
Abstract
Background
Numerous species within the genus Caragana have high ecological and medicinal value. However, species identification based on morphological characteristics is quite complicated in the genus. To address this issue, we analyzed complete plastid genome data for the genus.
Results
We obtained chloroplast genomes of two species, Caragana arborescens and Caragana opulens, using Illumina sequencing technology, with lengths of 129,473 bp and 132,815 bp, respectively. The absence of inverted repeat sequences in the two species indicated that they could be assigned to the inverted repeat-lacking clade (IRLC). The genomes included 111 distinct genes (4 rRNA genes, 31 tRNA genes, and 76 protein-coding genes). In addition, 16 genes containing introns were identified in the two genomes, the majority of which contained a single intron. Repeat analyses revealed 129 and 229 repeats in C. arborescens and C. opulens, respectively. C. arborescens and C. opulens genomes contained 277 and 265 simple sequence repeats, respectively. The two Caragana species exhibited similar codon usage patterns. rpl20-clpP, rps19-rpl2, and rpl23-ycf2 showed the highest nucleotide diversity (pi). In an analysis of sequence divergence, certain intergenic regions (matK-rbcL, psbM-petN, atpA-psbI, petA-psbL, psbE-petL, and rps7-rps12) were highly variable. A phylogenetic analysis showed that C. arborescens and C. opulens were related and clustered together with four other Caragana species. The genera Astragalus and Caragana were relatively closely related.
Conclusions
The present study provides valuable information about the chloroplast genomes of C. arborescens and C. opulens and lays a foundation for future phylogenetic research and molecular marker development.
Similar content being viewed by others


Background
The genus Caragana, which belongs to the subfamily Papilionoideae of the family Fabaceae, includes approximately 100 species, primarily found in arid and semiarid regions of Asia and Europe [1]. The majority of plants in this genus can withstand adverse environmental conditions, including sterile soil, drought, cold, high temperatures, strong winds, and insect and disease damage [1]. China is home to 66 species, 32 of which are endemic. In China, these species are primarily found at high altitudes and in harsh environments, such as shady and semi-shady areas in the northwest, southwest, northeast, and north [2]. Caragana is a deciduous undershrub with extensive adaptability and strong stress resistance, including resistance to wind; it also contributes to sand fixation [3]. In addition, the majority of species in the genus can fix nitrogen via nodules, thereby enhancing soil fertility and preventing dust cyclones and land desertification [4]. Caragana arborescens, also known as Siberian pea shrub, is typically found in Northeast China, North China, and Northwest China [5, 6]. It has a height of 4–5 m. The species, which blooms in May with yellow flowers and pods that mature in midsummer, is typically used as foliage and for garden decoration [7]. Caragana opulens is a shrub with a yellow corolla that thrives in the hills up to 3400 m above sea level in North China, Northwest China, and Southwest China and is distributed throughout these regions [8]. In addition, previous research has demonstrated that numerous species of this genus possess outstanding pharmacological properties, including protective effects against cancer, HIV, rheumatoid arthritis, and hypertension [1, 9, 10]. C. arborescens has been documented in traditional Chinese medicine and is a significant Mongolian medicine used to treat pulmonary hemorrhage and rheumatism [1].
Chloroplast genome data for Caragana plants are extremely limited, with only 14 reports to date. Although the phylogenetic relationships of Caragana plants have been studied based on nuclear ITS and plastid matK, trnL-F and psbA-trnH marker sequence data, the resolution of evolutionary analyses is relatively low, and there are unresolved issues regarding the classification of some medicinal plants, such as Caragana changduensis, Caragana frutex and Caragana polourensis [2, 11,12,13]. Therefore, it is crucial to discover a precise and convenient markers for identifying Caragana plants.
Our understanding of chloroplasts has improved substantially over the past decade, with research on their origin, structure, evolution, and genetic engineering [14,15,16]. It is hypothesized that the cp. is derived from bacterial endosymbiosis in eukaryotes; it is the site of photosynthesis in plants [17]. The chloroplast possesses its own DNA (cpDNA) and genetic system and exist as covalent double-stranded circular DNA in most species [18, 19]. With the rapid advancement of sequencing technology, scientists have discovered that the chloroplast genome contains effective molecular markers, facilitating the precise identification of species. The chloroplast genome is optimal for molecular identification, phylogenetic analyses, and species conservation research [20]. Unlike the nuclear genome, the chloroplast genome is characterized by unisexual inheritance, a simple structure, and more gene copies [19, 21]. Typically, the chloroplast genome is maternally inherited in angiosperms [22]. Its structure is comparatively stable and consists of a large single copy (LSC) and a small single copy (SSC) region separated by two inverted repeats (IRs) [23]. An inverted repeat-lacking clade (IRLC) has been described in leguminous plants [24,25,26,27,28]. Eight species of Caragana plants in the IRLC have been identified [2, 12, 13, 29]. With the refinement and expansion of Caragana chloroplast genome data, the genus will presumably represent a broad IRLC spectral system for scientific investigation. Moreover, chloroplast genome sequence data offer reliable information for analyses of genetic and phylogenetic relationships and population genomics [29,30,31].
In this study, the complete chloroplast genomes of C. arborescens and C. opulens were obtained using Illumina sequencing technology, and their structural properties and phylogenetic relationships were elucidated. This study enriches the chloroplast genomic database of Caragana, which is anticipated to serve as a foundation for systematic research in evolutionary biology and the protection and utilization of Caragana germplasm.
