
Abstract
Background
Intestinal microbes play significant roles in fish and can be possibly used as probiotics in aquaculture. In our previous study, Flaviramulus ichthyoenteri Th78T, a novel species in the family Flavobacteriaceae, was isolated from fish intestine and showed strong quorum quenching (QQ) ability. To identify the QQ enzymes in Th78T and explore the potential roles of Th78T in fish intestine, we sequenced the genome of Th78T and performed extensive genomic analysis.
Results
An N-acyl homoserine lactonase FiaL belonging to the metallo-β-lactamase superfamily was identified and the QQ activity of heterologously expressed FiaL was confirmed in vitro. FiaL has relatively little similarity to the known lactonases (25.2 ~ 27.9% identity in amino acid sequence). Various digestive enzymes including alginate lyases and lipases can be produced by Th78T, and enzymes essential for production of B vitamins such as biotin, riboflavin and folate are predicted. Genes encoding sialic acid lyases, sialidases, sulfatases and fucosidases, which contribute to utilization of mucus, are present in the genome. In addition, genes related to response to different stresses and gliding motility were also identified. Comparative genome analysis shows that Th78T has more specific genes involved in carbohydrate transport and metabolism compared to other two isolates in Flavobacteriaceae, both isolated from sediments.
Conclusions
The genome of Th78T exhibits evident advantages for this bacterium to survive in the fish intestine, including production of QQ enzyme, utilization of various nutrients available in the intestine as well as the ability to produce digestive enzymes and vitamins, which also provides an application prospect of Th78T to be used as a probiotic in aquaculture.
Similar content being viewed by others


Background
The family Flavobacteriaceae, a major phylogenetic lineage in the phylum Bacteroidetes [1,2], currently consists of over 110 genera (http://www.bacterio.cict.fr) comprising diverse bacteria which occur primarily in various marine environments from surface water to deep-sea sediment [3], as well as other temperate and polar habitats in terrestrial and freshwater ecosystems. Members of the Flavobacteriaceae are well-known for their capacity of degrading many different polysaccharides, proteins and other biopolymers [4,5]. Because of their roles as decomposers in marine environment, they have attracted considerable interest in recent years [6-9]. Whole genome analyses of some species of the Flavobacteriaceae have also revealed their special ecological niche adaptive strategies in the oligotrophic ocean [10,11] or in the extremely cold environment [12].
Flaviramulus ichthyoenteri Th78T is the type strain of the second established species in the genus Flaviramulus whose genomes have never been explored, and was isolated from the intestine of healthy flounder (Paralichthys olivaceus) in Qingdao, China, together with other four strains showed >99% 16S rRNA gene sequence similarity to Th78T [13,14]. No such isolates were found in the rearing water or other parts of the flounder, indicating that these strains may prefer the intestinal environment. These F. ichthyoenteri isolates have been demonstrated to degrade various N-acylhomoserine lactones (AHLs); AHLs are widely used by Gram-negative bacteria as signal molecules in cell-to-cell communication (quorum sensing, QS) [14]. However, the underlying mechanism of this quorum quenching (QQ) property remains undiscovered.
Intestinal microorganisms play an important role in the nutrition and well-being of fish, and the nutrient-rich intestine of fish provides a favorable growth environment for numerous bacteria [15-20]. These bacteria can be categorized as either indigenous or transient species depending on their ability to colonize and adhere to the mucus layer [21]. To elucidate the ecological niche adaptations of F. ichthyoenteri Th78T (including its QQ capability), its genome was sequenced and analyzed. Comparative genome studies with publicly available genome data of closely related flavobacterial strains were performed. Furthermore, the AHL degradation activity of candidate QQ enzymes was confirmed by recombinant expression in Escherichia coli. The results provide a first picture of the role of strain Th78T in interacting with the intestinal environment and other intestinal bacteria, and reveal its potential to be used as a probiotic in aquaculture.
Results and discussion
Genome properties
The genome of F. ichthyoenteri Th78T is 3,953,230 bp long (1 chromosome with no plasmid) with a G + C content of 32.06%. Of the 3,530 predicted genes, 3,451 are protein coding genes and 79 are RNAs. A total of six rRNA genes (two 5S rRNAs, two 16S rRNAs and two 23S rRNAs) and 40 tRNA genes were identified in the genome (Table 1). Among the predicted protein-coding genes, 925 (26.8%) can be assigned to COG categories and 1517 (43.9%), 3070 (88.9%), 844 (24.5%), and 3046 (88.3%) genes are found within the KEGG, NR, Swiss-Prot, and TrEMBL databases, respectively. The number of subsystems identified by RAST server is 331.
General metabolism
For central carbohydrate metabolism, the genome of strain Th78T encodes a full set of enzymes essential for carrying out Embden-Meyerhof-Parnas pathway which converts glucose into pyruvate and provide the precursors of metabolites for nucleotide and fatty acid biosynthesis. All the enzymes involved in the tricarboxylic acid cycle are also present. Pathways related to the utilization of various monosaccharides, disaccharides and aminosugars are predicted to be present in the genome of Th78T. Fructose and galactose can be converted to fructose-6-phosphate and glucose-1-phosphate respectively, and can be degraded through glycolysis. Sucrose can be converted to glucose-1-phosphate and fructose by a sucrose phosphorylase, and the ability to utilize sucrose has been confirmed experimentally in our previous study [13]. Consistent with the ability to use the amino sugar N-acetylglucosamine [13], two key enzymes, glucosamine-6-phosphate deaminase (NagB) and N-acetylglucosamine-6-phosphate deacetylase (NagA), are found in Th78T, as well as N-acetylglucosamine related transporters (NagX and NagP). For monosaccharide utilization, genes involved in ribose, L-fucose, mannose and xylose metabolism are present in the genome of Th78T. In addition, genes for D-galacturonate and D-glucuronate utilization are found (Additional file 1: Table S1). Most amino acids can be synthesized by strain Th78T, including animo acids essential for fish such as lysine, threonine, methionine, cysteine, leucine and histidine. All these features confer a metabolic versatility on strain Th78T and allow it to participate in the conversion of material and energy in the fish intestine.
