
Abstract
Background
Sporadic cases of infection with non-toxigenic Corynebacterium diphtheriae (C. diphtheriae) isolates have been reported in regions covered by the Diphtheria-Tetanus-Pertussis vaccine, but no information describing the whole genome of non-toxigenic strains collected in China is available. Therefore, in this work, the complete genome of a non-toxigenic strain of C. diphtheriae from a hospital located in southeastern China was performed.
Results
This non-toxigenic isolate belonged to the belfanti biotype and possessed a unique ST (assigned as ST799 in pubMLST). ErmX was present in the genome sequence and this isolate owned the resistance to erythromycin and clindamycin. Genes coding for virulence factors involved in adherence, iron-uptake and regulation of diphtheria toxin were also found. Two genes were involved in the interaction between pathogen and host. The phylogenetic analysis revealed that this newly isolated strain was similar to the strain NCTC10838, CMCNS703 and CHUV2995.
Conclusion
Non-toxigenic C. diphtheriae strain contained virulence factors, thus it is able to cause an infectious disease, aspect that could be clarified by performing the whole genome sequencing analysis.
Similar content being viewed by others

Background
Diphtheriae is usually caused by Coryneabacterium diphtheriae (C. diphtheriae) and it is a potentially lethal disease in children and adults when infected by toxin-producing strains [1]. It spreads among susceptible individuals, resulting in a high mortality in young children without vaccination [2]. Although the vaccine for protection against toxic C. diphtheriae has been available for a long time and infants are immunized with a combination of other vaccines such as Diphtheria-Tetanus-Pertussis (DTP) vaccine, sporadic cases or small outbreaks of diphtheriae still occur, especially in regions with low vaccine coverage [3,4,5,6,7].
The reported C. diphtheriae isolates are categorized as toxigenic and non-toxigenic according to the presence of the diphtheria toxin. The infection cases caused by the toxigenic strains declined after vaccine immunization program, but the current vaccines may not protect susceptible individuals from the non-toxigenic strains, which can also cause severe disease [8, 9]. Thus, the non-toxigenic strains with invasive ability including nontoxigenic but toxin-gene bearing strains should not be ignored [10]. The worst aspect is that the non-toxigenic strains may change to the toxigenic ones through lysogenic conversion [10]. Therefore, routine surveillance of both the toxigenic and non-toxigenic strains of C. diphtheriae is necessary to prevent potential outbreaks. There were four biotypes (mitis, gravis, intermedius and belfanti) in clinical C. diphtheriae isolates, but the belfanti biotype seemed to be rarely reported and appeared later than other biotypes [11].
The molecular genotyping of C. diphtheriae isolates is a useful approach to monitor the transmission or the original isolate during the outbreaks of infectious diseases. Multiple locus sequence typing based on seven housekeeping genes are generally used in C. diphtheriae studies. However, routine genotyping is not enough to evaluate its pathogenicity or possibility to infect host and transmission among individuals. Whole genome sequencing has become more suitable in the investigation of non-toxigenic C. diphtheriae isolates collected in regions covered by the DTP vaccine.
In this study, a non-toxigenic C. diphtheriae strain was collected from the bronchial alveolar lavage fluid collected from a patient aged 57 years [12] who showed symptoms including cough, expectoration and fever at diagnosis. Although non-toxigenic isolates were also reported in China, no information describing the whole genome is available [12,13,14]. Therefore, in this work, the complete genome of C. diphtheriae strain was sequenced, which could help researcher to understand the potential pathogenesis of a non-toxigenic strain.
Results
Whole genome assembly and gene annotation
The isolate contained a circular genome of 2,960,956 bp and a linear plasmid of 35,314 bp. According to the blast results, the linear plasmid showed a sequence identity greater than 99% compared to two C. diphtheriae strains (ChUV2995) and subspecies lausannense strain (CMCNS703). The strains C. sp. NML93–0612 possessed a sequence identity greater than 90% to our strain, but its coverages was 56%. Other strains showed less than 30% coverage (data not shown).
