
Abstract
Background
Corynebacterium diphtheriae complex was formed by the species C. diphtheriae, Corynebacterium ulcerans and Corynebacterium pseudotuberculosis in the recent past. In addition to C. diphtheriae, C. ulcerans and C. pseudotuberculosis species can carry the tox gene, which encodes diphtheria toxin. Currently, three new species have been included in the complex: Corynebacterium rouxii, Corynebacterium silvaticum, and Corynebacterium belfantii. C. rouxii is derived from the ancient Belfanti biovar of C. diptheriae. We provide the complete genome sequences of two non-toxigenic strains C. rouxii isolated from a cat with a purulent infection in Brazil. The taxonomic status and sequence type, as well as the presence of resistance and virulence genes, and CRISPR-Cas system were additionally defined.
Results
The genomes showed an average size of 2.4 Mb and 53.2% GC content, similar to the type strain of the species deposited in Genbank/NCBI. Strains were identified as C. rouxii by the rMLST database, with 95% identity. ANI and DDH in silico were consistent with values above the proposed cut-off points for species limit, corroborating the identification of the strains as C. rouxii. MLST analyses revealed a new ST, which differs from ST-537 only by the fusA allele. No horizontal transfer resistance gene was predicted in both genomes and no mutation was detected in the constitutive genes gyrA and rpoB. Some mutations were found in the seven penicillin-binding proteins (PBPs) detected. The tox gene was not found, but its regulatory gene dtxR was present. Among the predicted virulence genes are those involved in iron uptake and adherence, in addition to the DIP0733 protein involved in epithelial cell adhesion and invasion. The CRISPR-Cas type I-E system was detected in both genomes, with 16 spacer sequences each. Of them, half are unknown according to the databases used, indicating that there is an unexplored reservoir of corynebacteriophages and plasmids.
Conclusions
This is the first genomic study of C. rouxii reported in Brazil. Here we performed taxonomic analysis and the prediction of virulence factors. The genomic analyses performed in this study may help to understand the potential pathogenesis of non-toxigenic C. rouxii strains.
Similar content being viewed by others

Background
The genus Corynebacterium currently includes approximately 140 species [1]. The best-known species of the genus is the human pathogen Corynebacterium diphtheriae, the etiologic agent of diphtheria, a potentially fatal infection that affects the respiratory tract and occasionally skin [2]. Diphtheria toxin (DT) is the main virulence factor of C. diphtheriae codified by the tox gene. This gene is carried by corynebacteriophages which can lysogenize C. diphtheriae species, leading to the conversion of an atoxigenic to a toxigenic isolate [3]. It has already been described, however, that two other species of the genus, Corynebacterium ulcerans and Corynebacterium pseudotuberculosis can also be lysogenized, and therefore can also cause diphtheria. These two species are predominantly isolated from animals [4].
In the recent past, the species C. diphtheriae, C. ulcerans and C. pseudotuberculosis formed the C. diphtheriae complex with potential to cause different infectious processes in humans and animals [5]. The species C. ulcerans and C. pseudotuberculosis can also carry the tox gene. Recently, diphtheria caused by C. ulcerans is more common in industrialized countries, being increasingly recognized as an emerging pathogen [6]. C. pseudotuberculosis is the etiologic agent of caseous lymphadenitis in small ruminants, such as sheep and goats [7]. Although infections rarely affect humans, cases of lymphadenitis related to occupational infections have been reported, affecting rural workers who have frequent contact with the herd or who work in slaughterhouse [8].
Currently, the C. diptheriae complex is formed by three more recently described species: Corynebacterium belfantii, Corynebacterium silvaticum and Corynebacterium rouxii. C. silvaticum is a novel species of the nontoxigenic tox-gene-bearing (NTTB) strains, firstly isolated from lymph nodes of wild boars with severe lesions due to caseous lymphadenitis [2]. The species C. belfantii is derived from the ancient Belfanti biovar of C. diptheriae. Atypical strains of the same biovar gave origin to another species, C. rouxii [5].
C. rouxii strains were isolated between 2011 and 2017 in France from human infections of the skin or peritoneum and one isolated from a dog and described by Badell and collaborators in 2020, based on biochemical and genomic taxonomy [9]. Although biochemically similar to C. belfantii, C. rouxii strains are negative for maltose fermentation. The average nucleotide identity (ANI) between C. rouxii isolates and C. diphtheriae and C. belfantii was 92.4% and 91.4%, respectively, values lower than the currently recommended threshold values for species limit [10, 11].