F. ichthyoenteri Th78T is most likely to be a heterotrophic facultative member in the fish intestine, and it may have the potential for mixed acid fermentation in the anoxic intestine environment. According to KEGG pathway maps of Th78T, phosphoenolpyruvate (PEP) derived from glycolysis can be converted to oxaloacetate by a predicted phosphoenolpyruvate carboxylase and subsequently enter the TCA cycle to form succinate. The anaerobic dissimilation of pyruvate produced L- or D-lactate, acetate and acetyl-CoA; acetyl-CoA can be further fermented to butyrate-CoA. However, different from Formosa agariphila KMM 3901T which has also been predicted to be capable of mixed acid fermentation [9], no pyruvate formate-lyase (PFL) is identified in Th78T, indicating that other enzymes are possibly used for pyruvate cleavage [23]. In addition, although Th78T is unable to reduce nitrate to nitrite (due to the lack of nitrate reductase), a complete pathway of denitrification converting nitrite to nitrogen in the low oxygen environment can be found in its genome.
Quorum quenching properties
QS is a mechanism of gene regulation in response to the concentration of specific signal molecules [24], and group behaviors such as biofilm formation and production of other virulence factors are coordinated by QS [25,26]. In Gram-negative bacteria including several aquaculture pathogens, such as Edwardsiella tarda, Aeromonas spp. and Vibrio spp., AHLs are the main signal molecules [27,28]. Thus QQ, namely the enzymatic interruption of QS, may be important to maintain homeostasis in gut ecosystem and inhibit the virulence of pathogenic microbes in the complicated intestinal environment.
In our previous study [14], various QQ strains have been identified among the isolates from flounder. Among them, strain Th78T was demonstrated to degrade various AHLs with different acyl chains including C6-homoserine lactone (HSL), 3-oxo-C6-HSL, C8-HSL, 3-oxo-C8-HSL, C10-HSL, 3-oxo-C10-HSL, C12-HSL, 3-oxo-C12-HSL, C14-HSL and 3-oxo-C14-HSL. According to the experimental results, the degrading activity in strain Th78T was most likely attributed to AHL lactonases [14]. On that basis, bi-directional BLAST between all the predicted proteins of Th78T and several reported AHL lactonases was performed. Two predicted β-lactamases (encoded by GL001211 and GL001708) show more than 25% identity with the reported AHL lactonases from metallo-β-lactamase superfamily (Table 2), and conserved zinc binding motif HXHXDH region has been identified in these two proteins (Figure 1). Three other proteins (encoded by GL001057, GL001387 and GL003427) are similar to AidH, an AHL lactonase from the α/β-hydrolase fold family. No hits are found when proteins from Th78T were queried with QsdA [29] or AiiM [30].

Amino acid sequence comparison of six known AHL lactonases in metallo-β-lactamase superfamily and possible AHL lactones encoding by the genome of Th78 T . The two zinc-binding motifs are boxed with rectangles.
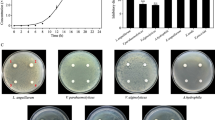
Although the identities between predicted proteins of Th78T and the reported AHL lactonases are relatively low, they still may be the active AHL lactonase considering the conserved zinc binding motif identified, so further experiments were performed to confirm the function of gene GL001211 and GL001708. The heterologous expression of gene GL001211 in E. coli led to a recombinant E.coli that is capable of degrading AHLs with different acyl-chain length and modifications (Figure 2). A protein with the expected mass (33 kDa) was produced (Figure 2). The name FiaL (F laviramulus i chthyoenteri N-acyl-homoserine lactonase) was proposed for this novel AHL-lactonase encoded by gene GL001211. However, the recombinant E.coli which expressed gene GL001708 did not show QQ activity.

Expression of FiaL and detection of its QQ activity. (A). SDS-PAGE analysis of overexpressed FiaL. Lane 1, protein molecular weight marker (Fermentas SM0431); Lane 2, supernatant of E. coli/pET 24a(+) after ultrasonication; Lane 3, resuspended cell pellet of E. coli/pET 24a(+) after ultrasonication; Lane 4, supernatant of E. coli/pET 24a(+)/GL001211 after ultrasonication; Lane 5, resuspended cell pellet of E. coli/pET 24a(+)/GL001211 after ultrasonication (the arrow indicates the position of FiaL). (B). AHL-degrading activity of recombinant E. coli BL21(DE3). 1, supernatant of E. coli/pET 24a(+)/GL001211 after ultrasonication; 2, supernatant of E. coli/pET 24a(+) after ultrasonication.
FiaL homologs can be found in various marine bacteria of the family Flavobacteriaceae. FiaL shows the highest identity (70%) to a predicted protein in Tenacibaculum ovolyticum (WP_028890039), followed by Polaribacter sp. Hel_I_88 (WP_026776245, 63% identity) and Aquimarina muelleri (WP_027414219, 61% identity). However, the QQ activity of these homologous proteins remains to be confirmed experimentally.
