
Abstract
Background
Severe equine asthma is a naturally occurring lung inflammatory disease of mature animals characterized by neutrophilic inflammation, bronchoconstriction, mucus hypersecretion and airway remodeling. Exacerbations are triggered by inhalation of dust and microbial components. Affected animals eventually are unable of aerobic performance. In this study transcriptomic differences between asthmatic and non-asthmatic animals in the response of the bronchial epithelium to an inhaled challenge were determined.
Results
Paired endobronchial biopsies were obtained pre- and post-challenge from asthmatic and non-asthmatic animals. The transcriptome, determined by RNA-seq and analyzed with edgeR, contained 111 genes differentially expressed (DE) after challenge between horses with and without asthma, and 81 of these were upregulated. Genes involved in neutrophil migration and activation were in central location in interaction networks, and related gene ontology terms were significantly overrepresented. Relative abundance of specific gene products as determined by immunohistochemistry was correlated with differential gene expression. Gene sets involved in neutrophil chemotaxis, immune and inflammatory response, secretion, blood coagulation and apoptosis were overrepresented among up-regulated genes, while the rhythmic process gene set was overrepresented among down-regulated genes. MMP1, IL8, TLR4 and MMP9 appeared to be the most important proteins in connecting the STRING protein network of DE genes.
Conclusions
Several differentially expressed genes and networks in horses with asthma also contribute to human asthma, highlighting similarities between severe human adult and equine asthma. Neutrophil activation by the bronchial epithelium is suggested as the trigger of the inflammatory cascade in equine asthma, followed by epithelial injury and impaired repair and differentiation. Circadian rhythm dysregulation and the sonic Hedgehog pathway were identified as potential novel contributory factors in equine asthma.
Similar content being viewed by others

Background
Severe equine asthma, formerly termed recurrent airway obstruction (RAO) or heaves, is a naturally occurring chronic lung inflammatory disease of horses exposed to airborne molds and particulate material [1]. The condition develops with repeated inhalation of molds and/or dusty air in barns or on pasture in hot and humid climates, as well as in environments with high concentration of fungal spores or grass pollen grains [2]. Once sensitized, affected horses cough and have nasal discharge associated with progressive airway obstruction from a neutrophilic exudate, mucus hyperproduction, airway hyperreactivity and bronchospasm. Recurrent episodes of inflammation lead to smooth muscle hyperplasia, fibrosis and irreversible airway remodeling [3,4,5,6]. Severe equine asthma is responsive to environmental improvement and anti-inflammatory and bronchodilatory therapy, but is considered to be irreversible once airway remodeling has started [3]. Nevertheless, affected animals may have periods of clinical remission followed by periods of exacerbation over months to years.
Development of severe equine asthma likely involves genetic predisposition in addition to environmental triggers [7]. Findings have suggested that inheritance patterns are complex, implying genetic heterogeneity [8]. A significant association between susceptibility and paternal haplotype was proposed [9, 10], and specific regions on equine chromosomes 13 and 15 containing genes such as the interleukin (IL)4 and IL21 receptors were associated with increased risk of developing severe asthma in some equine kindreds [10, 11]. Copy number loss of a region on chromosome 5 including the gene NME7 involved in ciliary function was more frequent in asthmatic than non-asthmatic horses [12]. However, strong evidence for a role of specific gene variations remains sparse.
The airway epithelium is the first barrier to inhaled substances, and includes multiple cell types such as ciliated and non-ciliated (club or Clara) cells, mucus producing goblet cells and precursor cells. It is thought that in severe equine asthma airway epithelial cells bind inhaled microbial components via pattern recognition receptors (PRR), which initiates an innate immune response with synthesis of inflammatory cytokines and chemokines [13]. In turn, inflammation of the epithelium results in generation of reactive oxygen metabolites, exosomes and proteases that injure epithelial cells and induce proliferation of airway smooth muscle cells, goblet cell hyperplasia, epithelial cell metaplasia and cell death [5, 14, 15]. In order for the epithelium to resume specialized barrier functions, cells need to regenerate with precise migration, proliferation and differentiation. Club cells, in particular, are markedly reduced in equine asthmatic airways resulting less anti-inflammatory secretoglobin 1A1 (SCGB1A1) in airway secretions [16, 17]. It is postulated here that repeated epithelial cell inflammation and injury results in progressively impaired regeneration of a fully functional epithelial barrier.
There are many proposed classification schemes for human asthma. According to most schemes, severe equine asthma is most similar to severe human adult or late onset asthma, which is distinct from childhood, allergic, exercise-induced and some other forms of human asthma [1, 18,19,20]. Phenotypes of human adult asthma are categorized according to age at onset, clinical characteristics, type of airway inflammation and response to therapy [19]. Severe human adult asthma is associated with airflow obstruction and most often neutrophilic inflammation, although eosinophilic and pauci-granulocytic inflammation is also observed [21]. Neither severe human adult asthma nor severe equine asthma is typically dominated by a Th2 immune response [3, 20]. It is difficult to investigate the pathogenesis of asthma in humans, and many inferences are based on nasal or sputum rather than bronchial or bronchiolar samples since the latter are difficult to obtain. Mice systemically sensitized to foreign antigen and then challenged by inhalation are widely used as models of human asthma, but recapitulate neither remission/exacerbation nor neutrophilic inflammation [22].
We hypothesized that the bronchial epithelial response to an inhaled challenge is different in asthmatic and non-asthmatic individuals. To address this hypothesis we designed a paired pre- and post-challenge study that accounts for individual variability in genetically heterogeneous animals, and obtained bronchial biopsy samples from affected and control animals that were processed for RNA sequencing and results analyzed.
Methods
Animals and procedures
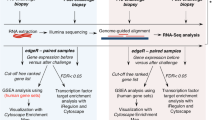
Details of study design and analysis are presented in Fig. 1. Six horses with and seven horses without asthma had similar mean ages of 15 and 12 years (p = 0.352, unpaired t test), respectively, and each group included a variety of breeds. All were maintained for >6 months outdoors prior to study. Horses with historical asthma had been affected for 2 to 6 years, and were free of clinical respiratory disease during at least 6 months prior to study. All animals were placed in a dust-free indoor environment for 24 h, and thereafter physical examination, pulmonary function test (PFT) and bronchoalveolar lavage (BAL) were performed. During physical examination, respiratory rate, nasal discharge, presence and severity of expiratory lift, nasal flaring, tracheal sounds, bronchial tones, crackles, wheezes, cough and chest resonance were assessed according to a preset scale yielding a clinical score between 0 and 26. Pulmonary function data were derived from integration and analysis of airflow data and corresponding transpulmonary pressure. For PFT, non-sedated horses were restrained in stocks and fitted with a mask attached to a heated pneumotachograph. Airflow data were captured and fed through a transducer to integrate the flow signal and derive volume measurements. An esophageal balloon catheter was placed midthorax and attached to a transducer at the proximal end to estimate pleural pressure. Volume and pressure data were analyzed via respiratory loop analysis to derive values for pleural pressure (PpI), dynamic compliance (Cdyn) and lung resistance (RL). During bronchoscopy, the appearance of the upper airways, trachea and main bronchi were visualized, and scored for presence and degree of erythema, edema, secretions, hemorrhagic exudate, and cough reflex. An endoscopic score between 0 and 15 was derived from these parameteres. Then, the bronchoscope was gently lodged in a 3rd to 5th generation bronchus, and two sequential aliquots of 200 mL of warmed saline were infused and re-aspirated. An aliquot of BAL fluid was analyzed by total nucleated cell counting and 200-cell differential counting of stained cytocentrifuge preparations. Between two and eight endoscopic biopsies were obtained for RNA-seq and histopathology. Horses were then exposed to dusty hay until respiratory impairment was apparent in asthmatic horses (range 1 to 3 days, average 2.2 days). Non-asthmatic horses were exposed to dusty hay for 3 days. At this time clinical examination, respiratory function testing and BAL were repeated. BAL and endoscopic biopsies were obtained from a contralateral lung lobe. At exacerbation, mean clinical and bronchoscopic scores in asthmatic horses had increased from a mean of 2.7 to 13.3, and from 2.2 to 9.7, respectively. Non-asthmatic horses had mean clinical and bronchoscopic scores of 1.6 and 2.6 prior to challenge, and 0.4 and 1.9 post-challenge, respectively (Fig. 3 and Additional file 1: Table S1). The average change in PpI was 7.92 cm H2O in asthmatic horses, and −0.82 cm H2O in non-asthmatic horses. All procedures were approved by the Institutional Animal Care Committee of the University of Guelph (protocol R10–031) and conducted in compliance with Canadian Council on Animal Care guidelines. Changes in pulmonary function and BAL fluid composition between the two groups following an asthmatic challenge were analyzed by taking the differences between “after” and “before” values for each horse and testing with Welch’s t test for significant association with presence of asthma.

