
The mitogenomes were annotated with MITOS v806 [71] and a circular alignment between the six available pangolin species were drawn in Circos v0.69 [72]. The GC content of the four African pangolin mitogenomes were calculated using GPMiner [73] with a sliding window of 300 bp. Arlequin v3.5.1 [74] was used to validate the GC scores obtained for the four mitogenomes using ANOVA analysis and the diagrams were plotted in R v3.3.1 [75].
The mitogenomes generated in this study comprised six animals from four African pangolin species and were combined and aligned with 11 other genomes using MAFFT v7 [76] (Table 1). The mitogenomes of Acinonyx jubatus [77], Crocuta crocuta [78], Canis lupus [79] and Arctocephalus pusillus [80] were used as out-groups, as the order Pholidota (pangolins) is reported to be evolutionary closer to carnivorans [56]. The phylogenetic program jModeltest v2.1.7 [81] was used to determine the best fit model of sequence evolution, under the Akaike Information Criterion (AIC) [82], Bayesian Information Criterion (BIC) [83] and Decision Theory Performance-Based Selection (DT) [84]. Partition analysis was also implemented using the program PartitionFinder v2 [85] to determine the best fit models of evolution for the different loci in the dataset. The partition was run using linked branch lengths and a greedy search for the models under the AIC. Phylogenetic analysis was conducted using MrBayes v3.2.6 [86] to infer relationships between the different species using Bayesian Inference (BI). The parameters used for MrBayes were two million generations after which 25% of the trees were discarded as burn-in. A Maximum Likelihood (ML) tree was constructed utilizing PhyML v3 [87] with the same models used for the Bayesian analysis and was run with 10,000 Bootstrap replications. Individual phylogenetic trees, for each loci, were also created with MrBayes v3.2.6 [86] and PhyML v3 [87].
Codon usage analysis for African and Asian species
The Relative Synonymous Codon Usage (RSCU) values for mitochondrial genes were established using the Mega v7 [88] software. This was performed on the four African pangolin species evaluated in this study as well as for the previously published M. pentadactyla (KT445978.1) and M. javanica (KT445979.1). The Principle Component Analysis (PCAs) generated from this data was performed using the FactoMineR package in R [89]. Codon usage bias (AT3 and GC3 content) was calculated using Mega v7, for each of the protein coding genes, where the A and T values at the third base were summed for the AT3 value. The same was performed with G and C for the GC3 content. The ratios were reported as percentages.
Confirmation of insertions observed in the control region of the pangolin mitogenome
Sanger sequencing of the control region of the mitochondrial genome was performed using five additional samples from each of the African pangolin species. The white-bellied and black-bellied pangolin samples were from Ghana and the Temminck’s ground pangolin samples from South Africa and Tanzania [58]. The giant ground pangolin samples were obtained from the collection of the Zoological Museum, University of Copenhagen. The protocol and cycle conditions outlined in Du Toit [56] were used for all the samples. Sequencing was conducted in order to verify the presence of insertions in the D-loop observed in the mitogenomes obtained from next-generation sequencing during this study. A sequence fragment of around 500 bp was targeted using the primer pair: PNG_Dloop forward 5′-CGTTCCTCTTAAATAAGACATCTCG-3′ and reverse 5′-TCTTGCTTTTGGGGTTTGAC-3′ for verification.
Results and discussion
Next-generation sequencing
The HiSeq rapid run resulted in approximately 22 million reads per sample, with an average read length of 250 nucleotides. These reads were used for a De Novo assembly of each sample (CLC Bio version 6.0). This resulted in 10,207 contigs for P. tricuspis with the largest contig being 16,565 nucleotides, consisting of 78,099 reads at an average coverage of 986×. For P. tetradactyla, there were 2801 contigs with the largest contig, 16,649 nucleotides consisting of 47,686 reads at an average coverage of 369×. For S. gigantea, there were 11,346 contigs with the largest contig, 16,540 nucleotides consisting of 13,076 reads and an average coverage of 98×. For the three S. temminckii samples (MF536685–MF536687), contigs ranged from 2742 to 5560. The largest contig in all three samples was 16,558 nucleotides consisting of 63,759; 29,702 and 6820 reads and an average coverage of 494×; 248× and 53× respectively. The contigs were identified as the mitogenomes of the pangolin species based on the (i) estimated length (≈16.5 kb); (ii) the occurrence of the proteins CoxI, Cob, NADH dehydrogenase V (NadV) and NADH dehydrogenase VI (NadVI) (Table 2) and (iii) correspondence with mitochondrial sequences from other Pholidota based on NCBI BLAST searches.
Genomic organisation
The mitogenome of the S. temminckii samples consisted of 16,558 bp while S. gigantea was 16,540 bp; P. tetradactyla was 16,649 bp and P. tricuspis was 16,565 bp in length (Table 2, Fig. 1). The light and heavy strands each contain their own arrangement of genes, proteins or loci respectfully located on each strand (Fig. 1). The heavy strand, or plus strand, comprises of the following loci: two Ribosomal RNAs (12S rRNA, 16S rRNA); 12 Protein-coding genes (NadI, NadII, CoxI, CoxII, AtpVIII, AtpVI, CoxIII, NdIII, NadIV-L, NadIV, NadV, Cob) and 14 Transfer RNAs (trnF, trnV, trnL2, trnI, trnM, trnW, trnD, trnK, trnG, trnR, trnH, trnS1; trnL1, trnT). The light or minus strand comprises one Protein-coding gene (NadVI) and eight Transfer RNAs (trnQ, trnA, trnN, trnC, trnY, trnS2, trnE, trnP). As indicated in Table 2, the mitogenome of the four African pangolins varied in terms of gene region size at several loci. Six regions [cytochrome oxidase II (CoxII), ATP synthase VI (AtpVI), cytochrome oxidase III (CoxIII), NADH dehydrogenase III (NadIII), NADH dehydrogenase IV-L (NadIV-L), cytochrome b (COB)] were found to be the same length in all four species and three loci [16S Ribosomal RNA, NADH dehydrogenase V (NadV), D-loop] each have different lengths in each of the four species. The remaining loci [12S Ribosomal RNA, NADH dehydrogenase I (NadI), NADH dehydrogenase II (NadII), cytochrome oxidase I (CoxI), ATP synthase VIII (AtpVIII), NADH dehydrogenase IV (NadIV), NADH dehydrogenase VI (NadVI)] had lengths that were generally consistent, with some pangolin species showing variation in length in comparison to the other species (Table 2).