Digestive enzyme and vitamin production
Gut microorganisms might have a beneficial effect in the digestive process of fish as these bacterial isolates have been demonstrated to break down protein, chitin, cellulose, lipid, starch and phytate [38,39].
Th78T was reported to be capable of hydrolyzing starch and sodium alginate in our previous study [13]. Starch may be hydrolyzed by a predicted 1, 4-α-glucan branching enzyme (GL001713) with an α-amylase domain, or other predicted glycoside hydrolase proteins. Four alginate lyases (GL000665, GL000671, GL000673 and GL000676) are identified, corresponding to the strong alginate hydrolysis ability in Th78T. Consistent with the lipase activity of strain Th78T, two putative lipases (GL002043 and GL003284) and two phospholipases (GL000298 and GL001418) are identified in its genome, and the breakdown of dietary lipids into fatty acids may promote the absorption of lipid in the intestine. Moreover, two xylanases (GL000440 and GL002553) in the genome of Th78T indicate the ability to hydrolyze xylan which is a major component of hemicellulose. Although two genes encoding for chitinases (GL001289 and GL002608) are present in Th78T, no obvious chitinase ability was detected in our previous experiment.
Intestinal bacteria are also great sources of vitamins needed by the host. Strain Th78T may be able to produce various B vitamins since homologs of most enzymes needed for the synthesis of biotin, riboflavin, pyridoxine, folate, nicotinate, thiamin and pantothenate are present in the genome of Th78T. This suggests that strain Th78T may have a beneficial effect on the growth of fish and could potentially be useful as a probiotic in aquaculture.
Utilization of substances in mucus
Mucus is a gel-like substance formed from glycoproteins (mucins) that contain negatively charged sugars (sialic acid or sulfosaccharide) [40]. It coats the surface of cells in digestive tract and has multiple roles in the intestinal environment [41].
Sialic acids comprise a family of nine-carbon amino sugars that are prevalent in mucus rich environments [42]. Several genes involved in sialic acid metabolism are found in the genome of Th78T, including a predicted N-acetyl neuraminate lyase (sialic acid lyase, NanA) and a tripartite ATP-independent periplasmic (TRAP) transporter for sialic acid. Catalyzed degradation of sialic acid by NanA yields pyruvate and N-acetyl-D-mannosamine (ManNAc), which makes sialic acid an attractive nutrient for microbes [43]. Sialidases are glycosylhydrolases that cleave the glycoketosidic linkages of Sia-O-acceptor substrates by an exohydrolytic reaction involving retention of configuration through a double-displacement mechanism [44]. Two genes encoding sialidases are identified, and this may allow Th78T to scavenge host sialoglycoconjugates in the intestinal environment. Moreover, these sialidases in strain Th78T may also be involved in regulation of host innate immunity, and play an important role in interspecies competition between sialidase-positive and sialidase-negative bacteria occupying the same niche [45,46].
Mucin in the intestine contained significant levels of sulfate covalently bound to the mucin oligosaccharide chains [47]. Sulfomucins may slow mucin degradation by mucin-degrading bacterial enzymes, and this role is thought to be particularly important in the colon. In the genome of strain Th78T, various kinds of sulfatases are predicted, including N-acetylgalactosamine-4-sulfatase, N-acetylgalactosamine-6-sulfatase and glucosamine-6-sulfatase, which could act on terminal or internal sulfated sugars of the mucin oligosaccharide chain and provide Th78T an easier access to mucin-derived carbohydrates.
In addition to N-acetyl glucosamine and N-acetyl galactosamine, galactose and fucose are also contained in typical mucins [48]. Six β-galactosidase-encoding genes are identified in strain Th78T, which was confirmed by the experimental result in the API 20E and 20NE strips [13]. Strain Th78T also has six genes coding for α-L-fucosidase (three of them are secretory), in agreement with the experimental result in API ZYM strip [13]. These α-L-fucosidases in Th78T are capable of hydrolyzing various types of fucosidic linkages as well as synthetic substrates. α-L-fucosyl residues are frequently found at the terminal of many oligosaccharide chains in intestinal mucins, and fucosidases may play an important role in the intestinal ecosystem. All genes involved in the utilization of substances in mucus are summarized in Additional file 1: Table S2.
General stress response
To successfully survive in the intestinal environment, F. ichthyoenteri Th78T needs the ability to respond to a variety of stress conditions, and this may also enhance the potential of Th78T to be used as a probiotic in the aquaculture [49]. The presence of bile acid is one of the main selective pressures in the intestine [50], and the mechanisms allowing intestinal bacteria to survive exposure to high bile acid concentrations include bile acid modifying enzymes and membrane transporters of these compounds [51,52]. One Na+ dependent transporter belonging to the sodium bile acid symporter family, which may contribute to bile acid resistance, was predicted by analysis of Th78T genome. Intestinal bacteria are likely to be exposed to osmotic stresses because of diet fluctuation. Th78T possesses two genes encoding for aquaporin Z which may increase the rate of water diffusion across cell membranes [53]. In addition, several enzymes which participate in the biosynthesis of the efficient osmoprotectant glycine betaine are identified, such as choline dehydrogenase (BetA) and choline sulfatase (BetC). Th78T also encodes various enzymes dealing with oxidative stress including catalase, alkyl hydroperoxide reductase and superoxide dismutase, as well as various key regulators of the oxidative stress response. Molecular chaperones related to various stress responses in Th78T include DnaK from the Hsp70 family, GroEL from the Hsp60 family, as well as ClpB and ClpC from the Hsp100 family. Cold shock proteins CspA and CspG are also present in Th78T.