Results
Chloroplast genome assembly and features
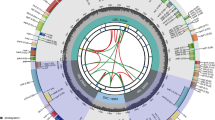
C. arborescens and C. opulens chloroplast genomes were sequenced using the Illumina NovaSeq platform. The whole genome sequence assemblies were 129,473 bp (Fig. 1A) and 132,815 bp (Fig. 1B). Due to the loss of the IR region, neither of the chloroplast genomes had the typical tetrad structure of most angiosperm chloroplast genomes, and their length was shortened accordingly. Nonetheless, the genetic structures were highly similar. In addition, we performed quality control on the sequencing data, and the Q20 and Q30 of C. arborescens and C. opulens were both greater than 90% (Additional file 1). To ensure the correctness of the assembly, we carried out quality control on the assembly results and verified that the two Caragana plants lacked the IR region. (Additional files 2 and 3)

Chloroplast gene maps of C. arborescens A and C. opulens B. The genes within the circle are transcribed in a clockwise direction, and the genes outside the circle are transcribed in the opposite direction. Different color codes are used to depict functionally distinct genomes. In addition, the GC content in the inner ring is shown in light gray, whereas the change in the AT content is shown in dark gray
In the chloroplast genomes of C. arborescens and C. opulens, there were 111 unique genes, including 76 protein-coding genes, 31 tRNA genes, and 4 rRNA genes. The GC contents of C. arborescens and C. opulens were very similar, with values of 34.30% and 34.71%, respectively (Table 1). Four chloroplast genomes in the Caragana genus (C. rosea, C. microphylla, C. kozlowii, and C. korshinskii) with missing IR regions were compared with C. arborescens and C. opulens chloroplast genomes. The total sequence length varied between 129,331 and 133,122 bp. Due to the absence of the IR region, the chloroplast genome length of C. korshinskii was the shortest, at only 129,311 bp, and that of C. rosea was the longest, at 133,122 bp. In addition, there was one more gene in C. arborescens and C. opulens than in other species (tRNA encoded by the trnN-GUU gene), whereas the numbers of protein-coding genes and rRNA genes were consistent among the six plants. C. rosea had the highest GC content in its chloroplast genome, at 34.84% (%), followed by C. kozlowii (34.5%), and C. microphylla had the lowest GC content (34.2%). We also examined variation in the GC content among the three gene types. The GC content was stable and highest in rRNA (over 50%), followed by tRNA, and the GC content of protein-coding genes was approximately 37%. In conclusion, the sequence lengths and gene numbers of the chloroplast genomes of the six Caragana species were generally similar, and the average GC content of the species was approximately 34%, suggesting that the genomes in Caragana were relatively conserved.
Comparable to the genomes of other species, the chloroplast genomes of C. arborescens and C. opulens encode three categories of genes (Table 2). There were 57 genes associated with self-replication, encompassing ribosomal RNA, transfer RNA, and three subunits (large, small, and DNA-dependent RNA polymerase) responsible for encoding chloroplast RNA polymerase. Additionally, there were 44 genes linked to photosynthesis, while the remaining genes were classified as other or unknown genes. In the chloroplast genomes of C. arborescens and C. opulens, 16 genes with introns were detected, one of which, ycf3, had two introns, and the remaining 15 genes (trnK-UUU, trnV-UAC, trnL-CAA, rpoC1, atpF, trnG-UCC, clpP, petB, petD, rpl16, rpl2, ndhB, trnI-GAU, trnA-UGC, and ndhA) had only one intron (Additional file 4). Among these 16 intron-containing genes, the intron lengths in the two species were remarkably similar.
Analyses of repetitive sequences and SSRs
Repeat sites are important in genomic evolution, such as in structural rearrangement and size-based evolution [32, 33]. In this study, we identified repetitive sequences in the chloroplast genomes of C. arborescens and C. opulens and analyzed their content. The chloroplast genome with a repeat length greater than or equal to 30 bp contained four types of repeats: forward (F), palindromic (P), reverse (R), and complementary (C) repeats. In the two plants, 129 (length range: 30–249 bp) and 229 (length range: 30–472 bp) repeats, respectively, were identified (Additional file 5). The length range of 30–49 bp had the highest frequency among all classes of repetitive sequences (C. arborescens: 68.22%, C. opulens: 52.40%).
Structural analysis of the repetitive sequences showed that the C. arborescens genome was composed of 85 forward repeats (65.89%), 36 palindromic repeats (27.91%), 7 reverse repeats (5.43%), and 1 complementary repeat (0.78%) (Additional file 6, Fig. 2). There were no complementary repeats in the C. opulens genome, which consisted of 165 forward repeats (72.05%), 62 palindromic repeats (27.07%), and 2 reverse repeats (0.87%) (Additional file 7, Fig. 2). The majority of repeat sequences existed in the intergenic spacer (IGS) region, and the majority were forward repeats.