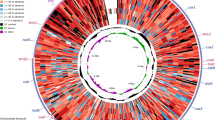
Circular diagram of six pangolin mitogenomes. The six coloured circles represent the six different pangolin species (four African and two Asian) aligned to each other to indicate the differences between individuals with the reference genome represented by P. tetradactyla. The outer ring represents the annotated loci located on the plus/heavy strand. The second ring represents the loci located on the minus/light strand of the mitogenome. Arrows are representative of the direction of the light and heavy strands; the heavy strand is located clockwise and the light strand anti-clockwise
Phylogenetic analysis of African pangolins
The best fit model of sequence evolution for the dataset under the AIC was the General Time Reversal model (GTR + I + G) with invariable site and gamma distribution of 0.822 [90, 91]. The best fit model under the BIC and DT was the Transition model two (TIM2 + I + G) with invariable site and gamma distribution values of 0.998 and 0.990 respectively [92]. From these two models three Bayesian phylogenetic trees along with three maximum likelihood trees were generated for the datasets. The different trees all showed consistent branching patterns, posterior probability values for the BI trees and bootstrap values for the ML trees. The trees were subsequently concatenated into a single consensus tree with the different support values indicated on the respective branches (Fig. 2). Partition analysis indicated a variety of models for the individual loci and was subjected to individual BI and ML analysis to confirm the results of the whole mitochondrial data. In the phylogenetic tree, using all available pangolin mitogenomes, it is evident that all the African pangolins group according to species, with the exception of the black-bellied pangolin genome (AJ421454.1) [54]. The latter mitogenome has previously been reported to be misclassified based on partial Cob analysis (Additional file 1: Figure S1) [58, 60] and was confirmed in this analysis as a white-bellied pangolin genome. In addition, the misclassified M. pentadactyla (JN411577.1) [55] grouped with M. javanica [58, 59], confirming an error also reported in previous studies. The phylogenetic tree (Fig. 2) which excluded the misclassified samples provided support for the Asian and African pangolin species separation into two distinct monophyletic clades with the latter consisting of all African pangolins species, P. tricuspis, P. tetradactyla, S. temminckii and S. gigantea. Within the African clade the giant and Temminck’s pangolin clustered separately from the white-bellied and black-bellied pangolins with significant Bayesian and ML support (Posterior Probabilities of 1). For the African pangolin species, the observed branching pattern thus provides support for the classification of the ground-dwelling and arboreal species into two separate genera; Phataginus and Smutsia. In addition, results from this analysis suggests the overall classification of pangolin into three genera; Manis (Asian pangolins), Phataginus (African tree pangolins) and Smutsia (African ground pangolins). However, further analysis should be undertaken for Asian pangolins to include the full mitochondrial genomes of the Philippine (M. culionensis) and Indian pangolin (M. crassicaudata). The above branching patterns were also confirmed using individual loci. The control region, rRNAs, light strand proteins, light strand tRNAs and heavy strand proteins (exclusive of COX2) BI and ML results were all concurrent with the whole mtDNA tree with high support. The heavy strand tRNAs showed differentiation in the tree pangolins and again in the ground pangolins of Africa for both BI and ML trees. Although the internal grouping differs, they still conform to the same three genera identified in the trees above namely Smutsia, Phataginus and Manis. The ML analysis for the heavy strand protein COX2 was in accordance with the results above. However, the BI analysis indicated that the black-bellied pangolin (MF509825) branched separately from the African pangolins, but still formed a monophyletic group with the African pangolin species adjacent to the Asian pangolins. Overall, the majority of the individual loci subject to different evolutionary rates and models along with a variety of phylogenetic analysis concurred with the results obtained from the whole mtDNA data.

Combined Bayesian Inference (BI) and Maximum Likelihood (ML) tree of pangolin species. Bayesian Posterior Probabilities are indicated on the bottom of each node whereas the Maximum Likelihood Bootstrap values are indicated on top of the node. Only Bootstrap values equal or greater than 70% (≥70%) were noted on the tree. The mitogenome of Acinonyx. jubatus, Crocuta crocuta, Canis lupus and Arctocephalus pusillus was selected as outgroups. * indicates the six mitogenomes sequenced during this study
Analysis of GC content and codon usage
Total GC content of the African pangolin species was observed to be similar between species (P. tetradactyla = 36.5%, S. gigantea = 36.9%, S. temminckii = 37.3% and P. tricuspis = 36.7%). These results confirm an AT-bias that has been reported in several other mammal species [93]. Analysis of codon usage and pattern of mitochondrial genes; AtpVI, AtpVIII, Cob, CoxI, CoxII, NadI, NadII, NadIII, NadIV, NadIV-L, NadV and NadVI provided evidence of bias in terms of the use of codons with A and C occurring most frequently at the third codon (Additional file 1: Table S1). Variation in base compositions within and among species has been suggested to occur as a result of two evolutionary processes namely biases in the process of mutation and/or natural selection [94]. Selective nucleotide compositional biases have been reported in chiropteran mitochondrial genomes [95]. Uddin and Chakraborty [96] similarly observed A or C as the most frequent codon at the 3rd position in a study of mitochondrial AtpVI in a variety of mammalian species. The authors attributed this bias to mutational pressure that can influence codon usage bias in mitochondria.