In this study, two non-toxigenic strains of C. rouxii were collected from a cat with a purulent infection. This is the first genomic study of C. rouxii reported in Brazil. We defined the taxonomic status and sequence type, as well as the presence of resistance and virulence genes, and the CRISPR-Cas system. These results may help to understand the potential pathogenesis of non-toxigenic strains of the species of C. diphtheriae complex.
Results
General features of genome sequencing
The average genome size is approximately 2.4 Mb. The genomes had predicted G + C contents of 53.2%. The assemblies of strains 70,862 and 70,863, as well N50, number of CDS, RNA and median coverage are shown in the Table 1.
Identification of species and genomic taxonomy
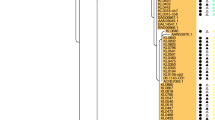
According to the rMLST database, the strains were identified as C. rouxii with 95% identity. The genomic sequencing results showed ANI values of 99.13% and 99.22% to C. rouxii 70,862 and 70,863 strains, respectively, when compared to C. rouxii FRC0190T (Fig. 1). The value of 100% was found when strain 70,862 was compared with strain 70,863 (Fig. 1). In relation to DDH in silico results, as expected, comparisons with the C. rouxii FRC0190T revealed values of 92.70% and 93.10% (Table 2).

Heatmap generated by OrthoANI Tool version 0.93.1 [12] indicating high ANI values (above 99%) between the Corynebacterium rouxii isolates and FRC0190 type strain
MLST characterization
The strains were classified as belonging to the new ST-899. The allelic profile is 37-25-91-26-61-21-17 for atpA, dnaE, dnaK, fusA, leuA, odhA and rpoB genes, respectively.
Phylogenetic analysis
C. rouxii strains 70,862 and 70,863 classifieds as ST-899 clustered with the only strain belonging to the ST-537, isolated from human, in Spain (Fig. 2). Both STs diverge only in one allele. One strain (FRC0810) from the MLST database was excluded because they did not present one of the 7 alleles.

Concatenated phylogenetic tree based on the sequences of 7 housekeeping genes (2544 positions) used in the MLST scheme of Corynebacterium diphtheriae. The distance was inferred based on the Maximum-Likelihood method (Kimura-2 parameters). Bootstrap values with 1000 replicates. The scale bar indicates a 0.01% divergence. Highlighted (bold) are the strains of this study. The type strain of Corynebacterium belfantii FRC0043 was used as an outgroup
Prediction of virulence factors
Pilus genes clusters such as spaA and spaD involved in adherence were also found in both genomes (Table 3). Furthermore, sapD gene, encoding surface-anchored pilus protein, and genes involved in ABC transporter of iron uptake were also found in both genomes. Gene clusters involved in iron uptake were found in addition to sigma A factor of Mycobacterium tuberculosis. The tox gene was not found, but the dtxR regulator gene was found in both strains.
CRISPR-Cas system
A total of two CRISPR-Cas systems were found, one in each strain. Both systems showed a high precision score (evidence level = 4; as high as possible) based on the parameters used by the CRISPRFinder database, which assigns evidence levels from 1 to 4 for spacer repetition and similarity [13]. The CRISPRFinder server identified the type I-E. A total of 16 spacer sequences were found in CRISPR arrays of both strains. Using the CRISPR-Cas + + database, only 1 spacer returned a match for CRISPR-Cas I-E C. diphtheriae bv. mitis PC0646 with 100% similarity. The CRISPRTarget server identified half of the spacers (8/16) with values above 80% identity with a match for C. diphtheriae (n = 4), C. ulcerans (n = 2), Marinobacter nauticus (n = 1) and Syntrophomonas wolfei (n = 1). Eight spacers are unknown according to the database used. Direct repeat consensus sequences in both systems were identical and their conservation is shown in Fig. 3.