Outline of study design and analysis
Changes in pulmonary function and BAL fluid (BALF) composition between the two groups following an asthmatic challenge were analyzed by taking the differences between “after” and “before” values for each horse and testing with Welch’s t test for significant association with presence of asthma. Graphs and p-values were generated using Prism 6.0a (GraphPad, La Jolla, CA) and unpaired t-tests with correction for multiple comparisons by the Holm-Sidak method with alpha = 5.000%. Significance threshold was set at p < 0.05. Complete information on horses and clinical tests is in Additional file 1: Table S1.
RNA extraction, library preparation and sequencing
Total RNA was extracted from endobronchial biopsies (Qiagen, Toronto, ON). Quality and concentration of RNA were determined with the Bioanalyzer RNA Nanochip (Agilent, ON) and gel electrophoresis. Only samples with RNA integrity number > 6.9 and little to no degradation apparent on electrophoretograms were accepted. RNA-seq unstranded library preparation and sequencing were performed at The Centre for Applied Genomics (TCAG; Toronto, ON) using the Illumina TruSeq RNA sample preparation and sequencing protocol following the manufacturer’s guidelines (Illumina, San Diego, CA). Briefly, for each sample, approximately 1 μg of non-degraded, high quality total RNA was enriched for poly-A RNA, fragmented into 200 to 300 bases, and converted to double stranded cDNA libraries. Illumina adapters were ligated to the ds-cDNA and PCR-amplified for 14 cycles. Barcoded primers were then added to each sample to allow sequencing in the same lane and detection of individual samples in the sequence data. Final RNA libraries were quantified (KAPA Library Quantification kit, Kapa Biosystems, Wilmington, MA) prior to pooling and sequencing. Illumina flow cell was prepared and sequenced on an Illumina HiSeq 2500 instrument in 5 lanes following the manufacturer’s instructions to generate paired-end reads of 100-bases.
Genome-guided RNA alignment
Raw read quality was assessed using FastQC software version 0.10.1 [23] and aligned to the horse reference genome [24] (Ensembl v70) with STAR version 2.4 [25]. The STAR_pass2 alignment protocol was followed including these adaptations: horse Ensembl version 70 GTF annotation file for first- and second-pass, and the junction SJ.tab file generated by STAR for the second-pass after non-canonical junctions were removed. Default settings were used except for: --runThreadN 8 --outFilterScoreMinOverLread 0.5 --outFilterMatchNminOverLread 0.5. Read counts were generated from STAR alignment files using HTSeq version 0.6.1p1 [26] with settings -s no -f bam -r name.
Differential gene expression
Differential expression (DE) analysis was performed in R, version 3.2.1 [27], with the edgeR package version 3.10.2 [28,29,30]. A paired DE analysis was performed to assess changes between groups (asthmatics versus non-asthmatics) and within groups (before versus after challenge). EdgeR analysis was based on section 3.5 of the edgeR user’s guide (last revised April 10, 2017). Briefly, the minimum count number was set at 1 read per million in at least 3 samples. Normalization factors and effective library size were applied, duplicates were removed and dispersion was estimated using the “estimateGLMCommonDisp”, “estimateGLMTrendedDisp” and “estimateGLMTagwiseDisp” functions. The model matrix was designed as: ~group + group:horse + group:challenge, where “group” refers to non-asthmatic and asthmatic groups, “horse” refers to each individual horse, and “challenge” refers to samples before and after the asthmatic challenge. Fit of the generalized linear model and tests for differences in expression were performed with the “glmFit” and “glmLRT” functions, respectively and the following contrast was used to compare asthmatic and non-asthmatic horses: glmLRT(fit, contrast = c(0,0,0,0,0,0,0,0,0,0,0,0,0,-1,1)). GC content bias was assessed using EDAseq [31], but need for normalization was not indicated. Statistical significance was set at a false discovery rate (FDR) <0.05.
Immunohistochemistry
The protein product of four genes with significant up- or down-regulation was assessed by immunohistochemistry (IHC). Confirmation of protein expression for a group of genes was deemed sufficient as a proxy to confirm the correctness of sequencing, alignment and statistical workflow. The genes were selected based on significant differential expression between asthmatic and non-asthmatic horses, availability of cross-reactive antibodies and potential roles in asthma pathogenesis. Antibodies were initially tested in Western blots with equine tissue samples to verify that a single protein product of appropriate size was detected (data not shown). Tumor necrosis factor receptor superfamily member 12A (TNFRSF12A or TWEAKR, tumor necrosis factor-like weak inducer of apoptosis receptor), patched-1 (PTCH1), cell division cycle 25 homolog A (CDC25A) and interleukin 8 (IL8) proteins were assessed in biopsies fixed in formalin and routinely sectioned and processed. Antibody reactivity was first assessed by western blot (WB) analysis against horse serum or lung protein extracts (Additional file 2: Figure S1). Proteins were separated in 12% (w/v) SDS-polyacrylamide gels (TGX Stain-Free FastCast premixed acrylamide solutions; Bio-Rad, Mississauga, ON) under reducing conditions. Proteins were then electro-transferred to PVDF membranes using the Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked in 5% BSA solution before immunoblotting with polyclonal rabbit anti-human TWEAKR (Biorbyt, Berkeley, CA), PTCH1 (C-terminal region; Aviva Systems Biology, San Diego, CA) and CDC25A (Abcam, Toronto, ON), and polyclonal rabbit anti-horse IL8 (MyBioSource, Inc., San Diego, CA). Membranes were then incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (DAKO, Mississauga, ON) and exposed with Clarity Western ECL Substrates (Bio-Rad). Images were captured with a ChemiDoc imaging system (Bio-Rad). If bands of expected size were present, antibodies were applied in immunohistochemistry (IHC) to 3–5 μm thick sections placed on charged glass slides, de-paraffinized in xylene, rehydrated in alcohol, incubated with dual endogenous enzyme blocker and serum-free protein blocker (both DAKO). Slides were then sequentially incubated with the above primary antibodies, Envision Dual Link System-HRP (DAKO) and Nova Red chromogen (Vector Laboratories, Burlingame, CA), and counterstained with hematoxylin. Negative control sections for each IHC analysis were prepared in the same manner except the primary antibody was omitted.
Protein network and gene ontology analysis
Gene products were searched for known and predicted protein interactions in Cytoscape version 3.4.0 [32] using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database [33] and string-db plugin [34] within Cytoscape. Horse Ensembl ID were converted to human ID using Biomart [35, 36] and to gene symbols directly in Cytoscape through the STRING database (Additional file 1: Table S2). When multiple horse Ensembl IDs had identical human symbols, redundant symbols were removed. The confidence score cut-off applied for interactions was 0.4 (medium confidence). Single nodes, doublets and triplets detached from the main network cluster were removed, and network analysis was performed. Node color and size was determined based on betweenness centrality (BC) and degree, respectively. Confidence of interactions was displayed with different intensity of edge color.
Gene ontology (GO) overrepresentation analysis of biological function was performed with Protein ANalysis THrough Evolutionary Relationships (PANTHER) software version 10.0 [37] with significance threshold set at p < 0.05 (including Bonferroni adjustment). The analysis was performed using annotations for Homo sapiens by converting the equine gene symbols into human gene symbols prior to analysis. Species to be analyzed was then identified as human, and information on protein function was assigned to candidates according to prediction in NCBI or UniProt databases.