Repetitive sequences in the chloroplast genomes of C. arborescens and C. opulens
Plant chloroplast genomes harbor numerous simple sequence repeats (SSRs). This form of sequence is transmitted from parents to offspring. It has a relatively basic structure and low variability. SSRs are therefore efficient molecular markers [34]. Using MISA v1.0, we identified 18 types in the two Caragana species. The chloroplast genomes of C. arborescens and C. opulens contained 277 and 265 SSR loci, respectively (Additional file 8). The proportion of SSRs classified as mononucleotides in the two Caragana plants was highest, with estimates of 57.04% and 63.40%, respectively. Dinucleotide and trinucleotide repeat sequences accounted for 7.58% and 29.24% in the former species, and tetranucleotide repeat sequences accounted for the smallest proportion (6.14%). In the latter, the proportions of dinucleotide, trinucleotide, and tetranucleotide repeat sequences were 4.91%, 28.68%, and 2.64%, respectively, while pentanucleotide repeats represented the smallest proportion, 0.38%.
In C. arborescens, the longest SSR was detected in the ycf1 gene of the chloroplast genome, which was a single nucleotide repeat sequence (A) with a length of 46 bp, whereas the longest SSR in C. opulens was a mononucleotide (T) with a length of 26 bp (Additional file 9). In addition, the distribution of SSRs in coding and noncoding regions was analyzed. Additional file 10 displays that the number of SSRs in the protein-coding region was significantly lower than that in the non-coding region. The majority of these SSRs were A/T single nucleotide repeats; 158 and 167 of the two Caragana species contained A/T, while only one contained C/G (Fig. 3A and B). Similarly, the majority of dinucleotide repeats consisted of AT/AT, resulting in a deviation in base composition, consistent with the finding that the overall AT content of plastids is greater than the GC content [35].

Simple sequence repeats (SSRs) identified in the plastid genomes of two Caragana species. A Number of different SSR types found in the two genomes; B Number of SSRs with different repetition types
Codon usage bias analysis
Plastids exhibit a prevalent codon usage bias. Codon usage bias may influence phylogenetic relationships and the molecular evolution of genes [36]; it can be used to study the origin, mutation model, and evolution of species. We analyzed the codon distribution in all protein-coding genes in these two plant species. The 76 protein-coding gene sequences of the two Caragana species were used to generate 44,033 codons in total. Leucine (Leu) was the amino acid with the highest content, accounting for 10.58% and 10.65%, respectively, followed by codons encoding isoleucine (Ile) (9% and 8.89%), while cysteine (Cys) had the lowest abundance in the two plants (Additional file 11).
We also independently calculated the relative synonymous codon usage (RSCU) values to evaluate the codon usage bias in the chloroplast genomes of the two species (Fig. 4). When the RSCU value is greater than 1.0, the codon is considered optimal. Among the 31 codons with RSCU values greater than 1.0, the AUG codon encoding methionine had the highest utilization bias (C. arborescens RSCU: 2.99 (Fig. 4A), C. opulens RSCU: 2.98 (Fig. 4B)). Tryptophane had no codon usage bias among these 31 codons (only one codon). Except for UUG, which encodes leucine, and AUG, which encodes methionine, the remaining codons terminated in A (12) or U (16) (Additional file 11).

Amino acid frequencies of the chloroplast genomes of C. arborescens A and C. opulens B. The squares below represent all of the codons that encode each type of amino acid; the height of the column above represents the sum of RSCU values for all codons; the height of each column represents the RSCU value for each codon
Sequence divergence analysis
Previous research has demonstrated that highly variable loci in the plastid genome can be used to develop molecular markers [13]. Therefore, DnaSP6 [37] was used to calculate nucleotide diversity (pi) in order to identify highly variable regions in the chloroplast genomes of six Caragana species. In a sliding window analysis, the Pi values of the six plants ranged from 0 to 0.14211, with an average value of approximately 0.01324 (Fig. 5), indicating that the chloroplast genome sequences within the genus had a high degree of similarity. rpl20-clpP, rps19-rpl2, and rpl23-ycf2 were the most highly variable regions based on the pi values. Furthermore, the rpl20-clpP region had the highest nucleotide diversity (pi = 0.1438), followed by the rpl23-ycf2 region (0.10584).

Nucleotide diversity (pi) for 111 genes in two Caragana species
Chloroplast genome sequence divergence was evaluated in C. arborescens and C. opulens. In particular, C. arborescens, C. opulens, C. kozlowii, C. rosea, C. microphylla, and C. korshinskii whole plastid genome sequences were compared with that of C. jubata (Fig. 6). Extremely low sequence divergence among species suggested that the chloroplast genome was conserved. IGS (matK-rbcL), IGS (psbM-petN), IGS (atpA-psbI), IGS (petA-psbL), IGS (psbE-petL), and IGS (rps7-rps1 2) exhibited significant differences among Caragana species. Additionally, the majority of protein-coding regions were highly conserved, with a few exceptions (accD, ycf2, and rps7). This indicates that IGS is responsible for the accelerated evolution of Caragana species.

Chloroplast genome differences in six Caragana species, as determined using mVISTA. The gray arrow indicates the direction of translation. The x-axis represents the coordinates in the chloroplast genome. The y-axis represents the percentage between 50% and 100%. Blue indicates protein coding (exon); light green indicates untranslated region (UTR); orange indicates conserved non-coding sequences (CNSs).
Phylogenetic analysis
To determine the phylogenetic position of Caragana in the family Fabaceae, we generated multiple sequence alignments based on 86 protein sequences commonly found in 23 plastids. In addition to Caragana sequences, additional sequences were obtained from Wisteria (1), Glycyrrhiza (2), Astragalus (1), Calophaca (1), Cicer (1), Medicago (3), Trifolium (3), and Lathyrus (4). Numbers in brackets indicate the number of species in the relevant group.