Conservation of the direct repeats in type I-E of CRISPR of Corynebacterium rouxii strains. The sequence logo was created by WebLogo 3.7.12 [14]. The height of the letters shows the relative frequency of the corresponding nucleotide at that position
Antimicrobial resistance genes
No antimicrobial resistance genes (ARGs) were detected in both genomes, as well as mutations in the gyrA and rpoB genes, responsible for resistance to quinolones and rifampicin in corynebacteria [15, 16]. Mutations were found within 6 chromosomal penicillin-binding proteins (PBPs) coding sequences comparing to the type strain FRC0190T: PBP1a, PBP1b, PBP2a, PBP2b, PBP2c, and PBP4b (Table 4). No mutations were found in the PBP4 gene.
Discussion
Non-toxigenic strains of C. rouxii were collected in this study and identified according to the complete genome sequences. C. rouxii strains have been isolated from cats, domestic dogs, and free-roaming red fox. Cases of infections with severe otitis in cats and purulent orbital cellulitis, otitis, rhinitis and ulcerative skin lesions in dogs have been reported [5, 9, 17].
The confirmation of species identification of the 2 strains was performed using taxonomic analysis, such as ANI and DDH in silico, in addition to identification by the rMLST database, a methodology based on 53 genes encoding the bacterial ribosome protein subunits (rps genes) [18]. The values obtained for ANI were consistent and above the proposed cut-off point for the species limit (95 ~ 96%) [11, 19]. The strains in this study showed DDH values above the limit (70%) for species definition [20].
MLST analysis revealed a new sequence type. ST-899 has been attributed to the two strains and is closer to ST-537, isolated from a bone biopsy of Homo sapiens in Spain, according to the MLST database. To date, there are only 10 strains of C. rouxii deposited in the MLST database, from France, the United States of America and Spain, with the following STs assigned: 74, 537, 694, 713 and 714. In the phylogenetic analysis using the MLST genes performed with the two Brazilian C. rouxii strains of this study plus nine C. rouxii strains available at the MLST database, it was possible to verify the distribution of Brazilian strains in the same clade as the representative of ST-537 (Fig. 2). ST-899 and ST-537 differ only in the fusA allele, 26 and 54, respectively, according to the MLST database.
Beta-lactam antimicrobials are antibacterial agents that inhibit bacterial cell wall synthesis as a result of their strong covalent binding to PBPs, which catalyze the last reaction of cell wall formation in Gram-positive and Gram-negative bacteria [21]. PBPs are target binding sites for β-lactams [22]. Seven PBPs reported in C. diphtheriae species were found in Brazilian C. rouxii genomes. Of these, six present amino acid mutations compared to the C. rouxii FRC0190T (Table 4), but the correlation between mutations and the phenotype of resistance or susceptibility to antimicrobials will be published in a subsequent study.
Some studies have shown an increase in the rate of antimicrobial resistance among Corynebacterium species [15, 16, 23], but horizontal transfer resistance genes were not predicted in both genomes, according to the Resfinder database. Furthermore, there are no mutations in housekeeping genes, such as gyrA and rpoB, which lead to resistance to quinolones and rifampicin, respectively [15, 16].
Potential virulence factors were also predicted in the genomes (Table 3). The diphtheria toxin was not found in the strains, but its regulatory gene dtxR was present [24]. Among the virulence factors, genes involved in regulation of diphtheria toxin, iron uptake and adherence were found in these non-toxigenic strains. The surface-anchored pilus protein sapD involved in the adherence was found. Sigma A factor of Mycobacterium tuberculosis was found too. This is the main sigma factor, indispensable for growth in both Mycobacterium tuberculosis and Mycobacterium smegmatis, being maintained at a constant level under various stress conditions [25]. All genes involved in iron uptake were found in both genomes. The DIP0733 protein was found in both genomes. This is a multi-functional virulence factor of C. diphtheriae involved in the adhesion, invasion of epithelial cells, and induction of apoptosis [26].