GC content and codon usage variation between pangolin species
The percentage of GC content and codon frequencies calculated for five regions (3100–3700 bp, 4500–4900 bp, 7500–7900 bp, 9700–9900 bp and 15,000–16,000 bp) of the mitogenome was significantly different between the four African pangolin species based on ANOVA analysis (Fig. 3a -e). The respective genes that correspond to these regions include NadI, NadII, CoxII, NadIII and Cob. Analysis of codon usage of three mitochondrial genes; CoxI, NadI and NadIII reveals a clear distinction between the African and Asian pangolins, as well as within the African clade (Additional file 1: Figure S2 a-c). The PCA plots for these three genes therefore identified a connection between codon usage and phylogeny and provide further support for the phylogenetic analysis based on the whole mitochondrial genome. The PCA plots of the remaining protein coding genes (AtpVIII, NadIV and NadVI), whilst not achieving the same resolution within the African clade, show a clear separation of the Asian and African pangolins (Additional file 1: Figure S2 d-f). Combined codon usage patterns of mitochondrial genes (AtpVI, AtpVIII, Cob, CoxI, CoxII, NadI, NadII, NadIII, NadIV, NadIV-L, NadV and NadVI) were plotted in order to perform a hierarchical clustering of each species to investigate the role of codon bias in the evolution of African pangolins. The resulting dendrogram is presented in Fig. 4. African pangolin species varied in the percentage of codon bias with Asian pangolins displaying a lower degree of AT3 bias. Variations in GC content and codon frequencies between pangolin species may indicate that two selective forces; mutational pressure and/or natural selection may play important roles in the molecular evolution of pangolins with different evolutionary forces acting to shape the mitochondrial genomes of the African and Asian pangolin species.

Representation of regions which display significant differences in terms of GC content among African pangolin species (a-e). Image (a) showing increased GC content (~3100–3700 bp) in S. temminckii; (b) showing an increased GC content (~4500–4900 bp) in M. tricuspis; (c) showing a decreased GC content (~7500–7900 bp) in M. tetradactyla; (d) showing an increased GC content (~9700–9900 bp) in M. tetradactyla and S. temminckii; (e) showing an increased GC content (~15,000–16,000 bp) in M. tetradactyla

Clustering of pangolin species according to the variation of codon usage and phylogeny. The dendrogram was inferred by hierarchical clustering with the AT3 frequency metric for each species. The value of the AT3 is indicated as a percentage
Mitogenome comparison between Smutsia and Phataginus
Two insertions (80 bp and 28 bp in length) in the D-loop region of the black-bellied pangolin were observed (Fig. 5) which was absent in the other African pangolin species. This insertion was validated with Sanger sequencing that included additional African pangolin species. When compared to the Asian pangolin species the first insertion was slightly shorter in length (62 bp). Length variation in the D-loop has been reported in various species including bats [97], rodents [98] and primates [99]. In addition, substantial nucleotide sequence differences within and between species have been identified in the D-loop [100, 101]. Lastly, heteroplasmy where individuals had more than one mtDNA form due to variation in numbers of tandem repeats in D-loop has been reported in shad [102], sturgeon [103], whiptail lizards [104] and rabbits [105]. Length variation has been proposed to occur via four different mechanisms including illegitimate elongation [103], intra- and intermolecular recombination [106], transposition [106] and slipped miss pairing [107]. The identification of the two insertions in the D-loop region may indicate that this region is under selection in black-bellied pangolins, which may demonstrate drift following the initial mutation event. However, further analysis among closely related species should be conducted in order to determine how selection impacts on the length and sequence variation within this region.

Alignment of a region of D-loop in six pangolin species. The insertion sequence for P. tetradactyla is indicated in a dashed box. The first insertion is 80 bp and the second 28 bp in length
Conclusions
In conclusion, our research study presents the mitogenomes for the four African pangolin species. These include two new reference genomes for the black-bellied pangolin and the giant ground pangolin. This study also presents the first phylogenetic assessment of six of the eight extant pangolin species using whole mitochondrial DNA genomes. The African and Asian pangolin species are shown to separate into two distinct monophyletic clades. Within the African pangolins it was further demonstrated that there is support for classification of the species into separate genera, representing the arboreal (P. tricuspis, P. tetradactyla) and ground-dwelling (S. temminckii and S. gigantea). The availability of these reference mitogenomes will, furthermore, contribute to a better understanding of the evolutionary processes of pangolin species globally, which in turn can contribute to essential conservation genetic studies.