Results
Induction of asthma
Following exposure to inhaled challenge material, severe bronchoconstriction and profound airway secretions were apparent in asthmatic but not non-asthmatic horses (Fig. 2a), and cell concentration and the proportion of neutrophils were increased in bronchoalveolar lavage (BAL) fluid (Fig. 2b). Bronchial biopsies showed submucosal edema and an influx of leukocytes (Fig. 2c). Changes in BAL fluid cell and tissue composition in non-asthmatics were mild or absent. As a group, asthmatic horses had significantly higher bronchoscopic scores, pleural pressure, BAL nucleated cell concentration and percent neutrophils, and significantly lower dynamic compliance after asthmatic challenge than non-asthmatics (Fig. 3). Complete data are in Additional file 1: Table S1.

Endoscopic appearance of bronchi (a), BAL lavage cell yield (b) and histological appearance of bronchial biopsies (c). Asthmatic horses had bronchoconstriction and increased secretions in airways after asthmatic challenge, while changes in horses without asthma consisted of mild bronchoconstriction (a). In asthmatic horses, cell concentration and proportion of neutrophils was increased in BAL fluid after challenge (b), and in bronchial biopsies epithelial basophilia and influx of submucosal leukocytes was evident (c)

The individual change from pre- to post-challenge bronchoscopic score, pleural pressure, alveolar leukocyte concentration, proportion of neutrophils, and dynamic compliance differed significantly between asthmatic and control horses, while the change in lung resistance did not have a uniform pattern
Differential expression analysis
The results of mapped RNA-seq reads for individual horses are summarized in Table 1. Analysis identified 111 genes differentially expressed (DE) between horses with and without asthma as a result of challenge (Fig. 4a). Significantly differentially expressed genes pertain to the epithelium and include keratin-related genes (identified as potential Keratin [KRT] 6 based on human homologues ENSG00000185479, and KRT17), genes linked to matrix metalloproteinases (MMPs), inflammation (Toll-like receptor 4 (TLR4), and others), neutrophil chemotaxis (IL8, C-X-C chemokine receptor type 2 (CXCR2)), coagulation and hemostasis (such as Pleckstrin (PLEK)), cell proliferation (CDC25A), apoptosis (such as BCL2 interacting killer [BIK]) and others. Six of 30 down-regulated genes function in regulation of the circadian rhythm. Identity and details of DE genes are in Additional file 1: Table S2.

EdgeR smear plot showing the log2 fold-change (FC, y-axis) versus the average log2 count per million (CPM, x-axis) of the change in gene expression due to challenge in all horses (a). Horizontal blue lines delineate 1-fold change and each point represents one gene. Differentially expressed genes are indicated in red (FDR <0.05). Genes with positive log2FC were up-regulated in asthmatic compared to non-asthmatic horses, while genes with negative FC were down-regulated. The majority of genes expressed differentially between the two groups are up-regulated. Leading logFC plot (b) shows individual horses with asthma before (black) and after (red) challenge, and horses without asthma before (blue) and after (green) challenge. No clustering was observed for non-asthmatic horses, but post- challenge samples from asthmatic horses are located distant from other samples
The relationship between paired samples from individual animals is shown in a multidimensional scaling (MDS) plot with the distance between pairs of samples corresponding to the average root mean square of the largest log2FC (leading logFC, Fig. 4b). Post-challenge samples from asthmatic animals were distinctly distant from those of pre-challenge asthmatic and non-asthmatic animals. This implies that within the asthmatic group differential expression of genes was greater than biological variance, and that most of the DE genes originated from asthmatic animals. The biological coefficient of variance (BCV) was calculated to determine how much the variance in counts exceeded that which would arise from Poisson counts alone [29]. The BCV for RNA-seq analysis of genetically identical organisms is typically around 0.1 [29] while in this study the BCV was 0.23 (data not shown). This high value reflects the biological variance as expected from outbred individuals, and also emphasizes the benefit of a paired sample design to correct for inter-individual variation.
A heat map of counts per million (CPM) of DE genes for each sample (Fig. 5) shows that there is a wide distribution of change in expression (logFC), an inverse relationship of CPM with log FC, and consistency of change across individuals. The magnitude of the log fold change of significantly up-regulated genes in asthmatic horses after challenge ranged from 0.6 to 5.6 (Fig. 6), and that of significantly down-regulated genes from −0.62 to −2.2 (Fig. 7).

A heat map of differentially expressed genes significantly up- (a) and down-regulated (b) between asthmatic and non-asthmatic horses after challenge shows consistent change across individuals. Genes with positive log2FC were up-regulated in asthmatic compared to non-asthmatic horses, while genes with negative FC were down-regulated. Level of expression is expressed as cpm and ordered from highest (top) to lowest log2 fold-change (logFC, bottom). Significance threshold was set at FDR <0.05

Stacked bar chart of positive log2 fold-change (logFC) for genes differentially expressed between asthmatic and non-asthmatic horses in response to challenge. a ENSECAG00000014899; b ENSECAG00000017229

Stacked bar chart of negative log2 fold-change (logFC) for genes differentially expressed between asthmatic and non-asthmatic horses in response to challenge
Protein expression
Specific RNA-seq results were further investigated in biopsy tissues by IHC. Expression of TNFRSF12A, CDC25A and IL8 mRNA was markedly increased in asthmatic and decreased in non-asthmatic horses after challenge, while PTCH1 mRNA was slightly decreased (Fig. 8a). Immunohistochemical results representative of each group showed more intense reactivity for TNFRSF12A, CDC25A and IL8 protein in tissues from asthmatic than non-asthmatic horses after challenge (Fig. 8b). TNFRSF12A staining was moderately intense throughout the epithelium of asthmatics after challenge and only present in individual epithelial and sub-epithelial cells from non-asthmatics. CDC25A reactivity was intense in epithelium of asthmatic animals, and less prominent in tissue from non-asthmatic animals, in particular after challenge. IHC results for IL8 also showed marked increase after challenge in asthmatic but not non-asthmatic animals. PTCH1 staining was less abundant in asthmatic than non-asthmatic animals after challenge.

Gene (a) and protein expression (b) of TNFRSF12A, PTCH1, CDC25A and IL8 in bronchial biopsies from horses with and without asthma. TNFRSF12A, CDC25A and IL8 mRNA, expressed as counts per million (CPM), was up-regulated in asthmatic and down-regulated in non-asthmatics following challenge, while PTCH1 was slightly down-regulated in asthmatics and up-regulated in non-asthmatics. (b) IHC results approximated gene expression with a relative increase in TNFRSF12A, CDC25A and IL8 immunoreactivity and decrease in PTCH1 immunoreactivity in asthmatic animals
Gene ontology analysis
PANTHER analysis of GO overrepresentation for biological processes (GOBP) using the Homo sapiens database identified significantly overrepresented gene sets among up- and down-regulated genes, listed in Table 2 with associated gene names. The most specifically involved gene sets concerned neutrophil chemotaxis (GO:0030593), immune response (GO:0006955), inflammatory response (GO:0006954), secretion (GO:0046903), positive regulation of blood coagulation (GO:0030194), positive regulation of apoptotic signaling pathway (GO:2,001,235), positive regulation of response to external stimulus (GO:0032103) and regulation of immune system process (GO:0002682) for up-regulated genes, and rhythmic process (GO:0048511) for down-regulated genes.