A total of 3108 and 11 pseudogenes were annotated. The characteristic of CRISPR was shown as number of spacers from CRISPR 1 to CRISPR 9: 1–1–1-11–1-2-6-2-1. A total of 79 non-coding RNAs were predicted from the complete genome, and included 15 rRNA, 53 tRNA and 11 other non-coding RNAs.
Identification of species and MLST
The C. diphtheriae strain was identified as C. diphtheriae biotype belfanti through the use of rMLST, with a 97% support. This isolate turned out to be a new type when analyzed by 7 housekeeping genes for determining the MLST type, nearest to ST612 and ST35 in the database. The detailed information for each locus is shown in Table 1. The locus atpA, leuA and rpoB in this study possessed mutations compared to the isolates in the database, when the remaining loci matched exactly to the alleles. The new mutation at locus atpA, leuA and rpoB had been submitted to pubMLST database and this new MLST type was assigned as ST799.
Resistance gene and phenotype of the collected C. diphtheriae
The complete genome analysis revealed that one gene conferring drug resistance (ErmX) coding an rRNA methyltransferase was found. The susceptibility to erythromycin and clincamycin was determined by disk diffusion method. We found this isolated C. diphtheriae was both resistant to erythromycin and clindamycin (supplementary Fig. 1).
Prediction of virulence factors
The gene encoding the diphtheria toxin was not found in this isolate, but the regulation dtxR gene existed. In addition, genes involved in adherence, iron uptake, and regulation of diphtheria toxin were also found in the genome (Table 2). In detail, two genes (srtB for encoding SpaD-type pili and sapD for encoding surface-anchored pilus protein, respectively) were present in genome. Moreover, more copy numbers of genes involved in ABC transporter were also found compared to C. diphtheriae NCTC 13129.
According to the results of the PHI database, two potential virulence factors were predicted, which were not in the database of the virulence factors. The sequence of GE1800 possessed a sequence identity of 99.4% with DIP0733 in the C. diphtheriae strain NCTC 13129. In addition, another gene such as GE2120 shared an identity of 95.5% with GE0813 in the strain CDCE8392.
Phylogenetic analysis based on the whole genome and housekeeping genes
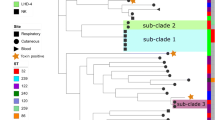
A total of 26 isolates with whole genome sequences were downloaded from NCBI to compare the similarity between the published C. diphtheriae strains and the isolate strain in this region (supplementary Table 1). Twenty-seven whole genome sequences were analyzed including the strain collected in our hospital and the results showed that 1519 genes belonged to the core genes. Then, the wgMLST tree was performed according to these core genes (Fig. 1). The C. diphtheriae isolate collected in this study was more similar to the strain NCTC10838 (Australia, throat swab, biotype belfanti), CMCNS703 (India, nasal swab) and CHUV2995 (Switzerland, broncho-alveolar lavage, biotype mitis or belfanti) than other isolates.

The wgMLST tree based on genomes from database and this C. diphtheriae isolate
A total of 57 C. diphtheriae were collected to extract the sequences from seven housekeeping genes and the evolutionary phylogenetic tree was constructed based on them (Fig. 2). The C. diphtheriae isolate collected in this study was distributed closer to the strains NCTC10838, CMCNS703, CHUV2995 and KL0479.

The evolutionary phylogenetic tree of 57 C. diphtheriae isolates based on 7 house-keeping genes
Discussion
One non-toxigenic C. diphtheriae was collected in this study and identified as C. diphtheriae belfanti according to the complete genome sequence. MLST analysis revealed this new sequence type and potential virulence factors were also predicted in this genome.