CRISPR-Cas is an adaptive immune system present in many bacteria and archaea, which allows cells to recognize and destroy invading genetic material, such as plasmids or phages. The CRISPR-Cas system is composed of two main parts: the CRISPR cluster (clustered regularly spaced short palindromic repeats) and the CRISPR-associated genes (cas). The CRISPR cluster contains repetitive DNA sequences interspersed by variable spaces that correspond to exogenous DNA sequences. Cas genes encode proteins that form the gene editing complex responsible for cutting invading DNA that corresponds to the variable spaces of the CRISPR cluster [27, 28]. In this study, we found Type I-E CRISPR-Cas systems in both strains. One of the sixteen spacers in each strain is known using the CRISPR-Cas + + database, matching with C. diphtheriae bv. mitis PC0646. Using the CRISPRTarget server, 7 spacers sequences are known. Of these, five matched with corynebacterial (C. diphtheriae and C. ulcerans). One spacer sequence matched with Marinobacter nauticus, a Gram-negative rod found in sea water able to degrade hydrocarbons. According to Carreira and coworkers (2018), this species is a moderate halophile and therefore could potentially be used in high saline wastewater, for which its role as a denitrifier is crucial to remove nitrate and nitrite content from industrial sources [29]. Another spacer sequence matched the Syntrophomonas wolfei, a Gram-negative, slightly helical rod, anaerobic, syntrophic, and fatty acid-oxidizing [30]. Of the 16 spacer sequences, half are unknown, indicating that there is an unexplored reservoir of corinebacteriophages and plasmids. C. rouxii strains share the same repeat consensus sequence shown in the Fig. 3.
Conclusions
This is the first genomic study of non-toxigenic C. rouxii strains isolated from purulent infection in the ear and head injuries of cat in Brazil. Resistance genes and diphtheria toxin gene were not predicted in both strains, but genes involved in the regulation of diphtheria toxin, iron uptake and adherence were found. The adaptive immune system named CRISPR-Cas was found. Analyzes of spacer sequences indicate previous contact with species of the genus Corynebacterium and environmental species such as Marinobacter nauticus and Syntrophomonas wolfei. The genomic analyses performed in this study may help to understand the potential pathogenesis of non-toxigenic C. rouxii strains.
Methods
Origin of bacterial strains
Two C. rouxii strains causing head and ear injuries in a 5-year-old cat were sent to the Laboratory of Diphtheria and Corynebacteria of Clinical Relevance of the State University of Rio de Janeiro. C. rouxii strains 70,862 and 70,863 were deposited at the Collection of Bacteria from Environment and Health (CBAS) of Oswaldo Cruz Foundation (Fiocruz) under the numbers CBAS 829 and CBAS 830, respectively.
Genome sequencing, assembly, and annotation
The genomic DNA extraction from the two strains was performed according to Kit Gen Elute Mammalian Mini-prep (Sigma-Aldrich). Next-generation sequencing was performed using Illumina HiSeq 2500 sequencer (Illumina Inc, USA). A library was constructed with the Nextera XT DNA Library Preparation Kit (Illumina). The reads were assembled de novo using the SPAdes v3.15.4 [31]. The quality control checks in FastQC software [32]. The genomes were annotated using the NCBI Prokaryotic Genome Annotation Pipeline.
Species identification by rps genes and genomic taxonomy
The genomes were submitted to the rMLST database for species identification [18]. The Average Nucleotide Identity (ANI) was calculated according to the OrthoANI algorithm using the OrthoANI tool v0.93.1 [12]. DNA-DNA hybridization (DDH) was determined in silico for these genomes using Genome-to-Genome Distance Calculator (GGDC) v.3.0 by the BLAST method. The results were based on recommended formula 2 (identities/HSP length), the most robust for incomplete draft genomes [33]. The strains were compared with the genomes of type strains of species belonging to the C. diphtheriae complex.
Determination of sequence type
For each strain, the MLST profile was determined by in silico extraction from WGS data using the Institut Pasteur MLST database (https://bigsdb.pasteur.fr/diphtheria/). MLST alleles of C. rouxii strains available in the MLST database were used to build a phylogenetic tree using MEGA version 11.0.10 [34].
Virulence factors and antimicrobial resistance genes
The presence of resistance genes acquired by horizontal transference genes was verified by upload of genomes in the Center for Genomic Epidemiology tool ResFinder version 4 [35]. Mutations in housekeeping genes gyrA and rpoB were investigated from the alignment of genes in the Clustal W program [36] using C. rouxii FRC0190T as a standard for alignment. The prediction of bacterial virulence factors was determined by the VFDB database and analyzed using VFAnalyser [37].