Up-regulated genes shared between the most specific gene-sets are shown in Table 3 along with evidence for their association with asthma and their known functions. S100 Calcium Binding Protein A9 (S100A9) was the only gene that contributed to all gene sets, and has been associated with asthma in mice [38]. All genes associated with 4 or more gene sets have also been associated with asthma in humans or mice and include S100A9, thrombospondin 1 (THBS1), TLR4, IL8, complement component 5a receptor 1 (C5AR1), MMP9, NLR Family, Pyrin Domain Containing 12 (NLRP12) and triggering receptor expressed on myeloid cells 1 (TREM1) [20, 38,39,40,41,42,43,44,45,46,47,48]. Other genes associated with 3 or fewer gene sets have also been associated with asthma such as Plasminogen Activator, Urokinase Receptor (PLAUR) and Serpin Family E Member 2 (SERPINE2), and several additional genes were first identified here.
Among down-regulated genes, only the rhythmic process (GO:0048511) gene set was overrepresented. This gene set includes genes associated with asthma in humans and mice such as adrenoceptor beta 2 (ADRB2), nuclear receptor subfamily 1 group D member 2 (NR1D2) and period circadian clock 3 (PER3), as well as genes that have not previously been linked to asthma such as D site of albumin promoter (albumin D-box) binding protein (DBP), circadian-associated repressor of transcription (CIART or CHRONO, ChIP-derived Repressor of Network Oscillator) and thyrotrophic embryonic factor (TEF).
Protein network analysis
Protein products of genes DE between groups in response to challenge identified multiple interactions with medium to high confidence (scores ranging from 0.4 to 1). The main protein interaction cluster derived from the 111 DE genes contained 51 nodes, each representing one protein and connected by 113 edges (Fig. 9). MMP1, IL8 and TLR4, followed by MMP9, had highest scores for betweenness centrality (BC), indicating they are most important for connections with other proteins. IL8, TLR4 and MMP9 had the highest number of direct connections (degree). S100A9, associated with all overrepresented gene sets (Table 3), is connected to the network through its predicted interaction with TLR4. In addition, MMP1 and THBS1 each connect two genes (Table 3) with potential role in severe equine asthma though not yet associated with asthma in humans or mice.

The main network cluster of genes DE between asthmatic and non-asthmatic horses after challenge. The network cluster is derived from 111 genes and contains 51 nodes each representing a protein, and 113 edges each representing an interaction between two proteins. Node color from white to green represents lowest to highest betweenness centrality (BC). The size of each node corresponds to the degree (number of connections). The color of edges represents the confidence of the interaction ranging from 0.4 (medium confidence, light gray) to 1 (highest confidence, black). Nodes with red borders have negative fold-change
Discussion
The goal of this study was to identify bronchial epithelial genes and pathways associated with severe asthma in horses. Although predisposition for development of severe asthma in horses is thought to be hereditary, inheritance is incompletely defined and thought to be complex [7]. Analysis of the bronchial epithelium aimed to capture the in situ genetic changes that characterize the pathogenesis of severe equine asthma, an approach difficult to apply in other species. Although components of the lung such as bronchioles, alveoli, interstitium and leukocytes are also affected in asthma, they were not specifically evaluated in this study. Biopsies consisted predominantly of epithelium, which in itself is a variable tissue composed of ciliated columnar cells, goblet cells, and in smaller bronchi occasional club cells. Subepithelial components of biopsies included cells such as leukocytes and fibroblasts, and extracellular matrix such as collagen and edema. Hence, cells other than epithelium contributed some RNA to analysis, which is a limit in this study. Nevertheless, assessment of in situ samples from a naturally occurring inducible model of asthma is previously unreported, and yielded unprecedented insight. Pre- and post-challenge bronchial biopsies from asthmatic and non-asthmatic horses were obtained, the transcriptome was derived from high throughput sequencing, and results were analyzed with a paired design to account for individual variation. Both groups of animals were of similar age, and as expected, the response to the inhaled challenge consisted of bronchoconstriction, marked airway neutrophilic inflammation, mucus accumulation and impaired airflow in asthmatic but not non-asthmatic animals.
RNA-seq data were analyzed using edgeR software. EdgeR and DESeq [49] are among the most widely used tools for RNA-seq analysis using raw counts. Both software tools use comparable methods with the exception of count normalization and dispersion estimation methods [50]. DESeq tends to be more conservative and edgeR more sensitive to outliers [51], but they yield overall highly similar results [51, 52]. Regardless of the method used, considering the genetic variability among horses, dispersion estimation would be more precise if data from a larger sample of individuals were analyzed. Therefore, within a limited sample size, the potential for false-positive results warrants caution for interpretation of individual findings.
Overall, the analysis identified 111 DE genes, which is a number similar to that identified in comparable studies in humans [53]. IHC analysis of representative bronchial biopsies confirmed that epithelial cell gene expression was representative of observed differential gene expression results. Transcriptome analysis of paired lung adenocarcinoma and non-neoplastic samples from non-smoking and smoking patients yielded 175 DE genes [53], while comparison of RNA-seq results of single bronchial biopsies from human asthmatics and healthy controls yielded 46 DE genes [53]. The latter study compared the bronchial transcriptome of asthmatics and non-asthmatics, rather than the difference in the response to an asthmatic challenge, as we did here. The workflow included different sequencing and statistical analyses, and the design did not account for inter-individual variability [54]. Nonetheless, similar to our results, up- and down-regulation of solute carrier (SLC) genes and up-regulation of an integrin-coding gene was detected. However, B-cell lymphoma 2 (BCL2) expression was lower, and SCGB1A1 was higher in asthmatics compared to control, which contrasts with results in severe asthma in horses [17, 55] and may be due to different experimental design and different phases of disease being assessed.
Gene ontology and network analysis were subsequently used to characterize the DE genes. Since limited annotation of the horse genome constrains species-specific gene network and gene set analyses, human databases were utilized to expand interpretation of the findings. This approach yielded results consistent with prior association in asthma, but specific function of such gene products in horses remains to be ascertained. Finally, expression of the protein product of 4 DE genes was investigated immunohistochemically. Semi-quantitative assessment affirmed a link between gene and protein expression but factors such as RNA transcript stability and cell-to-cell variability in gene expression are incompletely accounted for with this approach. Hence, linked rather than individual genes should convey greater confidence for a role in asthma pathogenesis.
Several genes within overrepresented gene sets have been linked to asthma in humans. S100A9 was the only gene shared across all gene sets, and is a calcium-binding protein highly conserved across species. S100A9 and related S100 proteins are highly expressed by neutrophils, and activate innate immune responses via interaction with TLR4 [56]. S100 proteins have also recently been identified to interact with airway epithelial cells to induce MUC5AC, the most abundant airway mucin [56]. Although MUC5AC was not differentially expressed in our study, it is known to be promoted by exposure to cytokines in both horses and humans in a concentration-and time-dependent manner [57, 58]. Our sampling time points may not have captured peak expression in all horses necessary reach significance in our stringent statistical analysis. Overexpression of MUC5A in horses with severe asthma [59] suggests a possible link of S100A9 with mucus hyperproduction.
Network analysis of genes DE between asthmatic and non-asthmatic horses intimated MMP1, MMP9, TLR4 and IL8 as responsible for many interactions, and therefore to link and influence several asthmatic pathways and processes. MMP9 was present in multiple significantly overrepresented GO gene sets. In addition, it had high BC and degree (number of direct connections) in the STRING network, meaning it accounted for many direct and indirect interactions within the network. MMP9 is increased in human asthma [60], has anti-apoptotic effects in kidney injury and neutrophils [61, 62] and may be a link between inflammation and tissue remodeling [60, 63]. MMP9 also links serglycin (SRGN) to the main interaction cluster. SRGN is a proteoglycan that forms complexes with proMMP9 [64, 65] and is expressed in a variety of hematopoietic and non-hematopoietic cells [66]. Presence in immature granules has suggested a role in neutrophil differentiation [67], which could also influence neutrophil function in the context of granule release and cell death associated formation of neutrophil extracellular traps (NETs), also called NETosis.