Based on the chloroplast genomes of 22 Fabaceae and Arabidopsis thaliana (outgroup), phylogenetic trees were constructed by Bayesian and maximum likelihood methods. The phylogenetic trees acquired by the two methods showed a similar topology. Phylogenetic analysis revealed that all samples were classified into three main branches. The following two pairs showed a close relationship: C. microphylla and C. korshinskii as well as C. opulens and C. rosea (Fig. 7). Of note, the genera Astragalus and Caragana were relatively closely related (bootstrap support: 100%) and categorized into Subtrib. Astragalinae. This result was consistent with those of previous studies [2]. From Fig. 7, it can be seen that the divergence of C. arborescens from C. opulens occurred at about 6.9585 Mya.

Phylogenetic tree based on 23 plant chloroplast genomes constructed using Bayesian inference and maximum likelihood. The number after the node represents the bootstrap support value. The GenBank accession numbers are shown after each species. C. arborescens and C. opulens are highlighted in red
Discussion
Caragana is a superb forage native to northwest China and certain plateau regions, with significant value for the enhancement of natural pastures and the establishment of forage bases. Caragana plants are cultivated extensively owing to their resistance to drought, aridity, cold, and heat. Using the Illumina platform, we sequenced the complete chloroplast genomes of two Caragana species in this study. By assembling and annotating these genomes, more detailed information was obtained. Two plastids ranged in size from 129,473 to 132,815 bp and were found in C. arborescens and C. opulens, respectively. Other Caragana genomes have comparable gene structures. In some species, the chloroplast genome reportedly lacks ycf2, rpl23, and accD [38,39,40], whereas these genes were present in Caragana. Throughout plant evolution, several genes have been lost from the plastid genome. For example, the rpl22 and infA genes were lost in some or all legumes [41], infA is an abnormally unstable flowering plant chloroplast gene, and rpl22 encoding ribosomal protein CL22 was lost in cpDNA and relocated to the nucleus [42, 43]. Similarly, in this study, the infA and rpl22 loci were not found in C. arborescens and C. opulens. Recent research has demonstrated, however, that the infA gene is present in the chloroplast genomes of C. jubata, C. erinacea, and C. bicolor [2]. These results suggest that infA exists in some Caragana genomes.
As with the majority of plant species, the plastids of two Caragana species were conserved and no rearrangements were detected. Multiple Caragana species, such as C. microphylla, C. erinacea, and C. intermedia, have reportedly lost their IR region [2, 11, 12]. Similarly, the chloroplast genomes of C. arborescens and C. opulens examined in this study lacked the IR region, and the two plants shared a high degree of similarity in terms of genomic structure, gene deletion, genomic size, gene types, repeat sequence distribution, etc. Moreover, the cpDNA G/C content is a key determinant of inter-specific affinity [2], and the DNA G/C content of the two Caragana species evaluated in this study was highly similar. Numerous repetitive sequences were identified in the plastid genomes of two plants. These sequences are significant genetic markers and are closely associated with the origin and evolution of species [44]. Four types of repeats were identified in this study: complement repeats, reverse repeats, forward repeats, and palindromic repeats. The dispersed repeats were longer than 30 base pairs, and the repeat sequence length in the two Caragana species varied between 30 and 472 base pairs. In addition, SSRs are regarded as essential molecular markers for population genetic research and are widely employed to assess genetic diversity, phylogenetic relationships, and evolution [45]. In total, the chloroplast genomes of two Caragana species contained between 265 and 277 SSRs with a significant A/T bias. In this study, the majority of SSR types were single nucleotide repeats and non-coding regions (IGSs) contained the most SSRs. Similar results have been observed in other Caragana species, including C. rosea, C. microphylla, C. korshinskii, and C. kozlowii [27]. These repeat sequences provide a crucial starting point for the development of genetic indicators for Caragana species and can be utilized for phylogenetic and ecological research.
Codon utilization preference is related to the species of origin and the mutational model. The study of codon bias patterns in chloroplast genomes can shed light on plant phylogenetic relationships, gene expression mechanisms, and molecular evolution [36]. Leucine (Leu) is the most abundant amino acid in C. arborescens and C. opulens (mean of 1,969), and the same trend has been observed in other Caragana species. In addition, our research revealed that the majority of synonymous codons preferred based on RSCU values terminated in A/U, resulting in a high AT content in the gene. This may be explained by natural selection and gene mutation. Codon preference and utilization patterns reflect the evolutionary relationships between species to a limited extent [46]; however, additional research is required.
We identified five IGS with relatively high diversity (pi > 0.01037): rpoC2-rps2, accD-cemA, rps18-clpP, rpoA-rpl36, and rpl2-rpl23. In addition, fragments of the ycf1, rps3, and rps7 genes exhibited comparatively high nucleotide diversity. These variable regions could potentially function as DNA barcode labels for phylogenetic relationships, species recognition, and population genetics research [47,48,49]. The sequence variation in six assembled Caragana plant genomes were then compared, revealing that the coding regions were more conserved than the untranslated regions, corroborating findings from other Caragana species.
A phylogenetic analysis of 23 Fabaceae species simultaneously revealed the phylogenetic position of Caragana and the relationships between Caragana and closely related species. The evolution of the plastid genome (nucleotide changes and structural changes) has been elucidated by phylogenetic methods [50, 51]. Caragana species were monophyletic, and C. arborescens, C. opulens, C. kozlowii, C. rosea, C. microphylla, and C. korshinskii were distinguishable from other species. Our findings may serve as a guide for future research on the evolution of Caragana and the creation of novel molecular markers. Our findings augment the chloroplast genome database for the genus Caragana.