CRISPR-Cas system identification
CRISPRCasFinder was applied to identify the CRISPR-Cas system of genomes. CRISPR arrays with low evidence (0 or 1) were not included in the analyses [13]. The type of CRISPR-Cas cassette was determined following the nomenclature and classification previously described [27]. Spacer sequences were analyzed for their identity using the CRISPRTarget database, and the cut-off score was the default parameters [38], and the CRISPR-Cas + + database with E-value = 0.01 [13]. Spacer hits were selected from the CRISPRTarget and CRISPR-Cas + + databases with a cut-off Identity Cover (IC) score of 0.80 [39]. The conservation of direct repeats was represented by WebLogo4 version 3.7.12 [14].
Phylogenetic relationship
The sequences of the seven housekeeping genes (atpA, dnaE, dnaK, fusA, leuA, odhA and rpoB) of all C. rouxii strains deposited in Institut Pasteur MLST database (https://bigsdb.pasteur.fr/diphtheria/), plus strain type FRC 0190 (Genbank accession number LR738855), were used in the construction of the phylogenetic tree, using the program MEGA 11.0.10 [34]. The sequences were aligned using Clustal W in the Bioedit program [40].
Availability of data and materials
All data generated or analyzed during this study are included in this published article. The whole genome sequences of Corynebacterium rouxii 70862 and 70863 strains were uploaded in NCBI with accession numbers of JARUHM010000000 and JASFAY010000000, respectively.
Abbreviations
- ANI:
-
Average Nucleotide Identity
- CRISPR-Cas:
-
Clustered Regularly Interspaced Short Palindromic Repeats
- DDH:
-
DNA-DNA hybridization
- MLST:
-
multilocus sequence typing
- ST:
-
sequence type
References
Parte AC, Carbasse JS, Meier-Kolthoff JP, Reimer LC, Göker M. List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int J Syst Evol Microbiol. 2020;70:5607–12. https://doi.org/10.1099/IJSEM.0.004332.
Prygiel M, Polak M, Mosiej E, Wdowiak K, Formińska K, Zasada AA. New Corynebacterium species with the potential to produce Diphtheria Toxin. Pathogens 2022;11. https://doi.org/10.3390/PATHOGENS11111264.
Sangal V, Hoskisson PA. Evolution, epidemiology and diversity of Corynebacterium diphtheriae: new perspectives on an old foe. Infect Genet Evol. 2016;43:364–70. https://doi.org/10.1016/J.MEEGID.2016.06.024.
Sharma NC, Efstratiou A, Mokrousov I, Mutreja A, Das B, Ramamurthy T. Diphtheria. Nat Rev Dis Primers. 2019;5:5. https://doi.org/10.1038/S41572-019-0131-Y.
Schlez K, Eisenberg T, Rau J, Dubielzig S, Kornmayer M, Wolf G, et al. Corynebacterium rouxii, a recently described member of the C. Diphtheriae group isolated from three dogs with ulcerative skin lesions. Antonie Van Leeuwenhoek. 2021;114:1361–71. https://doi.org/10.1007/S10482-021-01605-8.
Kawase J, Sakai T, Iwaki M, Umeda K, Fukuma A, Fujisawa N, et al. Rapid detection and discrimination of potentially toxigenic Corynebacterium ulcerans and Corynebacterium pseudotuberculosis by multiplex real-time PCR and amplicon melting curve analysis. J Microbiol Methods. 2022;195: 106454. https://doi.org/10.1016/J.MIMET.2022.106454.
Simpson-Louredo L, Ramos JN, Peixoto RS, Santos LS, Antunes CA, Ladeira EM, et al. Corynebacterium ulcerans isolates from humans and dogs: fibrinogen, fibronectin and collagen-binding, antimicrobial and PFGE profiles. Antonie Van Leeuwenhoek 2013. 2013;105:2. https://doi.org/10.1007/S10482-013-0080-5.
Guimarães AS, Carmo FB, Heinemann MB, Portela RWD, Meyer R, Lage AP, et al. High sero-prevalence of caseous lymphadenitis identified in slaughterhouse samples as a consequence of deficiencies in sheep farm management in the state of Minas Gerais. Brazil BMC Vet Res. 2011;7:68. https://doi.org/10.1186/1746-6148-7-68.
Badell E, Hennart M, Rodrigues C, Passet V, Dazas M, Panunzi L, et al. Corynebacterium rouxii sp. nov., a novel member of the diphtheriae species complex. Res Microbiol. 2020;171:122–7. https://doi.org/10.1016/J.RESMIC.2020.02.003.