Gliding motility and type IX secretion system
Gliding motility is commonly found in the members of the phylum Bacteroidetes and is important in the competition for nutrients and colonization on surfaces [54]. Corresponding with the gliding motility of Th78T [13], related genes were identified in its genome, including gldABCDEFGHIJKLMN, sprA, sprE and sprT. The amino acid sequences of Th78T and Flavobacterium johnsoniae motility proteins [55,56] are similar, with identities ranging from 42% to 74%. Potential homologous proteins of SprB and RemA, which are mobile cell surface adhesins involved in gliding of F. johnsoniae, may also be encoded by the genome of Th78T (33% and 30% identities, respectively). As a subset of gliding motility genes of Th78T, core genes for the type IX secretion system (gldK, gldL, gldM, gldN, sprA, sprE, and sprT) are present in the genome of Th78T [57]. By using the TIGRFAM database, 39 genes are predicted to encode proteins with conserved C-terminal domains which are found in proteins known to be secreted by type IX secretion system [58], but the functions of most of these genes are unknown.
Comparison with other Flavobacteriaceae genomes
Gaetbulibacter saemankumensis DSM 17032T and Lacinutrix sp. 5H-3-7-4, which show relatively higher 16S rRNA gene sequence similarities (96.4% and 95.8%, respectively) and closer phylogenetic relationships with Th78T among the genome-sequenced strains in Flavobacteriaceae (Additional file 2: Figure S1), were chosen for the comparative analysis with F. ichthyoenteri Th78T. G. saemankumensis DSM 17032T, the type species of Gaetbulibacter, was isolated from tidal flat sediment in Korea [59], while Lacinutrix sp. 5H-3-7-4 was reported as a polysaccharide-degrading strain from subseafloor sediment [22]. No in-depth genomic studies of these two strains have been performed yet. General features of these three genomes are compared in Table 1. The genome of Th78T is larger than that of the other two sediment isolates, and the genome size of Flavobacteriaceae bacteria varies from approximately 2 ~ 6 Mb. According to the subsystems features predicted by RAST server, the extra genes of Th78T mainly belong to subsystems such as carbohydrates, cell wall and capsule, membrane transport and sulfur metabolism.
Results of further comparative analysis based on all the predicted protein sequences of Th78T, DSM 17032T and 5H-3-7-4 are shown in Figure 3. Although these three strains belong to different species in Flavobacteriaceae, they share a large number of orthologous genes (1519), accounting for 44.0%, 56.5% and 51.2% of all genes of Th78T, DSM 17032T and 5H-3-7-4, respectively. Genes function in three COG categories, i.e. amino acid transport and metabolism (E), translation/ribosomal structure (J) and cell wall/membrane/envelope biogenesis (M), are highly conserved among three genomes. Corresponding with the larger genome of Th78T, it contains more specific genes (1358, 39.4%) than DSM 17032T (728, 27.1%) and 5H-3-7-4 (1066, 35.9%). The largest proportion of specific genes with certain function in Th78T are related to carbohydrate transport and metabolism (G) and cover genes encoding the following products: (i) α-L-fucosidases and fucose permeases, (ii) enzymes in aminosugar metabolism, (iii) enzymes in glucuronate metabolism, (iv) β-galactosidases, and (v) two complete TRAP-type C4-dicarboxylate transport systems which might be involved in sialic acid and glucuronate transportation (Additional file 1: Table S3). Th78T is exposed to much more multiple and abundant carbohydrates in the intestine when compared to DSM 17032T and 5H-3-7-4, thus evident distinctions can be found in carbohydrate utilization between Th78T and those two marine sediment isolates. In addition, many specific genes of Th78T are assigned to transcription (K), which encode for specific transcriptional regulators and AraC-type DNA-binding domain-containing proteins with unknown function. Generally there tends to be more transcriptional regulators in a larger genome [60], and this may help Th78T to develop a sophisticated system to coordinate gene expression and thus adapt to the changing and competitive environment in fish intestine.

Gene content comparison of Th78 T , G. saemankumensis DSM 17032 T and Lacinutrix sp. 5H-3-7-4. The Venn diagram shows the orthologous and specific genes in each strain, and the pie charts show the relative abundance compared to all COG categories of the orthologous and specific genes in each strain.
Conclusions
Current studies on intestinal microbes mainly focus on some well-known and dominating genera in humans including Bacteroides, Lactobacillus, Bifidobacterium, Fusobacterium, Enterobacteriaceae, etc. Here we explore the potential features of a novel isolate from fish intestine, Th78T, by genome analysis. The AHL lactonase named FiaL was identified and expressed in E.coli, and this QQ property may confer competitive advantages on Th78T in gut environment. Some signals used by aquaculture pathogens can be degraded by FiaL, such as C6-HSL (Aeromonas hydrophila and Edwardsiella tarda) [61,62], 3-oxo-C6-HSL (Edwardsiella tarda and Vibrio salmonicida) [63] and 3-oxo-C10-HSL (Vibrio anguillarum) [64]. Further analysis of Th78T genome unveils that it may be able to produce various digestive enzymes and vitamins, which can be beneficial to the host. Th78T also shows the ability to use some abundant nutrients in intestine, and this may be closely related to colonization and further influence the competitiveness. These results explained why Th78T is able to successfully survive in the fish intestine, and further efforts may be needed to explore its potential to be used as a probiotics in aquaculture.
Methods
Growth conditions and DNA extraction
F. ichthyoenteri Th78T was isolated from the intestine of cultured healthy flounder (Paralichthys olivaceus) in Qingdao, China and cultured on marine agar 2216 (MA; Becton Dickinson) at 28°C. Fish experiments were performed in compliance with Regulations for the Administration of Affairs Concerning Experimental Animals, and were approved by the Animal Ethics Committee of Shandong province. Genomic DNA of strain Th78T was isolated by phenol-chloroform method [65].