Conclusions
In this study, we assessed phylogenetic relationships based on the chloroplast genome sequences of C. arborescens, C. opulens, and 23 legume species belonging to the IRLC. The characterization of long repeats, SSRs, codon usage bias, and five hypervariable regions provides a basis for future work, such as the development of new molecular markers as well as population genetic and phylogenetic analyses. We analyzed the sequences and structures of the chloroplast genomes of two Caragana species as well as the evolutionary position and relationships with other species in the genus, providing a data basis for more in-depth and comprehensive studies aimed at Caragana species identification, analyses of genetic diversity, and phylogenetic research. Furthermore, our data expand the chloroplast genome database of Caragana.
Materials and methods
DNA extraction, library construction, and sequencing
C. arborescens and C. opulens leaves were collected in Qinghai Province (China) at the following coordinates: C. arborescens: 36° 43′ 24.80′′ N, 101° 44′ 54.11′′ E; C. opulens: 37° 36′ 53.34′′ N, 101° 19′ 18.63′′ E. An improved cetyltrimethylammonium bromide (CTAB) method was used to extract whole genome DNA from fresh leaves of Caragana plants [52]. Both the concentration and integrity of genomic DNA were measured via 1% agarose gel electrophoresis and Qubit 3.0 Fluorometer (Invitrogen, Ghent, Belgium). When the test was successful, the mechanical damage method was utilized to ultrasonically fragment the sample DNA, purify the genomic DNA, and stop the repair process. The DNA fragment size was subsequently determined using agarose gel electrophoresis, and the sequencing library was generated using PCR amplification. The qualified library was sequenced utilizing the Illumina NovaSeq platform to generate 150 bp pair-end reads.
Gene annotation and sequence analyses
Trimmomatic v0.39 [53] was used to remove low-quality reads from the original data. The chloroplast genome sequence was then assembled using SPAdes v3.10.1 (http://cab.spbu.ru/software/spades/) [54] to obtain its seed sequence and for a K-mer analysis of the seed sequences to obtain contigs. SSPACE v2 [55] was used to connect the contigs and obtain the scaffold sequences. GapFiller v2.1.1 [56] was used to supplement the gaps found in the scaffold sequence to assure the integrity of the pseudogenome sequence. After adjusting the corrected pseudogenome sequence, complete chloroplast genome sequences of C. arborescens and C. opulens were obtained. We use software Bowtie2 to align sequencing reads to the genome, and then use SAMtools to calculate genome coverage and insert size distribution. Blast (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to derive the chloroplast genome annotation results for the two plants. Hmmer (http://www.hmmer.org) and ARAGORN [57] (http://ogdraw.mpimp-golm.mpg.de/index.shtml) were used to obtain the rRNA and tRNA annotation information, respectively. OGDRAW [58] (http://ogdraw.mpimp-gol.m.mpg.de/index.html) was used to plot the chloroplast genome maps for C. arborescens and C. opulens. MT211962 and OQ656872 are the NCBI accession numbers for the newly obtained whole chloroplast genome data.
Repeat structure, SSRs, and codon usage analysis
Vmatch Web [59] was used to identify repeats (forward repeats, palindromic repeats, reverse repeats, and complementary repeats). MISA [60] was used to identify SSRs in two Caragana species with the following search parameters: mononucleotides set to ≥ 10 repeat units, dinucleotides ≥ 8 repeat units, trinucleotides, tetranucleotides, pentanucleotides and hexanucleotides ≥ 3 repeat units. CodonW v1.4.2 was used to calculate the relative synonymous codon usage (RSCU) values of protein-coding genes using the default settings.
Comparative genome analysis
C. arborescens and C. opulens whole chloroplast genome sequences were compared with those of C. kozlowii, C. rosea, C. microphylla, and C. korshinskii using mVISTA (Shuffle-LAGAN mode). As a reference, the C. jubata plastid was labeled. In total, 111 gene sequences in C. arborescens and C. opulens were aligned using MEGA7 [61]. To calculate the nucleotide diversity (Pi) values using DnaSP6, the following parameter configurations were utilized: Normal parameters, 200 bp step size and 600 bp window length [62].
Phylogenetic analysis
A phylogenic tree was established using plastome sequences of 20 species (pertaining to IRLC) downloaded from NCBI database and the two species sequenced in this study, with Arabidopsis thaliana as an outgroup. All 23 complete chloroplast genomes were aligned using MAFFT (default parameters), and the aligned sequences were optimized using MACSE. Bayesian inference (BI) and maximum likelihood (ML) methods were used to construct phylogenetic trees. MrBayes was used for BI analysis. The substitution model was selected using ModelFinder [63] and was set to GTR + F + I + G4. The MrBayes analysis was set to run for 1,000 cycles, and the first 25% of cycles were removed as burn-in. The average standard deviation of splitfrequencies was set to > 0.01 [64]. IQ-TREE was used for ML analysis, with the automatic partitioning module and bootstrap analysis set to 1,000 repetitions to evaluate branch support. In addition, we also calculated the divergence time of C. arborescens and C. opulens. Two time standard points were found through Timetree5 [65]: the estimated divergence time between Wisteria floribunda and Glycyrrhiza lepidota is at least 37 Mya, the estimated divergence time between Glycyrrhiza lepidota and Glycyrrhiza glabra is at least 8 Mya. In BEAST v1.8.4 [66], the random local clock and the Yule speciation prior set were used to estimate the divergence time. The posterior distributions of parameters were obtained using MCMC analysis for 10 million generations with a burn-in percentage of 10%. FigTree v1.4.2 [67] was used to visualize the resulting tree and to obtain the divergence time.