The C. diphtheriae isolate collected in this study was identified as the belfanti biotype, which is usually considered as non-toxigenic and proposed with the name C. belfanti [15]. The patient in this study did not show pseudo-membrane, but had symptoms related to an infection of C. diphtheriae including cough, fever and expectoration accompanied with ozena. A study from France revealed that C. belfanti can colonize susceptible individuals such as patients with cystic fibrosis, who can infect each other [16]. In addition, C. belfanti isolates from Algeria are phylogenetically grouped and associated with ozena, indicating that the infection site and symptoms may be specific for C. belfanti [17].
Whole genome sequencing and MLST analysis of isolated strains was essential in investigating the molecular prevalence of pathogens. Sharing the same ST type and core genes among isolates from temporospatial related patients indicated the potential ability of transmission of the non-toxigenic strains. However, this C. diphtheriae strain had unique ST (ST799) with mutations in atpA, leuA and rpoB, whichwas more similar to the ST612 and ST35 according to the published data. However, evidence regarding transmission events related to this isolate was not found during the follow-up [12].
Although the diphtheriae toxin was not found in the isolated strain, its regulatory gene dtxR was present. Once integrated into specific sites by the tox-encoding bacteriophage, the non-toxigenic strain might be converted into the toxigenic isolate in theory [10]. Among the virulence factors, genes involved in adherence, iron uptake and regulation of diphtheria toxin were also found in this non-toxigenic strain. The pili were essential for bacteria to adhere the epithelial cells and there were genes coding for different types of pili in the genome of C. diphtheriae. The spaA-type pili were prevalent in clinical isolates, but the genes for spaD or spaH-type pili were heterogenous as described in previous study [18]. In this isolate, only one gene (srtB) for spaD-type pili were found, indicating that the genes for spaABC-type pili might be absent in some non-toxigenic isolates [19, 20]. Moreover, more copies of genes involved in the ABC transporter were present in this isolate compared to the reference genome (NCTC 13129), suggesting its potential increase in the ability to uptake iron and nutrition [21, 22].
Two genes potentially involved in the interaction between host and pathogens were found in this study. DIP0733 (GE1800 in this isolate) could contribute to the binding of C. diphtheriae to the proteins of the extracellular matrix, thus potentially contributing its escape in immune response [23]. In addition, the DIP0733 protein could increase its ability to invade epithelial cell, as revealed by experiments in an animal model [23, 24]. The ability of C. diphtheriae to interact with epithelial cell is mainly dependent on the C-terminal coiled-coil domain structure of DIP0733, since mutant type strains showed a decreased virulence to invertebrate animals [25]. The C-terminal sequence of GE1800 in this study was completely identical to that of DIP0733, suggesting its potential ability of infection and consequent pathogenesis. Another gene GE2120, which was homologous to GE0813 in the strain CDCE8392, was involved in tellurite resistance. The presence of the GE0813 gene not only enhances the survival of pathogens in the natural environment, but increases the lethality of Caenorhabditis elegans and its survival inside human epithelial cells [26].
A gene encoding rRNA methyltransferase (ErmX) was found in the genome. ErmX can protect the ribosomes from inactivation because it binds to the antibiotics, and it was indeed involved in the resistance to macrolide, lincosamide and streptogramin. Previous studies reported that C. diphtheriae carrying ErmX is closely related to the resistance to macrolide, and the ErmX is the most common gene in macrolide-resistance corynebacterial strains [27,28,29], which was supported by the fact this isolate was resistant to erythromycin and clindamycin in this study.
Conclusions
Non-toxigenic C. diphtheriae strains could be pathogenic and cause sporadic disease. Thus, the analysis of the whole genome sequence could help the understanding of the molecular mechanism associated to the pathogenesis of the diseases.
Methods
Strain isolation and species identification
The C. diphtheriae was collected from the bronchial alveolar lavage fluid collected from a patient aged 57 years who had cough, expectoration, fever and white debris in the larynx at diagnosis. The sample was cultured on a blood agar plate and incubated at 35 °C under 5% CO2 for 24 h. At the end of the incubation time, white colony formed and was analyzed for species identification using IVD model by Matrix-Assisted Laser Desorption/Ionization Time of Flight Mass Spectrometry (VITEK, German).