Genome sequencing and assembly
The genome of F. ichthyoenteri Th78T was sequenced using Illumina Hiseq2000 sequencing platform with two libraries (500 bp and 6000 bp). A total of 907 Mb data was achieved constituting 229.4 fold coverage of the genome. The reads were assembled using SOAPdenovo assembler software [66], and a total of 17 contigs ranging from 246 bp to 1 394 011 bp (the N50 and N90 contig sizes were 878 943 bp and 267 282 bp, respectively) were obtained. By realigning the reads onto the contigs and using the paired-end information, these contigs were combined into five scaffolds. Intrascaffold gaps which were most likely comprised by repeats were closed using the pair-end extracted reads [66]. The largest scaffold was 3 944 211 bp, accounting for nearly 99.7% of the total length, and the other four scaffolds ranged from 637 bp to 6 595 bp.
Nucleotide sequence accession numbers
This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession AUYN00000000. The version described in this paper is version AUYN01000000.
Genome annotation
Putative coding sequences (CDSs) were identified by Glimmer 3.0 [67]. RNAmmer [68] and tRNAscan [69] were used to predict rRNAs and tRNAs respectively, and sRNA was identified using Rfam database [70]. Functional annotation was performed by similarity analysis using the KEGG (Kyoto encyclopedia of genes and genomes; http://www.genome.jp/kegg/) [71], COG (http://www.ncbi.nlm.nih.gov/COG/) [72], SwissProt and TrEMBL (http://www.uniprot.org/), GO (Gene Ontology; http://www.geneontology.org/) [73], TIGRFAM [74] and NR (NCBI non-redundant database; http://www.ncbi.nlm.nih.gov/RefSeq/) [75]. The functional annotation and subsystem prediction were also performed using the RAST server [76]. The draft metabolic model was constructed by SEED Viewer version 2.0 (theSEED.org) [77].
Expression of QQ enzymes and detection of QQ activity
The candidate QQ enzyme-encoding gene GL001211 was amplified by PCR with primer 1211-F (5'-CCGGAATTCATGAAAACAACAACTC-3') and 1211-R (5'-CCCTCGAGTTTCTTTAATAAGTTTTG-3') (EcoRI and XholI sites are italics). The amplified DNA fragment was digested with EcoRI and XholI and was inserted into similarly digested pET-24a (+) (Novagen). Protein expression was performed with E. coli BL21 (DE3) (Novagen). To induce protein expression, IPTG (isopropyl-β-D-thiogalacto-pyranoside) was added to E. coli cultures grown to an optical density at 590 nm (OD590) of 0.4 ~ 0.5 at a final concentration of 0.1 mM. The induction was allowed to proceed for 12 h at 16°C. After incubation, cells were harvested by centrifugation and resuspended in PBS buffer (pH 6.8) for ultrasonication. The overexpressed protein was assessed by SDS-PAGE and E. coli BL21 (DE3) harboring an empty pET-24a (+) was used as negative control. To detect the QQ activity, supernatant obtained after induction and ultrasonication was mixed with certain concentration of AHLs (C6 to C14-HSL and 3-oxo-C6 to 3-oxo-C14-HSL) and the reaction was carried out at 28°C for 24 h. C4-HSL, C6-HSL, 3-oxo-C6-HSL and C8-HSL were purchased from Cayman Chemical Company (Ann Arbor, Michigan, USA); 3-oxo-C8-HSL, C10-HSL, 3-oxo-C10-HSL, C12-HSL, 3-oxo-C12-HSL, C14-HSL and 3-oxo-C14-HSL were purchased from Sigma-Aldrich (St. Louis, Missouri, USA). The biosensor Agrobacterium tumefaciens A136, which contains the AHL-responsive transcription factor TraR (pCF218) and the TraR-regulated traI-lacZ reporter (pCF372) [78], was used to detect the residual AHLs. Briefly, to make the A136 X-gal assay solution, an overnight broth culture of A136 was inoculated (1:100) into AT minimal glucose medium with X-gal at a final concentration of 250 μg mL−1. Ten μL reaction mixture and 190 μL A136 X-gal assay solution were added to 96-well plates, and degradation of AHLs was indicated by failing to express β-galactosidase activity in the biosensor strain (no blue color reaction) after incubating at 28°C for 24 h.
Comparative genomics
The complete genome sequences of G. saemankumensis DSM 17032T and Lacinutrix sp. 5H-3-7-4 were retrieved from NCBI. The general information about the genome of DSM 17032T was obtained from IMG (http://img.jgi.doe.gov/). Proteins from Th78T were compared with those of DSM 17032T and 5H-3-7-4 using BLASTP with an E-value cutoff of 1e-5. Orthologous proteins are defined as reciprocal best hit proteins with a minimum 40% identity and 70% coverage, calculated by the BLAST algorithm [79]. Proteins without orthologs are considered to be strain-specific proteins. The COG function category was analyzed by searching all of the predicted proteins against the COG database on the basis of BLASTP.
References
Bernardet J. Family I. Flavobacteriaceae Reichenbach 1992. Bergey’s Manual of Systematic Bacteriology. 2011;4:106–11.
Bernardet J-F, Nakagawa Y. An introduction to the family Flavobacteriaceae. In: The Prokaryotes. New York: Springer; 2006. p. 455–80.