References
Gaudin TJ, Wible JR. The entotympanic of pangolins and the phylogeny of the Pholidota (Mammalia). J Mamm Evol. 1999;6:39–65. Kluwer Academic Publishers-Plenum Publishers. Available from: http://link.springer.com/10.1023/A:1020538313412
Skinner JD, Chimimba CT. The mammals of the southern African subregion. 3rd ed. Cambridge: Cambridge University Press; 2005. Available from: http://www.cambridge.org/za/academic/subjects/life-sciences/zoology/mammals-southern-african-sub-region-3rd-edition
Wilson DE, Reeder DM. Mammal species of the world: a taxonomic and geographic reference. 3rd ed. Baltimore: Johns Hopkins University Press; 2005. Available from: https://books.google.co.za/books?id=JgAMbNSt8ikC&redir_esc=y
Pietersen DW, Waterman C, Hywood L, Rankin P, Soewu D. Smutsia temminckii IUCN Red List Threat. Species 2014. 2014 [cited 2014 Aug 1]. p. e.T12765A45222717. Available from: https://doi.org/10.2305/IUCN.UK.2014-2.RLTS.T12765A45222717.en
Waterman C, Pietersen DW, Hywood L, Rankin P, Soewu D. Smutsia gigantea . IUCN Red List Threat. Species 2014. 2014 [cited 2016 Jul 12]. p. e.T12762A45222061. Available from: https://doi.org/10.2305/IUCN.UK.2014-2.RLTS.T12762A45222061.en
Waterman C, Pietersen DW, Soewu D, Hywood L, Rankin P. Phataginus tetradactyla . IUCN Red List Threat. Species 2014. 2014 [cited 2016 Jul 12]. p. e.T12766A45222929. Available from: https://doi.org/10.2305/IUCN.UK.2014-2.RLTS.T12766A45222929.en
Waterman C, Pietersen DW, Soewu D, Hywood L, Rankin P. Phataginus tricuspis. IUCN Red List Threat. Species 2014 . 2014 [cited 2016 Jul 12];e.T12767A45223135. Available from: https://doi.org/10.2305/IUCN.UK.2014-2.RLTS.T12767A45223135.en
Challender D, Baillie J, Ades G, Kaspal P, Chan B, Khatiwada A, et al. Manis pentadactyla . IUCN Red List Threat. Species 2014. 2014 [cited 2017 Jan 25]. Available from: https://doi.org/10.2305/IUCN.UK.2014-2.RLTS.T12764A45222544.en
Challender D, Nguyen Van T, Shepard C, Krishnasamy K, Wang A, Lee B, et al. Manis javanica . IUCN Red List Threat. Species 2014. 2014 [cited 2017 Jan 25]. p. e.T12763A45222303. Available from: https://doi.org/10.2305/IUCN.UK.2014-2.RLTS.T12763A45222303.en
Baillie J, Challender D, Kaspal P, Khatiwada A, Mohapatra R, Nash H. Manis crassicaudata . IUCN Red List Threat. Species 2014. 2014 [cited 2017 Jan 25]. p. e.T12761A45221874. Available from: https://doi.org/10.2305/IUCN.UK.2014-2.RLTS.T12761A45221874.en
Lagrada L, Schoppe S, Challender D. Manis culionensis . IUCN Red List Threat. Species 2014. 2014 [cited 2017 Jan 25]. Available from: https://doi.org/10.2305/IUCN.UK.2014- 2.RLTS.T136497A45223365.en.
Sodeinde OA, Adedipe SR. Pangolins in south-west Nigeria – current status and prognosis. Oryx . Cambridge University Press; 1994;28:43–50. Available from: https://www.cambridge.org/core/journals/oryx/article/div-classtitlepangolins-in-south-west-nigeria-current-status-and-prognosisdiv/5A318C23BE4A81F9F3B8F41BCFD08BCB
Yang CW, Chen S, Chang C-Y, Lin MF, Block E, Lorentsen R, et al. History and dietary husbandry of pangolins in captivity. Zoo Biol. 2007;26:223–30. Available from: http://onlinelibrary.wiley.com/doi/10.1002/zoo.20134/abstract;jsessionid=7EC208E94FB0C8044D536FA4C4735716.f04t03
Whiting MJ, Williams VL, Hibbitts TJ. Animals traded for traditional medicine at the faraday market in South Africa: species diversity and conservation implications. J Zool. 2011;284:84–96. Blackwell Publishing Ltd. Available from: http://doi.wiley.com/10.1111/j.1469-7998.2010.00784.x
Beck A. Electric fence induced mortality in South Africa. Johannesburg: University of the Witwatersrand; 2008. Available from: http://wiredspace.wits.ac.za/jspui/bitstream/10539/7980/1/Beck%20Final%20MSc%20Dissertation.pdf Final MSc Dissertation.pdf.
Pietersen DW. Ground pangolins and electric fences. In: Ferguson K, Hanks J, editors. Fenc. Impacts A Rev. Environ. Soc. Econ. impacts game Vet. Fenc. Africa with Part. Ref. to Gt. Limpopo Kavango-Zambezi Transfront. Conserv. Areas . Pretoria: Mammal Research Institute; 2010. p. 154–7. Available from: https://www.wcs-ahead.org/gltfca_grants/pdfs/ferguson_final_2010.pdf
Pietersen DW, McKechnie AE, Jansen R. A review of the anthropogenic threats faced by Temminck’s ground pangolin, Smutsia temminckii, in southern Africa. South African J Wildl Res. 2014;44:167–78. South African Wildlife Management Association. Available from: http://www.bioone.org/doi/abs/10.3957/056.044.0209
Challender DWS, Hywood L. African pangolins under increased pressure from poaching and intercontinental trade. TRAFFIC Bull. 2012;24:53–5. Available from: http://www.academia.edu/12488434/African_pangolins_under_increased_pressure_from_poaching_and_intercontinental_trade
Boakye MK, Pietersen DW, Kotzé A, Dalton DL, Jansen R. Ethnomedicinal use of African pangolins by traditional medical practitioners in Sierra Leone. J Ethnobiol Ethnomed. . BioMed Central; 2014;10:76–85. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4247607/
Boakye MK, Pietersen DW, Kotzé A, Dalton D-L, Jansen R. Knowledge and uses of African pangolins as a source of traditional medicine in Ghana. PLoS One. 2015;10:e0117199. Public Library of Science, Available from: http://dx.plos.org/10.1371/journal.pone.0117199
Challender DWS, Harrop SR, Macmillan DC. Understanding markets to conserve trade-threatened species in CITES. Biol Conserv. 2015;187:249–59. Available from: http://www.sciencedirect.com/science/article/pii/S0006320715001603
Challender D, Baillie JEM, Waterman C, Shepherd CR. On scaling up pangolin conservation. TRAFFIC Bull. 2016;28:19–21. Available from: http://www.pangolinsg.org/wp-content/uploads/sites/4/2016/06/Challender-et-al-2016-On-Scaling-Up-Pangolin-Conservation_TRAFFIC-Bulletin.pdf
Shepherd CR, Connelly E, Hywood L, Cassey P. Taking a stand against illegal wildlife trade: the Zimbabwean approach to pangolin conservation. Oryx . Cambridge: Cambridge University Press; 2016. pp. 1–6. Available from: http://journals.cambridge.org/article_S0030605316000119