Genome sequencing and assembly
The bacterium was collected from the blood agar plate, placed in an Eppendorf tube and stored in liquid nitrogen. The genome was extracted using QIAGEN Genomic-tip according to the manufacturer’s instructions (QIAGEN, German). The sequencing data was generated ONT PromethION by LC-Bio [30]. The reads were assembled into sequence by using Canu v1.5 / wtdbg v2.2 software as described previously [31, 32]. The genome sequence was available on NCBI (CP074413).
Determination of multiple loci sequence type
Species identification based on genome was performed using Ribosomal Multi-locus Sequence Typing (rMLST, https://pubmlst.org/species-id) as previously described [33, 34]. Sequence type based on atpA, dnaE, dnaK, fusA, leuA, odhA, and rpoB was analyzed in PubMLST (https://pubmlst.org/organisms/corynebacterium-diphtheriae) [35].
Phylogenetic tree construction based on core genes and housekeeping genes
Whole genome sequences were uploaded into PGAdg-builder (http://wgmlstdb.imst.nsysu.edu.tw/) [36] and a scheme consisting of core genes was established with a cut off value of the occurrence percentage of more than 95%. Then, the wgMLSTtree was established based on the core genes with default parameters (90% coverage and 90% identity).
A combination of 26 genome sequences mentioned above and 30 C. diphtheriae sequences from pubMLST database were analyzed to extract the sequences of seven housekeeping genes (updated by 4th Feb, 2021) to obtain a sequence of 2544 bp length consisting of fragments from atpA (378 bp), dnaE (354 bp), dnaK (345 bp), fusA (360 bp), leuA (384 bp), odhA (381 bp) and rpoB (342 bp). Then, the alignment of the sequences was constructed by clustaW in Mega X. The evolutionary history was analyzed using the Maximum Likelihood method and Tamura-Nei model in Mega X [37]. The bootstrap consensus tree performed from 1000 replicates [38] was used to represent the evolutionary history of the analyzed taxa [3]. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates were collapsed. The initial tree(s) for the heuristic search were automatically obtained by applying the Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura-Nei model, and then by selecting the topology with a superior log likelihood value.
Virulence factors analysis
The whole sequence with the annotated coding sequence was uploaded to the virulence factor database (VFDB, http://www.mgc.ac.cn/VFs/) and analyzed using VFanalyzer [39]. The C. diphtheriae NCTC 13129 was the reference genome used as comparison.
Drug resistance gene and phenotype determination
The assembled genome sequence was uploaded and analyzed using The Comprehensive Antibiotic Resistance Database (https://card.mcmaster.ca/) [40]. The potential gene conferring drug resistance in all bacteria was predicted by the protein homolog model.
The phenotype of antibiotic resistance was determined by disk diffusion method proposed by The European Committee on Antimicrobial susceptibility Testing (https://www.eucast.org/ast_of_bacteria/). In brief, 0.5 McFarland of bacterium was smeared on the blood culture plate. A 6 mm filter paper disk with 2 μg of clindamycin (OXOID, England) or 15 μg of erythromycin (CONT, China) was plated on the culture plates and incubated at 35 °C for 24 h. The inhibition zone diameters were obtained and phenotype was determined based on the breakpoints [41] (https://www.eucast.org/clinical_breakpoints/).
Availability of data and materials
All data generated or analyzed during this study are included in this published article. The whole genome sequence of newly isolated Corynebacterium diphtheriae was uploaded in NCBI with accession number of CP074413.
Abbreviations
- MLST:
-
multi-locus sequence typing
References
Dittmann S, Wharton M, Vitek C, Ciotti M, Galazka A, Guichard S, et al. Successful control of epidemic diphtheria in the states of the former Union of Soviet Socialist Republics: lessons learned. J Infect Dis. 2000;181(Suppl 1):S10–22. https://doi.org/10.1086/315534.