Bowman JP. The marine clade of the family Flavobacteriaceae: the genera Aequorivita, Arenibacter, Cellulophaga, Croceibacter, Formosa, Gelidibacter, Gillisia, Maribacter, Mesonia, Muricauda, Polaribacter, Psychroflexus, Psychroserpens, Robiginitalea, Salegentibacter, Tenacibaculum, Ulvibacter, Vitellibacter and Zobellia. The Prokaryotes. 2006;7:677–94. Proteobacteria: Delta, Epsilon Subclass.
Cottrell MT, Kirchman DL. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low-and high-molecular-weight dissolved organic matter. Appl Environ Microbiol. 2000;66(4):1692–7.
Kirchman DL. The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol Ecol. 2002;39(2):91–100.
Bauer M, Kube M, Teeling H, Richter M, Lombardot T, Allers E, et al. Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’reveals adaptations to degradation of polymeric organic matter. Environ Microbiol. 2006;8(12):2201–13.
McBride MJ, Xie G, Martens EC, Lapidus A, Henrissat B, Rhodes RG, et al. Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl Environ Microbiol. 2009;75(21):6864–75.
Qin Q-L, Zhang X-Y, Wang X-M, Liu G-M, Chen X-L, Xie B-B, et al. The complete genome of Zunongwangia profunda SM-A87 reveals its adaptation to the deep-sea environment and ecological role in sedimentary organic nitrogen degradation. BMC Genomics. 2010;11(1):247.
Mann AJ, Hahnke RL, Huang S, Werner J, Xing P, Barbeyron T, et al. The genome of the alga-associated marine flavobacterium Formosa agariphila KMM 3901T reveals a broad potential for degradation of algal polysaccharides. Appl Environ Microbiol. 2013;79(21):6813–22.
González JM, Fernández-Gómez B, Fernàndez-Guerra A, Gómez-Consarnau L, Sánchez O, Coll-Lladó M, et al. Genome analysis of the proteorhodopsin-containing marine bacterium Polaribacter sp. MED152 (Flavobacteria). Proc Natl Acad Sci U S A. 2008;105(25):8724–9.
Kwon S-K, Kim BK, Song JY, Kwak M-J, Lee CH, Yoon J-H, et al. Genomic makeup of the marine flavobacterium Nonlabens (Donghaeana) dokdonensis and identification of a novel class of rhodopsins. Genome Biol Evol. 2013;5(1):187–99.
Feng S, Powell SM, Wilson R, Bowman JP. Extensive gene acquisition in the extremely psychrophilic bacterial species Psychroflexus torquis and the link to sea-ice ecosystem specialism. Genome Biol Evol. 2014;6(1):133–48.
Zhang Y, Tang K, Shi X, Zhang X-H. Flaviramulus ichthyoenteri sp. nov., an N-acylhomoserine lactone-degrading bacterium isolated from the intestine of a flounder (Paralichthys olivaceus), and emended descriptions of the genus Flaviramulus and Flaviramulus basaltis. Int J Syst Evol Microbiol. 2013;63(Pt 12):4477–83.
Tang K, Zhang Y, Yu M, Shi X, Coenye T, Bossier P, et al. Evaluation of a new high-throughput method for identifying quorum quenching bacteria. Sci Rep. 2013;3:2935.
Austin B. The bacterial microflora of fish. Sci World J. 2002;2:558–72.
Belchior SGE, Vacca G. Fish protein hydrolysis by a psychrotrophic marine bacterium isolated from the gut of hake (Merluccius hubbsi). Can J Microbiol. 2006;52(12):1266–71.
De D, Ghoshal TK, Ananda Raja R. Characterization of enzyme-producing bacteria isolated from the gut of Asian seabass, Lates calcarifer and milkfish, Chanos chanos and their application for nutrient enrichment of feed ingredients. Aquacult Res. 2014;45(9):1573–80.
Ghosh K, Roy M, Kar N, Ring ØE. Gastrointestinal bacteria in rohu, Labeo rohita (Actinopterygii: Cypriniformes: Cyprinidae): Scanning electron microscopy and bacteriological study. Acta Ichthyol Pisc. 2010;40(2):129–35.
Gómez GD, Balcázar JL. A review on the interactions between gut microbiota and innate immunity of fish. FEMS Immunol Med Microbiol. 2008;52(2):145–54.
Luis Balcázar J, Decamp O, Vendrell D, De Blas I, Ruiz-Zarzuela I. Health and nutritional properties of probiotics in fish and shellfish. Microb Ecol Health Dis. 2006;18(2):65–70.
Ringø E, Olsen RE, Mayhew TM, Myklebust R. Electron microscopy of the intestinal microflora of fish. Aquaculture. 2003;227(1):395–415.
Klippel B, Lochner A, Bruce DC, Davenport KW, Detter C, Goodwin LA, et al. Complete genome sequences of Krokinobacter sp. strain 4H-3-7-5 and Lacinutrix sp. strain 5H-3-7-4, polysaccharide-degrading members of the family Flavobacteriaceae. J Bacteriol. 2011;193(17):4545–6.
Wagner N, Tran QH, Richter H, Selzer PM, Unden G. Pyruvate fermentation by Oenococcus oeni and Leuconostoc mesenteroides and role of pyruvate dehydrogenase in anaerobic fermentation. Appl Environ Microbiol. 2005;71(9):4966–71.
Waters CM, Bassler BL. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol. 2005;21:319–46.
Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton J, Greenberg E. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science. 1998;280(5361):295–8.
Gambello MJ, Iglewski BH. Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. J Bacteriol. 1991;173(9):3000–9.