Dixon M, Weiskotten C. CITES CoP17: Victory Today for Pangolins . WCS Newsroom. 2016 [cited 2016 Oct 20]. Available from: https://newsroom.wcs.org/News-Releases/articleType/ArticleView/articleId/9303/CITES-CoP17-Victory-Today-for-Pangolins.aspx
IUCN. What does the new trade ban mean for pangolin conservation? . IUCN Red List Threat. Species. 2016 [cited 2016 Oct 20]. Available from: http://www.iucnredlist.org/news/what-does-the-new-trade-ban-mean-for-pangolin-conservation
Pocock RI. The external characters of the pangolins (Manidae). Proc Zool Soc London. 1924;94:707–18. Available from: http://onlinelibrary.wiley.com/doi/10.1111/j.1096-3642.1924.tb03310.x/full
Emry RJ. A north American Oligocene pangolin and other additions to the Pholidota. Bull Am Museum Nat Hist. 1970;142:459–510. Available from: http://digitallibrary.amnh.org/handle/2246/1078
Mckenna MC, Bell SK. Classification of mammals above the species level. New York: Columbia University Press; 1997. Available from: https://books.google.co.za/books/about/Classification_of_Mammals.html?id=zS7FZkzIw-cC&redir_esc=y
Nowak RM. Walker’s Mammals of the World. 6th ed. Baltimore: Johns Hopkins University Press; 1999. Available from: https://www.amazon.co.uk/Walkers-Mammals-World-2-vol-set/dp/0801857899
Linnaeus C. Systema naturae per regna tria naturae, secundum classes, ordines, genera, species, cum characteribus, differentiis, synonymis, locis . Tomus I, editor. Holmiae, Laurentii Salvii; 1758. Available from: http://www.biodiversitylibrary.org/bibliography/542#/summary
Corbet GB, Hill JE. A world list of mammalian species. 3rd ed. London: Natural History Museum Publications & Oxford University Press; 1991. Available from: http://taxonomicon.taxonomy.nl/Reference.aspx?id=1594
Koenigswald W. Order Pholidota. In: Rössner GE, Heissig K, editors. Miocene L. Mamm. Eur. Munich: Verlag Dr. Friedrich Pfeil; 1999. p. 75–80. Available from: https://www.amazon.com/Miocene-Land-Mammals-Europe/dp/3931516504
Gaudin TJ, Emry RJ, Wible JR. The phylogeny of living and extinct pangolins (mammalia, pholidota) and associated taxa: a morphology based analysis. J Mamm Evol. 2009;16:235–305. Available from: http://link.springer.com/article/10.1007/s10914-009-9119-9
Kingdon J. The Kingdon field guide of African mammals. London: Academic Press; 1997.
Gaubert P, Antunes A. Assessing the taxonomic status of the Palawan pangolin Manis Culionensis (Pholidota) using discrete morphological characters. J Mammal. 2005;86:1068–74. Available from: http://jmammal.oxfordjournals.org/content/86/6/1068
Gaudin TJ, Emry RJ, Pogue B. A new genus and species of pangolin (Mammalia, Pholidota) from the late Eocene of Inner Mongolia. China J Vertebr Paleontol. 2006;26:146–59.
Botha J, Gaudin T. An early pliocene pangolin (Mammalia; Pholidota) from Langebaanweg. South Africa J Vertebr Paleontol. 2007;27:484–91. Available from: http://www.bioone.org/doi/abs/10.1671/0272-4634(2007)27%5B484%3AAEPPPF%5D2.0.CO%3B2
Gaubert P, Njiokou F, Ngua G, Afiademanyo K, Dufour S, Malekani J, et al. Phylogeography of the heavily poached African common pangolin (Pholidota, Manis Tricuspis) reveals six cryptic lineages as traceable signatures of Pleistocene diversification. Mol Ecol. 2016;25:5975–93. Available from: http://doi.wiley.com/10.1111/mec.13886
Yang L, Tan Z, Wang D, Xue L, Guan M-X, Huang T, et al. Species identification through mitochondrial rRNA genetic analysis. Sci Rep. 2014;4:4089–99. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24522485
Kia J-S, Hopb H, Kima S-J, Kimc I-C, Parkd HG, Leea J-S. Complete mitochondrial genome sequence of the Arctic gammarid, Onisimus Nanseni (Crustacea; Amphipoda): novel gene structures and unusual control region features. Comp Biochem Physiol Part D Genomics Proteomics. 2010;5:105–15. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20434417
Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–80. Oxford University Press. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10101183
Shadel GS, Clayton DA. Mitochondrial DNA maintenance in Vertebrates. Annu. Rev. Biochem. . Annual Reviews 4139 El Camino Way, P.O. Box 10139, Palo Alto, CA 94303-0139, USA; 1997;66:409–35. Available from: http://www.annualreviews.org/doi/10.1146/annurev.biochem.66.1.409
Hsieh H-M, Lee JC-I, Wu J-H, Chen C-A, Chen Y-J, Wang G-B, et al. Establishing the pangolin mitochondrial D-loop sequences from the confiscated scales. Forensic Sci Int Genet. 2011;5:303–7. Elsevier Ireland Ltd. Available from: http://dx.doi.org/10.1016/j.fsigen.2010.06.003
Mwale M, Dalton DL, Jansen R, Roelofse M, Pietersen DW, Mokgokong PS, et al. The forensic application of DNA barcoding for identification of illegally traded African pangolin scales. Genome. 2016;gen-2016-0144. Available from: http://www.nrcresearchpress.com/doi/10.1139/gen-2016-0144
Zhang H, Miller MP, Yang F, Chan HK, Gaubert P, Ades G, et al. Molecular tracing of confiscated pangolin scales for conservation and illegal trade monitoring in southeast Asia. Glob Ecol Conserv. 2015;4:414–22. Available from: http://www.sciencedirect.com/science/article/pii/S235198941500089X
Wirdateti GSY. Identifikasi trenggiling (Manis javanica) menggunakan penanda cytochrome B mitokondria DNA (IDENTIFICATION OF PANGOLIN (MANIS JAVANICA DESMAREST, 1822) USING CYTOCHROME B mtDNA MARKER). J Vet. 2013;14:467–74. Available from: http://ojs.unud.ac.id/index.php/jvet/article/view/7682
Kumar VP, Rajpoot A, Mukesh SM, Kumar D, Goyal SP. Illegal trade of Indian pangolin (Manis Crassicaudata): genetic study from scales based on mitochondrial genes. Egypt J Forensic Sci. 2016;6:524–33. Available from: http://www.sciencedirect.com/science/article/pii/S2090536X16300454
Cao Y, Adachi J, Janke A, Pääbo S, Hasegawa M. Phylogenetic relationships among eutherian orders estimated from inferred sequences of mitochondrial proteins: instability of a tree based on a single gene. J Mol Evol. 1994;39:519–27. Springer-Verlag. Available from: http://link.springer.com/10.1007/BF00173421
Cummings MP, Otto SP, Wakeley J. Sampling properties of DNA sequence data in phylogenetic analysis. Mol Biol Evol. 1995;12:814–822. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7476127.