World Health Organization. Diphtheria vaccine: WHO position paper, august 2017 - recommendations. Vaccine. 2018;36(2):199–201. https://doi.org/10.1016/j.vaccine.2017.08.024.
du Plessis M, Wolter N, Allam M, de Gouveia L, Moosa F, Ntshoe G, et al. Molecular characterization of Corynebacterium diphtheriae outbreak isolates, South Africa, march-June 2015. Emerg Infect Dis. 2017;23(8):1308–15. https://doi.org/10.3201/eid2308.162039.
Paveenkittiporn W, Sripakdee S, Koobkratok O, Sangkitporn S, Kerdsin A. Molecular epidemiology and antimicrobial susceptibility of outbreak-associated Corynebacterium diphtheriae in Thailand, 2012. Infect Genet Evol. 2019;75:104007. https://doi.org/10.1016/j.meegid.2019.104007.
Bhagat S, Grover SS, Gupta N, Roy RD, Khare S. Persistence of Corynebacterium diphtheriae in Delhi & National Capital Region (NCR). Indian J Med Res. 2015;142(4):459–61. https://doi.org/10.4103/0971-5916.169212.
Kitamura N, Le TTT, Le LT, Nguyen LD, Dao AT, Hoang TT, et al. Diphtheria outbreaks in schools in Central Highland districts, Vietnam, 2015-2018. Emerg Infect Dis. 2020;26(3):596–600. https://doi.org/10.3201/eid2603.191027.
Maramraj KK, Latha MLK, Reddy R, Sodha SV, Kaur S, Dikid T, et al. Addressing reemergence of diphtheria among adolescents through program integration in India. Emerg Infect Dis. 2021;27(3):953–6. https://doi.org/10.3201/eid2703.203205.
Wagner KS, White JM, Crowcroft NS, De Martin S, Mann G, Efstratiou A. Diphtheria in the United Kingdom, 1986-2008: the increasing role of Corynebacterium ulcerans. Epidemiol Infect. 2010;138(11):1519–30. https://doi.org/10.1017/S0950268810001895.
Indumathi VA, Shikha R, Suryaprakash DR: Diphtheria-like illness in a fully immunised child caused by Corynebacterium pseudodiphtheriticum. Indian J Med Microbiol 2014; 32(4):443–445, dio: https://doi.org/10.4103/0255-0857.142250.
Sharma NC, Efstratiou A, Mokrousov I, Mutreja A, Das B, Ramamurthy T. Diphtheria. Nat Rev Dis Primers. 2019;5(1):81. https://doi.org/10.1038/s41572-019-0131-y.
Czajka U, Wiatrzyk A, Mosiej E, Forminska K, Zasada AA. Changes in MLST profiles and biotypes of Corynebacterium diphtheriae isolates from the diphtheria outbreak period to the period of invasive infections caused by nontoxigenic strains in Poland (1950-2016). BMC Infect Dis. 2018;18(1):121. https://doi.org/10.1186/s12879-018-3020-1.
Yao PP, Wei JC, Mei LL, H.P. Z, Chen C, he HQ et al: pathogen characteristics of one patient carrying Corynebacterium diphtheriae in Zhejiang province (in Chinese). Chinese Journal Of Vaccines And Immunization. 2019;25(3):3.
Liu MZ, Zhang WZ, Shu J, Chen JD, Guan DW, Fu CX et al: [etiologic detection and epidemiological analysis of one suspected case of diphtheria in Guangdong province]. Zhonghua Yu Fang Yi Xue Za Zhi 2011; 45(10):909–911.
Zhou Y, Chen YW, Xie FQ, Jia HM, Zhang HR, Li QW, et al. Investigation on a case of Corynebacterium diphtheriae carriers in Fujian,2019 (in Chinese). Strait J Prev Med. 2020;26(3):3.