Availability of data and materials
The original sequencing data have been submitted to the NCBI database and received GenBank accession numbers MT211962 (C. arborescens), OQ656872 (C. opulens). The data used in this study are available in the public domain (https://www.ncbi.nlm.nih.gov).
References
Meng Q, Niu Y, Niu X, Roubin RH, Hanrahan JR. Ethnobotany, phytochemistry and pharmacology of the genus Caragana used in traditional Chinese medicine. J Ethnopharmacol. 2009;124(3):350–68.
Yuan M, Yin X, Gao B, Gu R, Jiang G. The chloroplasts genomic analyses of four specific Caragana species. PLoS ONE. 2022;17(9): e0272990.
Kang HM, Chen K, Bai J, Wang G. Antioxidative system’s responses in the leaves of six Caragana species during drought stress and recovery. Acta Physiol Plant. 2012;34(6):2145–54.
Fei M. Xiaofan, na, Tingting, Xu. Drought responses of three closely relatedCaraganaspecies: implication for their vicarious distribution. Ecol Evol. 2016;6(9):2763–73.
Moukoumi JL, Hynes RK, Dumonceaux TJ, Town J, Bélanger N. Characterization and genus identification of rhizobial symbionts from Caragana arborescens in western Canada. Can J Microbiol. 2013;59(6):399–406.
Sun X, Ma J, Li C, Zang Y, Tian J, Li L, Li Y, Ye F, Zhang D. Hypoglycemic oligostilbenes from the stems of Caragana sinica. Bioorg Chem. 2023;134:106458.
Kordyum E, Bilyavska N. Structure and biogenesis of ribonucleoprotein bodies in epidermal cells of Caragana arborescens L. Protoplasma. 2018;255(2):709–13.
Ma C, Gao Y, Li Q, Guo H, Zhang J, Shi Y. Water regulation characteristics and stress resistance of Caragana opulens population in different habitats of Inner Mongolia plateau. Ying Yong Sheng Tai Xue bao = J Appl Ecol. 2006;17(2):187–91.
Luo HF, Zhang LP, Hu CQ. ChemInform Abstract: five Novel oligostilbenes from the roots of Caragana sinica. ChemInform. 2010;32:37.
Pan L, Zhang T, Yu M, Shi M, Zou Z. Bioactive-guided isolation and identification of oligostilbenes as anti-rheumatoid arthritis constituents from the roots of Caragana Stenophylla. J Ethnopharmacol. 2021;280:114134.
Liu BB, Duan N, Zhang HL, Liu S, Shi JW, Chai BF. Characterization of the whole chloroplast genome of Caragana microphylla Lam (Fabaceae). Conserv Genet Resour. 2016;8(4):371–3.
Zhang ZL, Ma LY, Yao HB, Yang X, Luo JH, Gong X, Wei SY, Li QF, Wang W, Sun HB. Complete chloroplast genome of Caragana Intermedia (Fabaceae), an endangered shrub endemic to China. Conserv Genet Resour. 2016;8(4):1–3.
Mei J, Haimei C, Shuaibing H, Liqiang W, Amanda C, Chang L. Sequencing, characterization, and comparative analyses of the Plastome of Caragana Rosea var. Rosea. Int J Mol Sci. 2018;19(5):1419.
Kim K, Lee H. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004;11(4):247–61.
Peirong L, Shujiang Z, Fei L, Shifan Z, Hui Z, Xiaowu W, Rifei S, Guusje B, Borm TJA. A phylogenetic analysis of Chloroplast genomes elucidates the relationships of the six economically important Brassica Species comprising the triangle of U. Front Plant Sci. 2017;8:111.
Shen X, Wu M, Liao B, Liu Z, Bai R, Xiao S, Li X, Zhang B, Xu J, Chen S. Complete chloroplast genome sequence and phylogenetic analysis of the Medicinal Plant Artemisia annua. Molecules. 2017;22(8):133.
Neuhaus HE, Emes MJ. Nonphotosynthetic metabolism in plastids. Ann Rev Plant Physiol Plant Mol Biol. 2000;51(1):111–1.
Allen JF. Why chloroplasts and mitochondria contain genomes. Hindawi Publishing Corporation 2003; (1).
Zhang T, Xing Y, Xu L, Bao G, Kang T. Comparative analysis of the complete chloroplast genome sequences of six species of Pulsatilla Miller Ranunculaceae. Chin Med. 2019;14(1):1–4.
Somaratne Y, Guan DL, Wang WQ, Zhao L, Xu SQ. The complete chloroplast genomes of two Lespedeza species: insights into Codon usage Bias, RNA editing sites, and phylogenetic relationships in Desmodieae (Fabaceae: Papilionoideae). Plants. 2020;9(1):51.
Nai-hu HYLC-lMCW. Chloroplast DNA and its application to plant systematic studies. Chin Bull Bot. 1994;11(02):11–25.