Neutrophil infiltration was present in all asthmatic horses, consistent with the overrepresented neutrophil chemotaxis (GO:0030593) gene set. All genes within this gene set were previously associated with asthma pathogenesis except for CSF3R. CSF3 regulates production, differentiation and function of granulocytes, and overexpression is consistent with neutrophilic inflammation in equine severe asthma [68]. IL8 had among the highest BC and degree indicating a central role for linking components of the network. IL8 is a potent neutrophil attractant in the lung [69] and signals through CXCR1 and CXCR2 [70]. Increased IL8 expression likely initiates and perpetuates neutrophil influx into the airways, but IHC also identified epithelial cells as a source of IL8. Secretion of IL8 by human epithelial cells can be promoted by exposure to TWEAK and activation of its receptor TNFRSF12A [71]. TWEAK is up-regulated in multiple tissues with inflammation, and associated with tissue changes such as remodeling [72]. Hence it may be plausible that epithelial cells up-regulate TNFRSF12A early in response to challenge, which in turn enhances IL8 production and maintains neutrophilic inflammation, leading to eventual proteolytic and oxidative injury.
It has been reported that asthmatic horses have dysregulated apoptosis of BAL but not peripheral blood leukocytes [73, 74]. Conversely, higher expression of Immediate Early Response 3 gene (IER3) identified in another study suggested dysregulated apoptosis in peripheral blood mononuclear cells of asthmatic horses [75]. Hence, the importance of leukocyte apoptosis in asthma of horses is unresolved. Significant overrepresentation of the apoptotic signaling pathway (GO:2,001,235) gene set was identified in tissue biopsies in this study, which included some extravasated leukocytes. This gene set included S100A9, oncostatin M (OSM), THBS1, TNFRSF12A, stratifin (SFN), plasminogen activator urokinase receptor (PLAUR) and MMP9. Other genes, such as BIK, a pro-apoptotic protein [76], had lower expression in asthmatic compared to non-asthmatic horses. BIK interacts with BCL2 and may protect airway mucous cells from apoptosis during remission from asthmatic exacerbation [55, 77]. Although BCL2 was not DE, this may be a factor of the timing of biopsies and the lower proportion of mucous versus ciliated epithelial cells. Formation of neutrophil extracellular traps (NETs), another form of induced cell death, is prominent in BAL of horses with severe asthma [78]. The mechanism of NET formation is incompletely defined, but peptidyl arginine deiminase type IV (PADI4), differentially expressed in asthmatic and non-asthmatic horses, contributes through citrullination of histones [79]. PADI4 expressed during NET formation may also promote coagulation through the release of serine proteases [80]. The positive regulation of blood coagulation (GO:0030194) gene set was overrepresented and included S100A9, PLEK, THBS1 and TLR4. Chronic up-regulation of coagulation [81] and systemic inflammation were reported in horses with severe asthma [82], and activation of the coagulation cascade [81] together with impaired epithelial repair [83] are features of human asthma. Hence, several lines of evidence suggest concurrence of hemostatic, coagulative and tissue repair processes with neutrophil activation in severe asthma. Furthermore, apoptosis and NETosis appear to be component of asthma as suggested by differential expression and linkage of genes in these pathways.
Among the DE genes are several of potential interest that are not part of overrepresented gene sets or interaction networks. Differences in cell cycle-related gene expression in peripheral blood mononuclear cells (PBMCs) have been reported in asthmatic horses [75]. CDC25A, a cell cycle-related gene, was not part of any network, but was highly DE and likely influences cell cycle and differentiation in bronchial epithelium during inflammation, as it does in other contexts such as neoplasia [84, 85]. Genes such as ENSECAG00000014899 and ENSECAG00000017229 (potential orthologs of human KRT6 genes), KRT17 and ENSECAG00000007450 (potential ortholog of human SPRR1A/B) and transglutaminase 1 (TGM1) were not identified in network interactions, but are likely to function in squamous metaplasia [86]. Recent reports implicate hedgehog (HH) pathway-associated molecules in lung disease of humans [87]. Single nucleotide polymorphisms (SNPs) in Patched-1 (PTCH1), a DE gene, and hedgehog-interacting protein (HHIP), involved in the hedgehog pathway, have been associated with lung function in humans [88, 89]. In conjunction, PTCH1, HHIP and family with sequence similarity 13, member A (FAM13A) predicted lung function abnormalities in an asthmatic cohort [88].
Five differentially expressed genes linked to regulation of the circadian clock were consistently down-regulated in asthmatic animals: CIART (CHRONO), PER3, DBP, TEF, ADRB2 and NR1D2. CIART is part of a transcriptional repressor of the mammalian clock, and contributes to a suppressive glucocorticoid response that is dependent on physiological stress [90]. PER3 is expressed in a circadian pattern in the brain suprachiasmatic nucleus and also in peripheral tissues [91]. Changes in this group of genes may indicate disrupted circadian rhythm in the asthmatic lung. NR1D2 and PER3 have been associated with asthma in mice through bioinformatics analysis of genes and pathways [92]. ADRB2 has been directly linked to circadian leukocyte recruitment [93]. In addition, in mice club cells may have a role in the circadian regulation of the lung through rhythmic CXCL5 (orthologue gene to CXCL6 in horses and humans) responses and loss of this regulation leads to aberrant neutrophil influx [94]. SCGB1A1 is considered a key molecule for homeostasis in the lung, and club cells and SCGB1A1 are reduced in horses with severe asthma [78]. Club cell depletion may result from impaired epithelial precursor cell recruitment and differentiation, and trigger further dysregulated pulmonary circadian rhythm.
Conclusions
There were pronounced differences in the epithelial response to challenge in asthmatic and non-asthmatic horses. Genes identified include many with prior association in asthma, and novel genes that potentially link pathogenic mechanisms. For candidate genes of interest, further functional characterization should be undertaken. For example, a protein-protein interaction assay in BALF using recombinant versions of protein of interest might be informative. In addition, investigation of epigenetic markers may further characterize environmental influences on genes.
Abbreviations
- BAL:
-
Bronchoalveolar lavage
- BC:
-
Betweenness centrality
- BCL:
-
B-cell lymphoma
- BCV:
-
Biological coefficient of variance
- CDC25A:
-
Cell division cycle 25 homolog A
- CXCR:
-
X-C chemokine receptor
- DE:
-
Differentially expressed
- GO:
-
Gene ontology
- GOBP:
-
GO overrepresentation for biological processes
- IHC:
-
Immunohistochemistry
- IL:
-
Interleukin
- KRT:
-
Keratin
- MDS:
-
Multidimensional scaling
- MMP:
-
Matrix metalloproteinase
- PANTHER:
-
Protein ANalysis THrough Evolutionary Relationships
- PFT:
-
Pulmonary function testing
- PRR:
-
Pattern recognition receptor
- STRING:
-
Search Tool for the Retrieval of Interacting Genes/Proteins
- TLR:
-
Toll-like receptor
- TNFRSF:
-
Tumor necrosis factor receptor superfamily
- TWEAKR:
-
Tumor necrosis factor-like weak inducer of apoptosis receptor
References
Couëtil LL, Cardwell JM, Gerber V, Lavoie J-P, Léguillette R, Richard EA. Inflammatory airway disease of horses--revised consensus statement. J Vet Intern Med. 2016;30:503–15.
Costa LRR, Johnson JR, Baur ME, Beadle RE. Temporal clinical exacerbation of summer pasture-associated recurrent airway obstruction and relationship with climate and aeroallergens in horses. Am J Vet Res. 2006;67:1635–42.
Pirie RS. Recurrent airway obstruction: a review. Equine Vet J. 2014;46:276–88.
Martinez FD, Vercelli D. Asthma. Lancet. 2013;382:1360–72.
Vargas A, Roux-Dalvai F, Droit A, Lavoie J-P. Neutrophil-derived Exosomes: a new mechanism contributing to airway smooth muscle Remodeling. Am J Respir Cell Mol Biol. 2016;55:450–61.
Setlakwe EL, Lemos KR, Lavoie-Lamoureux A, Duguay J-D, Lavoie J-P. Airway collagen and elastic fiber content correlates with lung function in equine heaves. Am J Physiol Lung Cell Mol Physiol. 2014;307:L252–60.
Gerber V, Tessier C, Marti E. Genetics of upper and lower airway diseases in the horse. Equine Vet J. 2015;47:390–7.