Dazas M, Badell E, Carmi-Leroy A, Criscuolo A, Brisse S. Taxonomic status of Corynebacterium diphtheriae biovar Belfanti and proposal of Corynebacterium belfantii sp. nov. Int J Syst Evol Microbiol. 2018;68(12):3826–31. https://doi.org/10.1099/ijsem.0.003069.
Pivot D, Fanton A, Badell-Ocando E, Benouachkou M, Astruc K, Huet F, et al. Carriage of a Single Strain of Nontoxigenic Corynebacterium diphtheriae bv. Belfanti (Corynebacterium belfantii) in Four Patients with Cystic Fibrosis. J Clin Microbiol. 2019;57(5).
Benamrouche N, Hasnaoui S, Badell E, Guettou B, Lazri M, Guiso N, et al. Microbiological and molecular characterization of Corynebacterium diphtheriae isolated in Algeria between 1992 and 2015. Clin Microbiol Infect. 2016;22(12):1005 e1–7.
Broadway MM, Rogers EA, Chang C, Huang IH, Dwivedi P, Yildirim S, et al. Pilus gene pool variation and the virulence of Corynebacterium diphtheriae clinical isolates during infection of a nematode. J Bacteriol. 2013;195(16):3774–83. https://doi.org/10.1128/JB.00500-13.
Ramdhan ND, Blom J, Sutcliffe IC, Pereira-Ribeiro PMA, Santos CS, Mattos-Guaraldi AL, et al. Genomic analysis of a novel nontoxigenic Corynebacterium diphtheriae strain isolated from a cancer patient. New Microbes New Infect. 2019;30:100544. https://doi.org/10.1016/j.nmni.2019.100544.
Tagini F, Pillonel T, Croxatto A, Bertelli C, Koutsokera A, Lovis A et al: Distinct Genomic Features Characterize Two Clades of Corynebacterium diphtheriae: Proposal of Corynebacterium diphtheriae Subsp diphtheriae Subsp nov and Corynebacterium diphtheriae Subsp lausannense Subsp nov Front Microbiol 2018; 9:1743, doi: https://doi.org/10.3389/fmicb.2018.01743.
Draganova EB, Akbas N, Adrian SA, Lukat-Rodgers GS, Collins DP, Dawson JH, et al. Heme binding by Corynebacterium diphtheriae HmuT: function and Heme environment. Biochemistry. 2015;54(43):6598–609. https://doi.org/10.1021/acs.biochem.5b00666.
Sheldon JR, Heinrichs DE. Recent developments in understanding the iron acquisition strategies of gram positive pathogens. FEMS Microbiol Rev. 2015;39(4):592–630. https://doi.org/10.1093/femsre/fuv009.
Antunes CA. Sanches dos Santos L, hacker E, Kohler S, Bosl K, Ott L et al: characterization of DIP0733, a multi-functional virulence factor of Corynebacterium diphtheriae. Microbiology (Reading). 2015;161(Pt 3):639–47. https://doi.org/10.1099/mic.0.000020.
Sabbadini PS, Assis MC, Trost E, Gomes DL, Moreira LO, Dos Santos CS, et al. Corynebacterium diphtheriae 67-72p hemagglutinin, characterized as the protein DIP0733, contributes to invasion and induction of apoptosis in HEp-2 cells. Microb Pathog. 2012;52(3):165–76. https://doi.org/10.1016/j.micpath.2011.12.003.
Weerasekera D, Stengel F, Sticht H, de Mattos Guaraldi AL, Burkovski A, Azevedo Antunes C. The C-terminal coiled-coil domain of Corynebacterium diphtheriae DIP0733 is crucial for interaction with epithelial cells and pathogenicity in invertebrate animal model systems. BMC Microbiol. 2018;18(1):106. https://doi.org/10.1186/s12866-018-1247-z.