Dangel A, Berger A, Konrad R, Sing A. NGS-based phylogeny of diphtheria-related pathogenicity factors in different Corynebacterium spp. implies species-specific virulence transmission. BMC Microbiol. 2019;19:19. https://doi.org/10.1186/S12866-019-1402-1.
Kim M, Oh HS, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64:346–51. https://doi.org/10.1099/IJS.0.059774-0.
Lee I, Kim YO, Park SC, Chun J. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol. 2016;66:1100–3. https://doi.org/10.1099/IJSEM.0.000760/CITE/REFWORKS.
Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46:W246-251. https://doi.org/10.1093/NAR/GKY425.
Crooks G, Hon G, Chandonia J, Brenner S. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90. https://doi.org/10.1101/GR.849004.
Hennart M, Panunzi LG, Rodrigues C, Gaday Q, Baines SL, Barros-Pinkelnig M, et al. Population genomics and antimicrobial resistance in Corynebacterium diphtheriae. Genome Med. 2020;12:12. https://doi.org/10.1186/S13073-020-00805-7.
Ramos J, Valadão T, Baio P, Mattos-Guaraldi A, Vieira V. Novel mutations in the QRDR region gyrA gene in multidrug-resistance Corynebacterium spp. isolates from intravenous sites. Antonie Van Leeuwenhoek Intern J Gen Mol Microbiol. 2020;113:589–92. https://doi.org/10.1007/s10482-019-01353-w.
Hoefer A, Pampaka D, Herrera-León S, Peiró S, Varona S, López-Perea N, et al. Molecular and epidemiological characterization of toxigenic and nontoxigenic corynebacterium diphtheriae, corynebacterium belfantii, corynebacterium rouxii, and corynebacterium ulcerans isolates identified in Spain from 2014 to 2019. J Clin Microbiol. 2021;3:e02410-20. https://doi.org/10.1128/JCM.02410-20.
Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiol (Reading). 2012;158:1005–15. https://doi.org/10.1099/MIC.0.055459-0.
Dangel A, Berger A, Rau J, Eisenberg T, Kämpfer P, Margos G, et al. Corynebacterium silvaticum sp. nov., a unique group of NTTB corynebacteria in wild boar and roe deer. Int J Syst Evol Microbiol. 2020;70:3614–24. https://doi.org/10.1099/IJSEM.0.004195.
Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, da Costa MS, et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol. 2018;68:461–6. https://doi.org/10.1099/IJSEM.0.002516/CITE/REFWORKS.
Bush K, Bradford PA. β-Lactams and β-Lactamase inhibitors: an overview. Cold Spring Harb Perspect Med. 2016;6(8):a025247. https://doi.org/10.1101/CSHPERSPECT.A025247.
Forde BM, Henderson A, Playford EG, Looke D, Henderson BC, Watson C, et al. Fatal respiratory Diphtheria caused by ß-Lactam–resistant Corynebacterium diphtheriae. Clin Infect Dis. 2021;73:e4531-4538. https://doi.org/10.1093/CID/CIAA1147.
Leyton B, Ramos JN, Baio PVP, Veras JFC, Souza C, Burkovski A, et al. Treat me well or will resist: Uptake of Mobile genetic elements determine the Resistome of Corynebacterium striatum. Int J Mol Sci. 2021;22: 7499. https://doi.org/10.3390/ijms22147499.
Zakikhany K, Neal S, Efstratiou A. Emergence and molecular characterisation of non-toxigenic tox gene-bearing Corynebacterium diphtheriae biovar mitis in the United Kingdom, 2003–2012. Euro Surveill. 2014;19: 19. https://doi.org/10.2807/1560-7917.ES2014.19.22.20819.
Sachdeva P, Misra R, Tyagi AK, Singh Y. The sigma factors of Mycobacterium Tuberculosis: regulation of the regulators. FEBS J. 2010;277:605–26. https://doi.org/10.1111/J.1742-4658.2009.07479.X.
Antunes CA, Dos Santos LS, Hacker E, Köhler S, Bösl K, Ott L, et al. Characterization of DIP0733, a multi-functional virulence factor of Corynebacterium diphtheriae. Microbiol (Reading). 2015;161:639–47. https://doi.org/10.1099/MIC.0.000020.