Gerber V, Baleri D, Klukowska-Rötzler J, Swinburne JE, Dolf G. Mixed inheritance of equine recurrent airway obstruction. J Vet Intern Med. 2009;23:626–30.
Klukowska-Rötzler J, Swinburne JE, Drögemüller C, Dolf G, Janda J, Leeb T, Gerber V. The interleukin 4 receptor gene and its role in recurrent airway obstruction in Swiss Warmblood horses. Anim Genet. 2012;43:450–3.
Swinburne JE, Bogle H, Klukowska-Rötzler J, Drögemüller M, Leeb T, Temperton E, Dolf G, Gerber V. A whole-genome scan for recurrent airway obstruction in Warmblood sport horses indicates two positional candidate regions. Mamm Genome. 2009;20:504–15.
Jost U, Klukowska-Rötzler J, Dolf G, Swinburne JE, Ramseyer A, Bugno M, Burger D, Blott S, Gerber V. A region on equine chromosome 13 is linked to recurrent airway obstruction in horses. Equine Vet J. 2007;39:236–41.
Ghosh S, Das PJ, McQueen CM, Gerber V, Swiderski CE, Lavoie J-P, Chowdhary BP, Raudsepp T. Analysis of genomic copy number variation in equine recurrent airway obstruction (heaves). Anim Genet. 2016;47:334–44.
Beeler-Marfisi J, Clark ME, Wen X, Sears W, Huber L, Ackerley C, Viel L, Bienzle D. Experimental induction of recurrent airway obstruction with inhaled fungal spores, lipopolysaccharide, and silica microspheres in horses. Am J Vet Res. 2010;71:682–9.
Katavolos P, Ackerley CA, Clark ME, Bienzle D. Clara cell secretory protein increases phagocytic and decreases oxidative activity of neutrophils. Vet Immunol Immunopathol. 2011;139:1–9.
Greene CM, McElvaney NG. Proteases and antiproteases in chronic neutrophilic lung disease - relevance to drug discovery. Br J Pharmacol. 2009;158:1048–58.
Katavolos P, Ackerley CA, Viel L, Clark ME, Wen X, Bienzle D. Clara cell secretory protein is reduced in equine recurrent airway obstruction. Veterinary Pathology Online. 2009;46:604–13.
Côté O, Lillie BN, Hayes MA, Clark ME, van den Bosch L, Katavolos P, Viel L, Bienzle D. Multiple secretoglobin 1A1 genes are differentially expressed in horses. BMC Genomics. 2012;13:712.
Ray A, Raundhal M, Oriss TB, Ray P, Wenzel SE. Current concepts of severe asthma. J Clin Invest. 2016;126:2394–403.
Ilmarinen P, Tuomisto LE, Kankaanranta H. Phenotypes, risk factors, and mechanisms of adult-onset asthma. Mediat Inflamm. 2015;2015:514868.
Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18:716–25.
Thomson NC. Novel approaches to the management of noneosinophilic asthma. Ther Adv Respir Dis. 2016;10:211–34.
Kumar RK, Herbert C, Foster PS. Mouse models of acute exacerbations of allergic asthma. Respirology. 2016;21:842–9.
Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 2 Dec 2015.
Wade CM, Giulotto E, Sigurdsson S, Zoli M, Gnerre S, Imsland F, Lear TL, Adelson DL, Bailey E, Bellone RR. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science. 2009;326:865–7.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Anders S, Pyl PT, Huber W. HTSeq--a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9.
R: The R Project for Statistical Computing. https://www.r-project.org/. Accessed 23 Feb 2016.
Zhou X, Lindsay H, Robinson MD. Robustly detecting differential expression in RNA sequencing data using observation weights. Nucleic Acids Res. 2014;42:e91.
McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–97.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinformatics. 2011;12:480.
Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, Hanspers K, Isserlin R, Kelley R, Killcoyne S, Lotia S, Maere S, Morris J, Ono K, Pavlovic V, Pico AR, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2:2366–82.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52.
stringApp: Importing and augmenting Cytoscape networks from string-db. http://www.cgl.ucsf.edu/cytoscape/stringApp/index.shtml. Accessed 12 Oct 2016.
Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W. BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–40.
Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/bioconductor package biomaRt. Nat Protoc. 2009;4:1184–91.
Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–86.
Chen B, Miller AL, Rebelatto M, Brewah Y, Rowe DC, Clarke L, Czapiga M, Rosenthal K, Imamichi T, Chen Y, Chang C-S, Chowdhury PS, Naiman B, Wang Y, Yang D, Humbles AA, Herbst R, Sims GP. S100A9 Induced inflammatory responses are mediated by distinct damage associated molecular patterns (DAMP) receptors in vitro and in vivo. PLoS One. 2015;10:e0115828.
Freishtat RJ, Benton AS, Watson AM, Wang Z, Rose MC, Hoffman EP. Delineation of a gene network underlying the pulmonary response to oxidative stress in asthma. J Investig Med. 2009;57:756–64.
Park S-M, Park JS, Park H-S, Park C-S. Unraveling the genetic basis of aspirin hypersensitivity in asthma beyond arachidonate pathways. Allergy Asthma Immunol Res. 2013;5:258–76.
Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15:410–6.
Khan MA, Assiri AM, Broering DC. Complement mediators: key regulators of airway tissue remodeling in asthma. J Transl Med. 2015;13:272.
Cundall M, Sun Y, Miranda C, Trudeau JB, Barnes S, Wenzel SE. Neutrophil-derived matrix metalloproteinase-9 is increased in severe asthma and poorly inhibited by glucocorticoids. J Allergy Clin Immunol. 2003;112:1064–71.
McMillan SJ, Kearley J, Campbell JD, Zhu X-W, Larbi KY, Shipley JM, Senior RM, Nourshargh S, Lloyd CM. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J Immunol. 2004;172:2586–94.
Allen IC, Lich JD, Arthur JC, Jania CM, Roberts RA, Callaway JB, Tilley SL, Ting JP-Y. Characterization of NLRP12 during the development of allergic airway disease in mice. PLoS One. 2012;7:e30612.
Li Q, Baines KJ, Gibson PG, Wood LG. Changes in Expression of Genes Regulating Airway Inflammation Following a High-Fat Mixed Meal in Asthmatics. Nutrients. 2016;8(1). pii: E30. doi:10.3390/nu8010030. PubMed PMID: 26751474; PubMed Central PMCID: PMC4728644.
Chen Y-C, Statt S, Wu R, Chang H-T, Liao J-W, Wang C-N, Shyu W-C, Lee C-C. High mobility group box 1-induced epithelial mesenchymal transition in human airway epithelial cells. Sci Rep. 2016;6:18815.
Bucova M, Suchankova M, Dzurilla M, Vrlik M, Novosadova H, Tedlova E, Urban S, Hornakova E, Seligova M, Durmanova V, Penz P, Javor J, Paulovicova E. Inflammatory marker sTREM-1 reflects the clinical stage and respiratory tract obstruction in allergic asthma bronchiale patients and correlates with number of neutrophils. Mediat Inflamm. 2012;2012:628754.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106.
Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. Count-based differential expression analysis of RNA sequencing data using R and bioconductor. Nat Protoc. 2013;8:1765–86.
Charlotte Soneson MD. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics. 2013;14:91.
Nookaew I, Papini M, Pornputtapong N, Scalcinati G, Fagerberg L, Uhlén M, Nielsen J. A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces Cerevisiae. Nucleic Acids Res. 2012;40:10084–97.
Li Y, Xiao X, Ji X, Liu B, Amos CI. RNA-seq analysis of lung adenocarcinomas reveals different gene expression profiles between smoking and nonsmoking patients. Tumour Biol. 2015;36:8993–9003.
Yick CY, Zwinderman AH, Kunst PW, Grünberg K, Mauad T, Dijkhuis A, Bel EH, Baas F, Lutter R, Sterk PJ. Transcriptome sequencing (RNA-Seq) of human endobronchial biopsies: asthma versus controls. Eur Respir J. 2013;42:662–70.