Santos LS, Antunes CA, Santos CS, Pereira JA, Sabbadini PS, Luna M, et al. Corynebacterium diphtheriae putative tellurite-resistance protein (CDCE8392_0813) contributes to the intracellular survival in human epithelial cells and lethality of Caenorhabditis elegans. Mem Inst Oswaldo Cruz. 2015;110(5):662–8. https://doi.org/10.1590/0074-02760140479.
Szemraj M, Kwaszewska A, Pawlak R, Szewczyk EM. Macrolide, lincosamide, and streptogramin B resistance in lipophilic Corynebacteria inhabiting healthy human skin. Microb Drug Resist. 2014;20(5):404–9. https://doi.org/10.1089/mdr.2013.0192.
Chagina IA, Borisova O, Mel'nikov VG, Ivashinnikova GA, Pimenova AS. Donskikh EE et al: [sensitivity of Corynebacterium diphtheriae strains to antibacterial preparations]. Zh Mikrobiol Epidemiol Immunobiol. 2014;4:8–13.
Ortiz-Perez A, Martin-de-Hijas NZ, Esteban J, Fernandez-Natal MI, Garcia-Cia JI, Fernandez-Roblas R. High frequency of macrolide resistance mechanisms in clinical isolates of Corynebacterium species. Microb Drug Resist. 2010;16(4):273–7. https://doi.org/10.1089/mdr.2010.0032.
Murigneux V, Rai SK, Furtado A, Bruxner TJC, Tian W, Harliwong I, et al. Comparison of long-read methods for sequencing and assembly of a plant genome. Gigascience. 2020;9(12).
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–36. https://doi.org/10.1101/gr.215087.116.
Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2020;17(2):155–8. https://doi.org/10.1038/s41592-019-0669-3.
Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology (Reading). 2012;158(Pt 4):1005–15. https://doi.org/10.1099/mic.0.055459-0.
Ribosomal Multi-locus Sequence Typing [https://pubmlst.org/species-id]. Access 4 Feb 2021.
Public databases for molecular typing and microbial genome diversity [https://pubmlst.org/organisms/corynebacterium-diphtheriae]. Access 4 Feb 2021.
The web server for building microbial pangenome allele database for molecular fine typing [http://wgmlstdb.imst.nsysu.edu.tw/]. Access 4 Feb 2021.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–26. https://doi.org/10.1093/oxfordjournals.molbev.a040023.
Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39(4):783–91. https://doi.org/10.1111/j.1558-5646.1985.tb00420.x.
Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019;47(D1):D687–D92. https://doi.org/10.1093/nar/gky1080.
The Comprehensive Antibiotic Resistance Database [https://card.mcmaster.ca/]. Access 4 Feb 2021.
Barberis CM, Sandoval E, Rodriguez CH, Ramirez MS, Famiglietti A, Almuzara M, et al. Comparison between disk diffusion and agar dilution methods to determine in vitro susceptibility of Corynebacterium spp. clinical isolates and update of their susceptibility. J Glob Antimicrob Resist. 2018;14:246–52. https://doi.org/10.1016/j.jgar.2018.05.009.
Acknowledgements
Not applicable.
Funding
No funding was received in this study.
Author information
Authors and Affiliations
Contributions
LG performed the isolation of the strain and genomic data analysis. WS and ZS conducted the species identification and genomic extraction. ZY collected the whole genome sequences from database and wrote the draft. PX analyzed the virulent factor, multi-locus sequencing typing and phylogenetic tree. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The informed consent was obtained from the patient. All methods were performed in accordance with the relevant guidelines and regulations. This study was approved by the Ethics Committee and Institutional Review Board of Dongyang People’s Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1 Supplementary Fig. 1
The inhibition zone diameters of tested antibiotics. (A) erythromycin; (B) clindamycin.
Additional file 2.
The accession number information of 26 whole genome sequences involved in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, G., Wang, S., Zhao, S. et al. Whole genome sequence of a non-toxigenic Corynebacterium diphtheriae strain from a hospital in southeastern China. BMC Genom Data 22, 42 (2021). https://doi.org/10.1186/s12863-021-00998-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-021-00998-9