Makarova K, Wolf Y, Iranzo J, Shmakov S, Alkhnbashi O, Brouns S, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18:67–83. https://doi.org/10.1038/S41579-019-0299-X.
Makarova K, Koonin E. Annotation and classification of CRISPR-Cas systems. Methods Mol Biol. 2015;1311:47–75. https://doi.org/10.1007/978-1-4939-2687-9_4.
Carreira C, Mestre O, Nunes RF, Moura I, Pauleta SR. Genomic organization, gene expression and activity profile of Marinobacter hydrocarbonoclasticus denitrification enzymes. PeerJ. 2018;6. https://doi.org/10.7717/PEERJ.5603.
McInerney MJ, Bryant MP, Hespell RB, Costerton JW. Syntrophomonas wolfei gen. nov. sp. nov., an anaerobic, syntrophic, fatty acid-oxidizing bacterium. Appl Environ Microbiol. 1981;41:1029. https://doi.org/10.1128/AEM.41.4.1029-1039.1981.
Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. Using SPAdes De Novo Assembler. Curr Protoc Bioinformatics. 2020;70: e102. https://doi.org/10.1002/CPBI.102.
Andrews S. FastQC - A quality control tool for high throughput sequence data. 2010. https://www.Bioinformatics.Babraham.Ac.Uk/Projects/Fastqc/.
Meier-Kolthoff JP, Carbasse JS, Peinado-Olarte RL, Göker M. TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022;50:D801-807. https://doi.org/10.1093/NAR/GKAB902.
Tamura K, Stecher G, Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol. 2021;38:3022–7. https://doi.org/10.1093/MOLBEV/MSAB120.
Bortolaia V, Kaas RS, Ruppe E, Roberts MC, Schwarz S, Cattoir V, et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother. 2020. https://doi.org/10.1093/jac/dkaa345.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of Progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. https://doi.org/10.1093/NAR/22.22.4673.
Liu B, Zheng D, Zhou S, Chen L, Yang J. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022;50:D912-917. https://doi.org/10.1093/NAR/GKAB1107.
Biswas A, Gagnon J, Brouns S, Fineran P, Brown C. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013;10:817–27. https://doi.org/10.4161/RNA.24046.
Sangal V, Fineran P, Hoskisson P. Novel configurations of type I and II CRISPR–Cas systems in Corynebacterium diphtheriae. Microbiol (N Y). 2013;159:2118–26. https://doi.org/10.1099/MIC.0.070235-0.
Hall TA. BioEdit. A user-friendly Biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
Acknowledgements
Not applicable.
Funding
This study was funded by the Fundação Oswaldo Cruz (Programa Inova Fiocruz/VPPCB-007-FIO-18-2-103), Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (309948/2018-5), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro – FAPERJ (E-26/202.088/2020; E-26/205.901/2022; E26/211.554/2019; E-26/210.285/2021), Sub-Reitoria de Pós-Graduação e Pesquisa da Universidade do Estado do Rio de Janeiro (SR-2/UERJ). The funding body played no role in the design of the study and collection, analysis, interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
MRBA and MABS performed the isolation of the strains, identification by MALDI-TOF and contributed to data generation; LBA performed the extractions; EMDV performed the sequencing of 16 S rRNA and rpoB genes; JNR and PVPB contributed to data generation and analysis, and article writing; JFCV assembled the genomes; LOS and LSS contributed to data collection and interpretation; CHC, CTS and KRC contributed to critical appraisal and article review; MBNS and SB contributed to study design and interpretation; ALMG and VVV contributed to study design, data collection and article review.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The project was submitted to the Research Ethics Committee through Plataforma Brasil (a unified national database of research records involving human subjects) and was approved according to the registry CAAE no. 25847614.8.0000.5259 and technical opinion number 3.728.797.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ramos, J.N., Araújo, M.R.B., Baio, P.V.P. et al. Molecular characterization and phylogenetic analysis of the first Corynebacterium rouxii strains isolated in Brazil: a recent member of Corynebacterium diphtheriae complex. BMC Genom Data 24, 65 (2023). https://doi.org/10.1186/s12863-023-01167-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-023-01167-w