Bartner LR, Robinson NE, Kiupel M, Tesfaigzi Y. Persistent mucus accumulation: a consequence of delayed bronchial mucous cell apoptosis in RAO-affected horses? Am J Phys Lung Cell Mol Phys. 2006;291:L602–9.
Kang JH, Hwang SM, Chung IY. S100A8, S100A9 And S100A12 activate airway epithelial cells to produce MUC5AC via extracellular signal-regulated kinase and nuclear factor-κB pathways. Immunology. 2015;144:79–90.
Fujisawa T, Velichko S, Thai P, Hung L-Y, Huang F, Wu R. Regulation of airway MUC5AC expression by IL-1beta and IL-17A; the NF-kappaB paradigm. J Immunol. 2009;183:6236–43.
Oslund KL, Adamson G, Wu R. Evaluation of MUC5AC expression and upregulation in airway epithelial cells of horses. Am J Vet Res. 2010;71:690–6.
Gerber V, De Feijter-Rupp H, Wagner J, Venta P, Harkema JR, Robinson NE. Differential association of MUC5AC and CLCA1 expression in small cartilaginous airways of RAO-affected and control horses. Equine Vet J. 2009;41:817–23.
Ohbayashi H, Shimokata K. Matrix metalloproteinase-9 and airway remodeling in asthma. Current drug targets-inflammation & amp. Allergy. 2005;4:177–81.
Bengatta S, Arnould C, Letavernier E, Monge M, de Préneuf HM, Werb Z, Ronco P, Lelongt B. MMP9 And SCF protect from apoptosis in acute kidney injury. J Am Soc Nephrol. 2009;20:787–97.
Kolaczkowska E, Koziol A, Plytycz B, Arnold B, Opdenakker G. Altered apoptosis of inflammatory neutrophils in MMP-9-deficient mice is due to lower expression and activity of caspase-3. Immunol Lett. 2009;126:73–82.
Chaudhuri R, McSharry C, Brady J, Grierson C, Messow CM, Spears M, Miele G, Nocka K, MacNee W, Connell M, Murchison JT, Sproule M, Hilmi OJ, Miller DK, Thomson NC. Low sputum MMP-9/TIMP ratio is associated with airway narrowing in smokers with asthma. Eur Respir J. 2014;44:895–904.
Malla N, Berg E, Theocharis AD, Svineng G, Uhlin-Hansen L, Winberg J-O. In vitro reconstitution of complexes between pro-matrix metalloproteinase-9 and the proteoglycans serglycin and versican. FEBS J. 2013;280:2870–87.
Winberg JO, Kolset SO, Berg E, Uhlin-Hansen L. Macrophages secrete matrix metalloproteinase 9 covalently linked to the core protein of chondroitin sulphate proteoglycans. J Mol Biol. 2000;304:669–80.
Korpetinou A, Skandalis SS, Labropoulou VT, Smirlaki G, Noulas A, Karamanos NK, Theocharis AD. Serglycin: at the crossroad of inflammation and malignancy. Front Oncol. 2014;3:327.
Niemann CU, Cowland JB, Klausen P, Askaa J, Calafat J, Borregaard N. Localization of serglycin in human neutrophil granulocytes and their precursors. J Leukoc Biol. 2004;76:406–15.
Palande K, Meenhuis A, Jevdjovic T, Touw IP. Scratching the surface: signaling and routing dynamics of the CSF3 receptor. Front Biosci (Landmark Ed). 2013;18:91–105.
Kunkel SL, Standiford T, Kasahara K, Strieter RM. Interleukin-8 (IL-8): the major neutrophil chemotactic factor in the lung. Exp Lung Res. 1991;17:17–23.
Owen C. Chemokine receptors in airway disease: which receptors to target? Pulm Pharmacol Ther. 2001;14:193–202.
Xu H, Okamoto A, Ichikawa J, Ando T, Tasaka K, Masuyama K, Ogawa H, Yagita H, Okumura K, Nakao A. TWEAK/Fn14 interaction stimulates human bronchial epithelial cells to produce IL-8 and GM-CSF. Biochem Biophys Res Commun. 2004;318:422–7.
Burkly LC, Michaelson JS, Hahm K, Jakubowski A, Zheng TS. TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine. 2007;40:1–16.
Niedzwiedz A, Jaworski Z, Tykalowski B, Smialek M. Neutrophil and macrophage apoptosis in bronchoalveolar lavage fluid from healthy horses and horses with recurrent airway obstruction (RAO). BMC Vet Res. 2014;10:29.
Turlej RK, Fiévez L, Sandersen CF, Dogné S, Kirschvink N, Lekeux P, Bureau F. Enhanced survival of lung granulocytes in an animal model of asthma: evidence for a role of GM-CSF activated STAT5 signalling pathway. Thorax. 2001;56:696–702.
Pacholewska A, Jagannathan V, Drögemüller M, Klukowska-Rötzler J, Lanz S, Hamza E, Dermitzakis ET, Marti E, Leeb T, Gerber V. Impaired cell cycle regulation in a natural equine model of asthma. PLoS One. 2015;10:e0136103.
Chinnadurai G, Vijayalingam S, Rashmi R. BIK, the founding member of the BH3-only family proteins: mechanisms of cell death and role in cancer and pathogenic processes. Oncogene. 2008;27(Suppl 1):S20–9.
Hardwick JM, Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5
Côté O, Clark ME, Viel L, Labbé G, Seah SYK, Khan MA, Douda DN, Palaniyar N, Bienzle D. Secretoglobin 1A1 and 1A1A differentially regulate neutrophil reactive oxygen species production, phagocytosis and extracellular trap formation. PLoS One. 2014;9:e96217.
Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–13.
Massberg S, Grahl L, von Bruehl M-L, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner KT, Engelmann B. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16:887–96.
Lambrecht BN, Hammad H. Asthma and coagulation. N Engl J Med. 2013;369:1964–6.
Leclere M, Bédard C, Cortes-Dubly M-L, Lavoie J-P. Blood hypercoagulability and systemic inflammation in horses with heaves. Vet J. 2015;206:105–7.
Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Phys Lung Cell Mol Phys. 2010;298:L715–31.
Sullivan C, Liu Y, Shen J, Curtis A, Newman C, Hock JM, Li X. Novel interactions between FOXM1 and CDC25A regulate the cell cycle. PLoS One. 2012;7:e51277.
Wu W, Fan YH, Kemp BL, Walsh G, Mao L. Overexpression of cdc25A and cdc25B is frequent in primary non-small cell lung cancer but is not associated with overexpression of c-myc. Cancer Res. 1998;58:4082–5.
Tesfaigzi J, Th'ng J, Hotchkiss JA, Harkema JR, Wright PS. A small proline-rich protein, SPRR1, is upregulated early during tobacco smoke-induced squamous metaplasia in rat nasal epithelia. Am J Respir Cell Mol Biol. 1996;14:478–86.
Kugler MC, Joyner AL, Loomis CA, Munger JS. Sonic hedgehog signaling in the lung. From development to disease. Am J Respir Cell Mol Biol. 2015;52:1–13.
Li X, Howard TD, Moore WC, Ampleford EJ, Li H, Busse WW, Calhoun WJ, Castro M, Chung KF, Erzurum SC, Fitzpatrick AM, Gaston B, Israel E, Jarjour NN, Teague WG, Wenzel SE, Peters SP, Hawkins GA, Bleecker ER, Meyers DA. Importance of hedgehog interacting protein and other lung function genes in asthma. J Allergy Clin Immunol. 2011;127:1457–65.
Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, Marciante KD, Franceschini N, van Durme YMTA, Chen T-H, Barr RG, Schabath MB, Couper DJ, Brusselle GG, Psaty BM, van Duijn CM, Rotter JI, Uitterlinden AG, Hofman A, Punjabi NM, Rivadeneira F, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42:45–52.