Whitehead NA, Barnard AM, Slater H, Simpson NJ, Salmond GP. Quorum-sensing in Gram-negative bacteria. FEMS Microbiol Rev. 2001;25(4):365–404.
Defoirdt T. Virulence mechanisms of bacterial aquaculture pathogens and antivirulence therapy for aquaculture. Rev Aquac. 2014;6(2):100–14.
Uroz S, Oger PM, Chapelle E, Adeline MT, Faure D, Dessaux Y. A Rhodococcus qsdA-encoded enzyme defines a novel class of large-spectrum quorum-quenching lactonases. Appl Environ Microbiol. 2008;74(5):1357–66.
Wang WZ, Morohoshi T, Ikenoya M, Someya N, Ikeda T. AiiM, a novel class of N-acylhomoserine lactonase from the leaf-associated bacterium Microbacterium testaceum. Appl Environ Microbiol. 2010;76(8):2524–30.
Dong YH, Xu JL, Li XZ, Zhang LH. AiiA, an enzyme that inactivates the acylhomoserine lactone quorum-sensing signal and attenuates the virulence of Erwinia carotovora. Proc Natl Acad Sci U S A. 2000;97(7):3526–31.
Carlier A, Uroz S, Smadja B, Fray R, Latour X, Dessaux Y, et al. The Ti plasmid of Agrobacterium tumefaciens harbors an attM-paralogous gene, aiiB, also encoding N-acyl homoserine lactonase activity. Appl Environ Microbiol. 2003;69(8):4989–93.
Zhang HB, Wang LH, Zhang LH. Genetic control of quorum-sensing signal turnover in Agrobacterium tumefaciens. Proc Natl Acad Sci U S A. 2002;99(7):4638–43.
Park S-Y, Lee SJ, Oh T-K, Oh J-W, Koo B-T, Yum D-Y, et al. AhlD, an N-acylhomoserine lactonase in Arthrobacter sp., and predicted homologues in other bacteria. Microbiology. 2003;149(6):1541–50.
Morohoshi T, Tominaga Y, Someya N, Ikeda T. Complete genome sequence and characterization of the N-acylhomoserine lactone-degrading gene of the potato leaf-associated Solibacillus silvestris. J Biosci Bioeng. 2012;113(1):20–5.
Wang WZ, Morohoshi T, Someya N, Ikeda T. AidC, a novel N-acylhomoserine lactonase from the potato root-associated cytophaga-flavobacteria-bacteroides (CFB) group bacterium Chryseobacterium sp. strain StRB126. Appl Environ Microbiol. 2012;78(22):7985–92.
Mei GY, Yan XX, Turak A, Luo ZQ, Zhang LQ. AidH, an alpha/beta-hydrolase fold family member from an Ochrobactrum sp. strain, is a novel N-acylhomoserine lactonase. Appl Environ Microbiol. 2010;76(15):4933–42.
Ray AK, Roy T, Mondal S, Ringø E. Identification of gut-associated amylase, cellulase and protease-producing bacteria in three species of Indian major carps. Aquacult Res. 2010;41(10):1462–9.
Ray A, Ghosh K, Ringø E. Enzyme-producing bacteria isolated from fish gut: a review. Aquacult Nutr. 2012;18(5):465–92.
Perez-Vilar J, Hill RL. The structure and assembly of secreted mucins. J Biol Chem. 1999;274(45):31751–4.
Shephard KL. Functions for fish mucus. Rev Fish Biol Fisher. 1994;4(4):401–29.
Almagro-Moreno S, Boyd EF. Insights into the evolution of sialic acid catabolism among bacteria. BMC Evol Biol. 2009;9(1):118.
Vimr ER, Kalivoda KA, Deszo EL, Steenbergen SM. Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev. 2004;68(1):132–53.
Vimr ER. Microbial sialidases: does bigger always mean better? Trends Microbiol. 1994;2(8):271–7.
Shakhnovich EA, King SJ, Weiser JN. Neuraminidase expressed by Streptococcus pneumoniae desialylates the lipopolysaccharide of Neisseria meningitidis and Haemophilus influenzae: a paradigm for interbacterial competition among pathogens of the human respiratory tract. Infect Immun. 2002;70(12):7161–4.
Vimr E, Lichtensteiger C. To sialylate, or not to sialylate: that is the question. Trends Microbiol. 2002;10(6):254–7.
Amerongen AN, Bolscher J, Bloemena E, Veerman EC. Sulfomucins in the human body. Biol Chem. 1998;379(1):1–18.
Roussel P, Delmotte P. The diversity of epithelial secreted mucins. Curr Org Chem. 2004;8(5):413–37.
Mills S, Stanton C, Fitzgerald GF, Ross RP. Enhancing the stress responses of probiotics for a lifestyle from gut to product and back again. Microb Cell Fact. 2011;10 Suppl 1:S19.
Bernstein C, Bernstein H, Payne CM, Beard SE, Schneider J. Bile salt activation of stress response promoters in Escherichia coli. Curr Microbiol. 1999;39(2):68–72.
Philipp B. Bacterial degradation of bile salts. Appl Microbiol Biotechnol. 2011;89(4):903–15.
Paul S, Alegre KO, Holdsworth SR, Rice M, Brown JA, McVeigh P, et al. A single-component multidrug transporter of the major facilitator superfamily is part of a network that protects Escherichia coli from bile salt stress. Mol Microbial. 2014;92(4):872–84.
Soupene E, King N, Lee H, Kustu S. Aquaporin Z of Escherichia coli: reassessment of its regulation and physiological role. J Bacteriol. 2002;184(15):4304–7.