Zardoya R, Meyer A. Phylogenetic performance of mitochondrial protein-coding genes in resolving relationships among vertebrates. Mol Biol Evol. 1996;13:933–942. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8752002.
Cao Y, Fujiwara M, Nikaido M, Okada N, Hasegawa M. Interordinal relationships and timescale of eutherian evolution as inferred from mitochondrial genome data. Gene. 2000;259:149–58. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11163972
Ingman M, Kaessmann H, Pääbo S, Gyllensten U. Mitochondrial genome variation and the origin of modern humans. Nature. 2000;408:708–13. Nature Publishing Group. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11130070
Miya M, Kawaguchi A, Nishida M. Mitogenomic exploration of higher teleostean phylogenies: a case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol Biol Evol. 2001;18:1993–2009. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11606696
Arnason U, Adegoke JA, Bodin K, Born EW, Esa YB, Gullberg A, et al. Mammalian mitogenomic relationships and the root of the Eutherian tree. Proc Natl Acad Sci U S A. 2002;99:8151–6. National Academy of Sciences. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12034869
Qin X-M, Dou S-R, Guan Q-X, Qin P-S, She Y. Complete mitochondrial genome of the Manis Pentadactyla (Pholidota, Manidae): comparison of M. Pentadactyla and M. Tetradactyla. Mitochondrial DNA. 2012;23:37–8. Available from: http://www.tandfonline.com/doi/full/10.3109/19401736.2011.643881
Du Toit Z, Grobler JP, Kotzé A, Jansen R, Brettschneider H, Dalton D-L. The complete mitochondrial genome of Temminck’s ground pangolin (Smutsia temminckii; smuts, 1832) and phylogenetic position of the Pholidota (Weber, 1904). Gene. 2014;551:49–54. Elsevier B.V. Available from: http://dx.doi.org/10.1016/j.gene.2014.08.040
Hari R, Siew Woh C. Complete mitogenome of M. javanica and M. pentadactyla obtained from next generation sequencing data. Unpublished . 2015; Available from: https://www.ncbi.nlm.nih.gov/nuccore/KT445979.1
Hassanin A, Hugot J-P, Jansen van Vuuren B. Comparison of mitochondrial genome sequences of pangolins (Mammalia, Pholidota). C R Biol. 2015;338:260–5. ScienceDirect. Available from: http://dx.doi.org/10.1016/j.crvi.2015.02.003
Hari R, Paterson IC, Choo SW. A new complete mitogenome of the critically endangered Chinese pangolin Manis Pentadactyla. Conserv Genet Resour. Springer Netherlands. 2016:1–4. Available from: http://link.springer.com/10.1007/s12686-016-0586-z
Gaubert P, Njiokou F, Olayemi A, Pagani P, Dufour S, Danquah E, et al. Bushmeat genetics: setting up a reference framework for the DNA typing of African forest bushmeat. Mol Ecol Resour. 2015;15:633–51. Available from: http://doi.wiley.com/10.1111/1755-0998.12334
Smith DR. Not seeing the genomes for the DNA. Brief Funct Genomics. 2012;11:289–90. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22408189
King JL, LaRue BL, Novroski NM, Stoljarova M, Seo SB, Zeng X, et al. High-quality and high-throughput massively parallel sequencing of the human mitochondrial genome using the Illumina MiSeq. Forensic Sci Int Genet. 2014;12:128–35. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24973578
Seo SB, Zeng X, King JL, Larue BL, Assidi M, Al-Qahtani MH, et al. Underlying data for sequencing the mitochondrial genome with the massively parallel sequencing platform ion torrent™ PGM™. BMC Genomics. 2015;16:S4. BioMed Central. Available from: http://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-16-S1-S4
Smith DR. The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief. Funct. Genomics. 2015;15:1–8. Available from: http://bfg.oxfordjournals.org/content/early/2015/06/27/bfgp.elv027.abstract
Raz T, Kapranov P, Lipson D, Letovsky S, Milos PM, Thompson JF. Protocol dependence of sequencing-based gene expression measurements. El-Sayed NM, editor. PLoS One. Public Library of Science; 2011;6:e19287. Available from: http://dx.plos.org/10.1371/journal.pone.0019287
Smith DR. RNA-Seq data: a goldmine for organelle research. Brief. Funct. Genomics. 2013;12:454–6. Oxford University Press. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23334532
Boakye MK, Kotzé A, Dalton DL, Jansen R. Unravelling the pangolin bushmeat commodity chain and the extent of trade in Ghana. Hum Ecol. 2016;44:257–64. Available from: http://link.springer.com/10.1007/s10745-016-9813-1
Mwale M, Dalton DL, Roelofse M, Radebe T, Labuschagne K, Kloppers A, et al. Progress in the barcoding of illegally traded South African wildlife species at the National Zoological Gardens of South Africa. Genome. Canadian Science Publishing, NRC Research Press, 65 Auriga Dr, Suite 203, Ottawa, ON K2E 7W6, Canada; 2015. p. 259–60.