Goriki A, Hatanaka F, Myung J, Kim JK, Yoritaka T, Tanoue S, Abe T, Kiyonari H, Fujimoto K, Kato Y, Todo T, Matsubara A, Forger D, Takumi T. A novel protein, CHRONO, functions as a core component of the mammalian circadian clock. PLoS Biol. 2014;12:e1001839.
Pendergast JS, Friday RC, Yamazaki S. Distinct functions of Period2 and Period3 in the mouse circadian system revealed by in vitro analysis. PLoS One. 2010;5:e8552.
Riba M, Garcia Manteiga JM, Bošnjak B, Cittaro D, Mikolka P, Le C, Epstein MM, Stupka E. Revealing the acute asthma ignorome: characterization and validation of uninvestigated gene networks. Sci Rep. 2016;6:24647.
Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang J-E, Zhang D, Hashimoto D, Merad M, Frenette PS. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity. 2012;37:290–301.
Gibbs J, Ince L, Matthews L, Mei J, Bell T, Yang N, Saer B, Begley N, Poolman T, Pariollaud M, Farrow S, DeMayo F, Hussell T, Worthen GS, Ray D, Loudon A. An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat Med. 2014;20:919–26.
Ge XN, Bahaie NS, Kang BN, Hosseinkhani MR, Ha SG, Frenzel EM, Liu F-T, Rao SP, Sriramarao P. Allergen-induced airway remodeling is impaired in galectin-3-deficient mice. J Immunol. 2010;185:1205–14.
López E, del Pozo V, Miguel T, Sastre B, Seoane C, Civantos E, Llanes E, Baeza ML, Palomino P, Cárdaba B. Inhibition of chronic airway inflammation and remodeling by galectin-3 gene therapy in a murine model. J Immunol. 2006;176:1943–50.
Gao P, Simpson JL, Zhang J, Gibson PG. Galectin-3: its role in asthma and potential as an anti-inflammatory target. Respir Res. 2013;14:136.
Gao P, Gibson PG, Baines KJ, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, Jenkins C, Peters MJ, Zhang J, Simpson JL. Anti-inflammatory deficiencies in neutrophilic asthma: reduced galectin-3 and IL-1RA/IL-1β. Respir Res. 2015;16:5.
Simpson JL, Baines KJ, Boyle MJ, Scott RJ, Gibson PG. Oncostatin M (OSM) is increased in asthma with incompletely reversible airflow obstruction. Exp Lung Res. 2009;35:781–94.
Poole A, Urbanek C, Eng C, Schageman J, Jacobson S, O'Connor BP, Galanter JM, Gignoux CR, Roth LA, Kumar R, Lutz S, Liu AH, Fingerlin TE, Setterquist RA, Burchard EG, Rodriguez-Santana J, Seibold MA. Dissecting childhood asthma with nasal transcriptomics distinguishes subphenotypes of disease. J Allergy Clin Immunol. 2014;133:670–8.e12.
Chan IHS, Tang NLS, Leung TF, Ma SL, Zhang YP, Wong GWK, Wong CK, Lam CWK. Association of prostaglandin-endoperoxide synthase 2 gene polymorphisms with asthma and atopy in Chinese children. Allergy. 2007;62:802–9.
Planagumà A, Domènech T, Pont M, Calama E, García-González V, López R, Aulí M, López M, Fonquerna S, Ramos I, de Alba J, Nueda A, Prats N, Segarra V, Miralpeix M, Lehner MD. Combined anti CXC receptors 1 and 2 therapy is a promising anti-inflammatory treatment for respiratory diseases by reducing neutrophil migration and activation. Pulm Pharmacol Ther. 2015;34:37–45.
Zhang J, Shan L, Koussih L, Redhu NS, Halayko AJ, Chakir J, Gounni AS. Pentraxin 3 (PTX3) expression in allergic asthmatic airways: role in airway smooth muscle migration and chemokine production. PLoS One. 2012;7:e34965.
McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, Dong X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature. 2015;519:237–41.
Karlsson T, Glogauer M, Ellen RP, Loitto V-M, Magnusson K-E, Magalhães MAO. Aquaporin 9 phosphorylation mediates membrane localization and neutrophil polarization. J Leukoc Biol. 2011;90:963–73.
Liu H, Liu J, Toups M, Soos T, Arendt C. Gene signature-based mapping of immunological systems and diseases. BMC Bioinformatics. 2016;17:171.
Dellambra E, Patrone M, Sparatore B, Negri A, Ceciliani F, Bondanza S, Molina F, Cancedda FD, De Luca M. Stratifin, a keratinocyte specific 14-3-3 protein, harbors a pleckstrin homology (PH) domain and enhances protein kinase C activity. J Cell Sci. 1995;108(Pt 11):3569–79.
Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature. 2006;441:362–5.
Stewart CE, Nijmeh HS, Brightling CE, Sayers I. uPAR regulates bronchial epithelial repair in vitro and is elevated in asthmatic epithelium. Thorax. 2012;67:477–87.
Zhao L, Yang W, Yang X, Lin Y, Lv J, Dou X, Luo Q, Dong J, Chen Z, Chu Y, He R. Chemerin suppresses murine allergic asthma by inhibiting CCL2 production and subsequent airway recruitment of inflammatory dendritic cells. Allergy. 2014;69:763–74.
Kawano T, Morimoto K, Uemura Y. Partial purification and properties of urokinase inhibitor from human placenta. J Biochem. 1970;67:333–42.
Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, Ellwanger A, Sidhu SS, Dao-Pick TP, Pantoja C, Erle DJ, Yamamoto KR, Fahy JV. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104:15858–63.
Leonard B, McCann JL, Starrett GJ, Kosyakovsky L, Luengas EM, Molan AM, Burns MB, McDougle RM, Parker PJ, Brown WL, Harris RS. The PKC/NF-κB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 2015;75:4538–47.
Munitz A, Bachelet I, Levi-Schaffer F. Reversal of airway inflammation and remodeling in asthma by a bispecific antibody fragment linking CCR3 to CD300a. J Allergy Clin Immunol. 2006;118:1082–9.
Nakahashi-Oda C, Tahara-Hanaoka S, Shoji M, Okoshi Y, Nakano-Yokomizo T, Ohkohchi N, Yasui T, Kikutani H, Honda S-I, Shibuya K, Nagata S, Shibuya A. Apoptotic cells suppress mast cell inflammatory responses via the CD300a immunoreceptor. J Exp Med. 2012;209:1493–503.
Acknowledgements
Not applicable.
Funding
Sources of support were Natural Sciences and Engineering Research Council of Canada (Canada Research Chair, DB, all aspects of study), Equine Guelph (data collection and analysis), Ontario Ministry of Agriculture and Food (research animals), and the Ontario Veterinary College Scholarship Program (stipend LT, all aspects of study).
Availability of data and materials
The raw sequences have been submitted to NCBI sequence read archive (SRA) on May 2nd, 2017, and are publicly available under study PRJNA384774 (SRP106023).
Author information
Authors and Affiliations
Contributions
LT, OC, MEC, LV, ADM and DB participated at the conception and design of the work and acquisition of data. LT and DB performed the analysis and interpretation of data, and drafting of the work. SA critically revised and participated to the analysis and interpretation of data. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
All procedures were approved by the Institutional Animal Care Committee of the University of Guelph (protocol R10–031) and conducted in compliance with Canadian Council on Animal Care guidelines. Animals used in this research belong to the University of Guelph.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Clinical parameters, bronchoalveolar lavage and pulmonary function results. Table S2. Top genes differentially expressed between horses with and without asthma after challenge, ranked by logFC. (XLSX 31 kb)
Additional file 2: Figure S1.
Immunoblots assessing antibody reactivity for (A) TNFRSF12A, (B) PTCH1, (C) CDC25A and (D) IL8. Only antibodies yielding a single band of expected size were used in subsequent immunohistochemical assays. (TIFF 69 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tessier, L., Côté, O., Clark, M.E. et al. Impaired response of the bronchial epithelium to inflammation characterizes severe equine asthma. BMC Genomics 18, 708 (2017). https://doi.org/10.1186/s12864-017-4107-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4107-6