Yun S, Yan C, Lv J, Jin X, Zhu S, Li MF, Chen N. Development of Chloroplast genomic resources for Oryza species discrimination. Front Plant ence. 2017;8:1854.
Jiao Y, Ming Y, Chuan N, Xiong-Feng M, Zhong-Hu L. Comparative analysis of the Complete Chloroplast Genome of Four Endangered Herbals of Notopterygium. Genes. 2017;8(4):124.
Palmer JD, Thompson WF. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell. 1982;29(2):537–50.
Sabir J, Schwarz E, Ellison N, Zhang J, Baeshen NA, Mutwakil M, Jansen R, Ruhlman T. Evolutionary and biotechnology implications of plastid genome variation in the inverted-repeat‐lacking clade of legumes. Plant Biotechnol J. 2014;12(6):743–54.
Lei W, Ni D, Wang Y, Shao J, Liu C. Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus Membranaceus. Sci Rep. 2016;6: 21669.
Moghaddam M, Ohta A, Shimizu M, Terauchi R, Kazempour-Osaloo S. The complete chloroplast genome of Onobrychis gaubae (Fabaceae-Papilionoideae): comparative analysis with related IR-lacking clade species. BMC Plant Biol. 2022;22(1):75.
Zhu S, Liu A, Xie X, Xia M, Chen H. Wisteriopsis reticulataCharacterization of the complete chloroplast genome of (Fabaceae): an IRLC legumes. Mitochondrial DNA Part B Resour. 2022;7(6):1137–9.
Zhumanova K, Lee G, Baiseitova A, Shah AB, Park KH. Inhibitory mechanism of O-methylated quercetins, highly potent β-secretase inhibitors isolated from Caragana balchaschensis (Kom.) Pojark. J Ethnopharmacol. 2021;272(421):113935.
Raman G, Park KT, Kim JH, Park SJ. Characteristics of the completed chloroplast genome sequence of Xanthium spinosum: comparative analyses, identification of mutational hotspots and phylogenetic implications. BMC Genom. 2020;21:1–14.
Jianguo Z, Yingxian C, Xinlian C, Ying L, Zhichao X, Baozhong D, Yonghua L, Jingyuan S, Hui Y. Complete chloroplast genomes of Papaver rhoeas and Papaver orientale: Molecular structures, comparative analysis, and Phylogenetic Analysis. Molecules. 2018;23(2):437.
Jo YD, Park J, Kim J, Song W, Hur CG, Lee YH, Kang BC. Complete sequencing and comparative analyses of the pepper (Capsicum annuum L.) plastome revealed high frequency of tandem repeats and large insertion/deletions on pepper plastome. Plant Cell Rep. 2011;30(2):217–29.
Sloan D, Triant D, Forrester N, Bergner L, Wu M, Taylor D. A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol Phylogenet Evol. 2014;72:82–9.
Ebert D, Peakall R. Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol Ecol Resour. 2009;9(3):673–90.
Kuang DY, Wu H, Wang YL, Gao LM, Lu L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome. 2011;54(8):663–73.
Parvathy S, Udayasuriyan V, Bhadana V. Codon usage bias. Mol Biol Rep. 2022;49(1):539–65.
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio J, Guirao-Rico S, Librado P, Ramos-Onsins S, Sánchez-Gracia A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34(12):3299–302.
Jansen RK, Cai Z, Raubeson LA, Daniell H, Boore JL. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci. 2008;104(49):19369–74.
Oliver M, Murdock A, Mishler B, Kuehl J, Boore J, Mandoli D, Everett K, Wolf P, Duffy A, Karol K. Chloroplast genome sequence of the moss Tortula ruralis: gene content, polymorphism, and structural arrangement relative to other green plant chloroplast genomes. BMC Genomics. 2010;11:143.
Wicke S, Schneeweiss GM, Depamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3–5):273–97.
Yen L, Kousar M, Park J. Desmodium stryacifoliumComparative Analysis of Chloroplast Genome of with closely related Legume Genome from the Phaseoloid Clade. Int J Mol Sci. 2023;24(7):6072.
Gantt JS, Baldauf SL, Calie PJ, Weeden NF, Palmer JD. Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO J. 1991;10(10):3073–8.
Millen RS, Olmstead RG, Adams KL, Palmer JD, Lao NT, Heggie L, Kavanagh TA, Hibberd JM, Gray JC, Morden CW. Many parallel losses of infA from Chloroplast DNA during Angiosperm evolution with multiple independent transfers to the Nucleus. Plant Cell. 2001;13(3):645–58.
Xie D, Yu Y, Deng Y, Li J, Liu H, Zhou S, He X. UrophysaComparative Analysis of the Chloroplast genomes of the Chinese endemic Genus and their contribution to Chloroplast Phylogeny and adaptive evolution. Int J Mol Sci. 2018;19(7):1847.
Varshney RK, Sigmund R, Borner A, Korzun V, Stein N, Sorrells ME, Langridge P, Graner A. Interspecific transferability and comparative mapping of barley EST-SSR markers in wheat, rye and rice. Plant Sci. 2005;168(1):195–202.
Chen M, Zhang M, Liang Z, He Q. UncariaCharacterization and Comparative Analysis of Chloroplast Genomes in Five Species Endemic to China. Int J Mol Sci. 2022;23(19):11617.
Henriquez CL, Abdullah, Ahmed I, Carlsen MM, Mckain MR. Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics. 2020;112(3):2349–60.