McBride MJ. Bacterial gliding motility: multiple mechanisms for cell movement over surfaces. Annu Rev Microbiol. 2001;55(1):49–75.
Braun TF, Khubbar MK, Saffarini DA, McBride MJ. Flavobacterium johnsoniae gliding motility genes identified by mariner mutagenesis. J Bacteriol. 2005;187(20):6943–52.
Rhodes RG, Samarasam MN, Shrivastava A, van Baaren JM, Pochiraju S, Bollampalli S, et al. Flavobacterium johnsoniae gldN and gldO are partially redundant genes required for gliding motility and surface localization of SprB. J Bacteriol. 2010;192(5):1201–11.
McBride MJ, Zhu Y. Gliding motility and Por secretion system genes are widespread among members of the phylum Bacteroidetes. J Bacteriol. 2013;195(2):270–8.
Glew MD, Veith PD, Peng B, Chen Y-Y, Gorasia DG, Yang Q, et al. PG0026 is the C-terminal signal peptidase of a novel secretion system of Porphyromonas gingivalis. J Biol Chem. 2012;287(29):24605–17.
Jung SY, Kang SJ, Lee MH, Lee SY, Oh TK, Yoon JH. Gaetbulibacter saemankumensis gen. nov., sp. nov., a novel member of the family Flavobacteriaceae isolated from a tidal flat sediment in Korea. Int J Syst Evol Microbiol. 2005;55(Pt 5):1845–9.
Cases I, de Lorenzo V, Ouzounis CA. Transcription regulation and environmental adaptation in bacteria. Trends Microbiol. 2003;11(6):248–53.
Swift S, Karlyshev AV, Fish L, Durant EL, Winson MK, Chhabra SR, et al. Quorum sensing in Aeromonas hydrophila and Aeromonas salmonicida: identification of the LuxRI homologs AhyRI and AsaRI and their cognate N-acylhomoserine lactone signal molecules. J Bacteriol. 1997;179(17):5271–81.
Han Y, Li X, Qi Z, Zhang XH, Bossier P. Detection of different quorum-sensing signal molecules in a virulent Edwardsiella tarda strain LTB-4. J Appl Microbiol. 2010;108(1):139–47.
Bruhn JB, Dalsgaard I, Nielsen KF, Buchholtz C, Larsen JL. Quorum sensing signal molecules (acylated homoserine lactones) in gram-negative fish pathogenic bacteria. Dis Aquat Org. 2005;65:43–52.
Milton DL, Hardman A, Camara M, Chhabra SR, Bycroft BW, Stewart G, et al. Quorum sensing in Vibrio anguillarum: characterization of the vanI/vanR locus and identification of the autoinducer N-(3-oxodecanoyl)-L-homoserine lactone. J Bacteriol. 1997;179(9):3004–12.
Moore E, Arnscheidt A, Krüger A, Strömpl C, Mau M. Simplified protocols for the preparation of genomic DNA from bacterial cultures. Molecular microbial ecology manual. 1999;1(1):1–15.
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–72.
Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23(6):673–9.
Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–8.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):0955–64.
Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37 suppl 1:D136–40.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278(5338):631–7.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9.
Haft DH, Loftus BJ, Richardson DL, Yang F, Eisen JA, Paulsen IT, et al. TIGRFAMs: a protein family resource for the functional identification of proteins. Nucleic Acids Res. 2001;29(1):41–3.
Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35 suppl 1:D61–5.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9(1):75.
Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang H-Y, Cohoon M, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33(17):5691–702.
Zhu J, Chai Y, Zhong Z, Li S, Winans SC. Agrobacterium bioassay strain for ultrasensitive detection of N-acylhomoserine lactone-type quorum-sensing molecules: detection of autoinducers in Mesorhizobium huakuii. Appl Environ Microbiol. 2003;69(11):6949–53.
Yu M, Tang K, Liu J, Shi X, Gulder TA, Zhang X-H. Genome analysis of Pseudoalteromonas flavipulchra JG1 reveals various survival advantages in marine environment. BMC Genomics. 2013;14(1):707.
Acknowledgments
This work was supported by the International Science and Technology Cooperation Programme of China (no. 2012DFG31990).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
X-HZ and TC designed and oversighted the study. YZ performed the laboratory work, analyzed the data and drafted the manuscript. JL, KT and MY analyzed the data. All authors read and approved the final manuscript.
Additional files
Additional file 1: Table S1.
Genes predicted to be involved in the general metabolism of Flaviramulus ichthyoenteri Th78T. Table S2. Genes predicted to be involved in the utilization of substances in mucus in Flaviramulus ichthyoenteri Th78T. Table S3. Specific genes predicted to be related to carbohydrate transport and metabolism (G) in Flaviramulus ichthyoenteri Th78T when compared with Gaetbulibacter saemankumensis DSM 17032T and Lacinutrix sp. 5H-3-7-4.
Additional file 2: Figure S1.
Neighbour-joining phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic position of strain Th78T and closely related members of the family Flavobacteriaceae. Bootstrap percentages (>70%) based on 1000 replicates are shown at branch points. Cryomorpha ignava ACAM 647T (GenBank accession no. NR027184) was used as an outgroup (not shown). Bar, 0.02 substitutions per nucleotide position. *indicates the strains whose genome sequence is available.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zhang, Y., Liu, J., Tang, K. et al. Genome analysis of Flaviramulus ichthyoenteri Th78T in the family Flavobacteriaceae: insights into its quorum quenching property and potential roles in fish intestine. BMC Genomics 16, 38 (2015). https://doi.org/10.1186/s12864-015-1275-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-1275-0