Andrews S. FastQC: a quality control tool for high throughput sequence data . Babraham Bioinforma. 2010 [cited 2016 Jul 18]. Available from: http://dnacore.missouri.edu/PDF/FastQC_Manual.pdf
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. Oxford University Press. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4103590/
Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–9. Available from: http://www.sciencedirect.com/science/article/pii/S1055790312003326
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res; 2009;19:1639–1645. Cold Spring Harbor Laboratory Press. Available from: http://genome.cshlp.org/cgi/doi/10.1101/gr.092759.109
Lee T-Y, Chang W-C, Hsu JB-K, Chang T-H, Shien D-M. GPMiner: an integrated system for mining combinatorial cis-regulatory elements in mammalian gene group. BMC Genomics BioMed Central; 2012;13:S3–14. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22369687
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour. 2010;10:564–7. Blackwell Publishing LtdAvailable from: http://doi.wiley.com/10.1111/j.1755-0998.2010.02847.x
Development Core Team R. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2008. Available from: http://www.r-project.org
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. Oxford University Press. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3603318/
Burger PA, Steinborn R, Walzer C, Petit T, Mueller M, Schwarzenberger F. Analysis of the mitochondrial genome of cheetahs (Acinonyx Jubatus) with neurodegenerative disease. Gene. 2004;338:111–9. Available from: http://www.sciencedirect.com/science/article/pii/S0378111904003117
Bon C, Berthonaud V, Maksud F, Labadie K, Poulain J, Artiguenave F, et al. Coprolites as a source of information on the genome and diet of the cave hyena. Proc R Soc B Biol Sci. 2012;279:2825–30. The Royal Society. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22456883
Koblmüller S, Vilà C, Lorente-Galdos B, Dabad M, Ramirez O, Marques-Bonet T, et al. Whole mitochondrial genomes illuminate ancient intercontinental dispersals of grey wolves ( Canis Lupus ). J Biogeogr. 2016;43:1728–38. Available from: http://doi.wiley.com/10.1111/jbi.12765
Arnason U, Gullberg A, Janke A, Kullberg M, Lehman N, Petrov EA, et al. Pinniped phylogeny and a new hypothesis for their origin and dispersal. Mol Phylogenet Evol. 2006;41:345–54. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1055790306001977
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772–5. Nature Publishing Group. Available from: http://www.faculty.biol.ttu.edu/olson/Molecular_Ecology_and_Evolution_files/nmeth.2109.pdf
Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr. 1974;19:716–23. IEEE. Available from: http://ieeexplore.ieee.org/lpdocs/epic03/wrapper.htm?arnumber=1100705
Hirose K, Kawano S, Konishi S, Ichikawa M. Bayesian information criterion and selection of the number of factors in factor analysis models. J Data Sci. 2011;9:243–259. Available from: https://www.google.co.za/url?sa=t&rct=j&q=&esrc=s&source=web&cd=3&cad=rja&uact=8&ved=0ahUKEwi99JCMlvDNAhUlJ8AKHZ67DY8QFgg0MAI&url=http://www.jds-online.com/file_download/306/JDS-682.pdf&usg=AFQjCNFMZ4eDW9vlQ6CE9hwwpIL8K1nVqw&bvm=bv.126993452,d.d24.
Minin V, Abdo Z, Joyce P, Sullivan J. Performance-based selection of likelihood models for phylogeny estimation. Syst Biol. 2003;52:674–83. Available from: http://sysbio.oxfordjournals.org/content/52/5/674.long
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2017;34:772–3. Oxford University Press. Available from: https://academic.oup.com/mbe/article-abstract/34/3/772/2738784/PartitionFinder-2-New-Methods-for-Selecting?redirectedFrom=fulltext
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, et al. Mrbayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3329765/
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21. Oxford University Press. Available from: https://academic.oup.com/sysbio/article-lookup/doi/10.1093/sysbio/syq010
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4. Available from: http://mbe.oxfordjournals.org/lookup/doi/10.1093/molbev/msw054
Lê S, Josse J, Husson F. FactoMineR: an R package for multivariate analysis. J Stat Softw. 2008;25:1–18. Available from: http://www.jstatsoft.org/v25/i01/
Lanave C, Preparata G, Saccone C, Serio G. A new method for calculating evolutionary substitution rates. J Mol Evol. 1984;20:86–93. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6429346
Rodríguez F, Oliver JL, Marín A, Medina JR. The general stochastic model of nucleotide substitution. J Theor Biol. 1990;142:485–501. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2338834
Posada D. Using MODELTEST and PAUP* to select a model of nucleotide substitution. Curr. Protoc. Bioinforma. John Wiley and Sons, Inc; 2003;00:6.5:6.5.1–6.5.14. Available from: http://onlinelibrary.wiley.com/doi/10.1002/0471250953.bi0605s00/abstract
Dou H, Zhang Y, Feng L. Complete mitochondrial genome of the Himalayan serow (Capricornis Thar) and its phylogenetic status within the genus Capricornis. Biochem Syst Ecol. 2016;65:115–23. Available from: http://www.sciencedirect.com/science/article/pii/S0305197816300357
Mooers AØ, Holmes EC. The evolution of base composition and phylogenetic inference. Trends Ecol Evol. 2000;15:365–9. Available from: http://www.sciencedirect.com/science/article/pii/S0169534700019340
Meganathan PR, Pagan HJT, McCulloch ES, Stevens RD, Ray DA. Complete mitochondrial genome sequences of three bats species and whole genome mitochondrial analyses reveal patterns of codon bias and lend support to a basal split in Chiroptera. Gene. 2012;492:121–9. Elsevier B.V.. Available from: http://dx.doi.org/10.1016/j.gene.2011.10.038
Uddin A, Chakraborty S. Analysis of codon usage pattern in mitochondrial ATPase 6 in some mammalian species. Int J Recent Sci Res. 2014;5:883–8. Available from: https://www.researchgate.net/publication/283681777_ANALYSIS_OF_CODON_USAGE_PATTERN_IN_MITOCHONDRIAL_ATPASE_6_IN_SOME_MAMMALIAN_SPECIES
Wilkinson GS, Mayer F, Kerth G, Petri B. Evolution of repeated sequence arrays in the D-loop region of bat mitochondrial DNA. Genetics. 1997;146:1035–48. Genetics Society of AmericaAvailable from: http://www.ncbi.nlm.nih.gov/pubmed/9215906
Larizza A, Pesole G, Reyes A, Sbisà E, Saccone C. Lineage specificity of the evolutionary dynamics of the mtDNA D-loop region in rodents. J Mol Evol. 2002;54:145–55. Springer-VerlagAvailable from: http://link.springer.com/10.1007/s00239-001-0063-4
Saitou N, Ueda S. Evolutionary rates of insertion and deletion in noncoding nucleotide sequences of primates. Mol Biol Evol. 1994;11:504–12. Available from: https://www.ncbi.nlm.nih.gov/pubmed/8015443
Aquadro CF, Greenberg BD. Human mitochondrial DNA variation and evolution: analysis of nucleotide sequences from seven individuals. Genetics. 1983;103:287–312. Genetics Society of AmericaAvailable from: http://www.ncbi.nlm.nih.gov/pubmed/6299878
Foran DR, Hixson JE, Brown WM, Arbor A. Comparisons of ape and human sequences that regulate mitochondrial DNA transcription and D-loop DNA synthesis. Nucleic Acids Res. 1988;16:5841–61.