Zheng G, Wei L, Ma L, Wu Z, Gu C, Chen K. Comparative analyses of chloroplast genomes from 13 Lagerstroemia (Lythraceae) species: identification of highly divergent regions and inference of phylogenetic relationships. Plant Mol Biol. 2020;102(6):659–76.
Park I, Song J, Yang S, Choi G, Moon B. A comprehensive study of the Genus Sanguisorba (Rosaceae) based on the Floral Micromorphology, Palynology, and Plastome Analysis. Genes. 2021;12(11): 1764.
Saski C, Lee SB, Daniell H, Wood TC, Tomkins J, Kim HG, Jansen RK. Complete chloroplast genome sequence of Glycine max and comparative analyses with other Legume genomes. Plant Mol Biol. 2005;59(2):309–22.
Haberle RC, Fourcade HM, Boore JL, Jansen RK. Extensive rearrangements in the Chloroplast Genome of Trachelium Caeruleum are Associated with repeats and tRNA genes. J Mol Evol. 2008;66(4):350–61.
Dierckxsens N, Mardulyn P, Smits G, NOVOPlasty. De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45(4):e18.
Bolger AM, Marc L, Bjoern U. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;15:2114–20.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27(4):578–9.
Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13(6):R56.
Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Oxford University Press 2004 (1).
Stephan G, Pascal L, Ralph B. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nuclc Acids Res. 2019;W1:W59–64.
Kurtz S. The Vmatch large scale sequence analysis software-A Manual. Cent Bioinf. 2010;170(24):391–2.
Beier S, Thiel T, Münch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinf (Oxford England). 2017;33(16):2583–5.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2015;33:1870–4.
Xu F, He L, Gao S, Su Y, Xu L. Comparative analysis of two sugarcane ancestors Saccharum officinarum and S. Spontaneum based on complete chloroplast genome sequences and photosynthetic ability in cold stress. Int J Mol Sci. 2019;20(15): 3828.
Kalyaanamoorthy S, Minh BQ, Wong T, Haeseler AV, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14(6):587–9.
Hu G, Wang Y, Wang Y, Zheng S, Dong N. New Insight into the phylogeny and taxonomy of cultivated and related species of Crataegus in China, based on complete chloroplast genome sequencing. Horticulturae. 2021;7(9): 301.
Kumar S, Suleski M, Craig JM, Kasprowicz AE, Sanderford M, Li M, Stecher G, Hedges SB. TimeTree 5: an expanded resource for species divergence Times. Mol Biol Evol. 2022;39(8): msac174.
Drummond AJ, Rambaut A. BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214–21.
Song J, Cui BK. Phylogeny, divergence time and historical biogeography of Laetiporus (Basidiomycota, Polyporales). BMC Evol Biol. 2017;17(1):102.
Acknowledgements
We would like to thank editor of MogoEdit for his assistance with English language and grammatical editing of the manuscript.
Statement
Our experimental research and field studies on plants comply with relevant institutional, national, and international guidelines and legislation.
Funding
Innovation Platform for the Development and Construction of Special Project of Qinghai Province (Grant Number 2021-ZJ-T05) and National Natural Science Foundation (No. 32160386) of China.
Author information
Authors and Affiliations
Contributions
L.L. designed and executed experiments, completed data analysis, and wrote the first draft of the paper. H.L., J.L., and X.L. contributed to the experimental design and analysis. N.H. and H.W. assisted in sample collection and species identification. W.Z. was the project developer and leader, guiding the experimental design, data analysis, and paper writing and revision. The final text has been read and approved by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Caragana arborescens and Caragana opulens were collected in September 2019 from non-private land, and anyone is permitted to collect these wild plants for research purposes without causing ecological harm. Voucher specimens of Caragana arborescens and Caragana opulens are stored in the herbarium of the School of Ecological and Environmental Engineering at Qinghai University. The botanical identification was performed by the corresponding author, Dr. Zhou. The voucher specimen number for Caragana arborescens is QhST20190078 and for Caragana opulens is QhST20190079.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Statistical table of sequencing data.
Additional file 2: Fig. S1.
Genome coverage of chloroplast genome assembly sequence of Caragana arborescens.
Additional file 3: Fig. S2.
Genome coverage of chloroplast genome assembly sequence of Caragana opulens.
Additional file 4: Table S2.
The intron-containing genes and the length of exons and introns in the chloroplast genomes of two Caragana species.
Additional file 5: Table S3.
Types and numbers of Repeats in chloroplast genome of C. arborescens and C.opulens.
Additional file 6: Fig. S3.
Numbers of four types of repeats found in C. arborescens.
Additional file 7: Fig. S4.
Numbers of four types of repeats found in C. opulens.
Additional file 8: Table S4.
Types and numbers of SSR in chloroplast genome of C. arborescens and C.opulens.
Additional file 9: Table S5.
Distribution of SSRs in cp genome of C. arborescens and C.opulens.
Additional file 10: Fig. S5.
The number of SSRs was found in coding (CDS), and intronic regions, intergenic (IGS), Respectively.
Additional file 11: Table S6.
Analysis of coding ability and codon preference of chloroplast genome.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, L., Li, H., Li, J. et al. Chloroplast genome analyses of Caragana arborescens and Caragana opulens. BMC Genom Data 25, 16 (2024). https://doi.org/10.1186/s12863-024-01202-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-024-01202-4