Bentzen P, Leggett WC, Brown GG. Length and restriction site heteroplasmy in the mitochondrial DNA of american shad (Alosa Sapidissima). Genetics. 1988;118:509–18. Available from: http://www.genetics.org/content/118/3/509
Buroker NE, Brown JR, Gilbert TA, O’Hara PJ, Beckenbach AT, Thomas WK, et al. Length heteroplasmy of sturgeon mitochondrial DNA: an illegitimate elongation model. Genetics. 1990;124:157–63. Available from: http://www.genetics.org/content/124/1/157
Moritz C, Brown WM. Tandem duplications in animal mitochondrial DNAs: variation in incidence and gene content among lizards. Proc Natl Acad Sci U S A. 1987;84:7183–7. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=299254&tool=pmcentrez&rendertype=abstract
Mignotte F, Gueride M, Champagne A-M, Mounolou J-C. Direct repeats in the non-coding region of rabbit mitochondrial DNA. Involvement in the generation of intra- and inter-individual heterogeneity. Eur J Bochemistry. 1990;194:561–71. Available from: http://0-onlinelibrary.wiley.com.wagtail.ufs.ac.za/doi/10.1111/j.1432-1033.1990.tb15653.x/epdf
Rand DM, Harrison RG. Ecological genetics of a mosaic hybrid zone : mitochondrial, nuclear, and reproductive differentiation of crickets by soil type. Evolution (N Y). 1989;43:432–49.
Streisinger G, Okada Y, Emrich J, Newton J, Tsugita A, Terzaghi E, et al. Frameshift mutations and the genetic code. Cold Spring Harb Symp Quant Biol. 1966;31:77–84. Available from: http://symposium.cshlp.org/cgi/doi/10.1101/SQB.1966.031.01.014
Acknowledgements
We thank the African Pangolin Working Group (APWG) for sharing their specimen database. We also thank the Zoological Museum, University of Copenhagen for providing us with museum samples of their pangolin specimens (Specimens CN191 and CN339).
Funding
This study was financially supported by the National Zoological Gardens of South Africa (NZG) and partially funded by the National Research Foundation (NRF) grant no: (86125).
Availability of data and materials
The reference mitogenomes generated and/or analysed during the current study are available in the GENBANK repository, [https://www.ncbi.nlm.nih.gov/].
All data generated or analysed during this study are included in this published article [and its additional information file].
Author information
Authors and Affiliations
Contributions
Conceptualised idea for research: AK; RJ. Responsible for data collection/analysis/design: ZDT; MDP; DLD; JPG. Lead author writing up of article: ZDT. Editorial input: ZDT; MDP; DLD; JPG; RJ; AK. Postgraduate supervisor of the lead author: DLD; JPG; AK; RJ. Co-developed and executed research: ZDT. Project leader/budget owner: DLD; AK. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Ethical approval was obtained from the Animal Research Ethics Committee, University of the Free State, South Africa (UFS-AED2015/0070) and the NZG Research, Ethics and Scientific Committee (NZG/RES/P/001/F/02). Samples were obtained under a Section 20 permit (12/11/1/1/18) from the Department of Agriculture, Forestry and Fisheries, South Africa. Sample collection was also approved under a CPC5 permit (02437) from the Department of Agriculture and Rural Development, South Africa and a Biodiversity permit (FAUNA 714/2012) from the Department of Environment and Nature Conservation, South Africa.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Figure S1.
Bayesian mitogenome phylogenetic tree of all available pangolin mitogenomes. Posterior probabilities are indicated on the respective branches. A. jubatus was selected as an outgroup as pangolins are more closely related to the order Carnivora. Asterisks indicate the misidentified mitogenomes. Figure S2. Principal Component Analysis (PCA) of Relative Synonymous Codon Usage values (RSCU) for six pangolin species. Three distinct genera is present (Manis, Phataginus and Smutsia) in the (a) CoxII; (b) NadI, (c) NadIII genes. The two sub-families (Smutsiinae and Maninae) are distinguished in the (d) AtpVIII; (e) NadIV; (f) NadVI genes. Table S1. List of nucleotide percentages and its 3rd codon position percentage (%) (PDF 426 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
du Toit, Z., du Plessis, M., Dalton, D.L. et al. Mitochondrial genomes of African pangolins and insights into evolutionary patterns and phylogeny of the family Manidae. BMC Genomics 18, 746 (2017). https://doi.org/10.1186/s12864-017-4140-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4140-5