
Abstract
Background
Maize kernel filling, which is closely related to the process of double fertilization and is sensitive to a variety of environmental conditions, is an important component of maize yield determination. Silk is an important tissue of maize ears that can discriminate pollen and conduct pollination. Therefore, investigating the molecular mechanisms of kernel development and silk senescence will provide important information for improving the pollination rate to obtain high maize yields.
Results
In this study, transcript profiles were determined in an elite maize inbred line (KA105) to investigate the molecular mechanisms functioning in self-pollinated and unpollinated maize kernels and silks. A total of 5285 and 3225 differentially expressed transcripts (DETs) were identified between self-pollinated and unpollinated maize in a kernel group and a silk group, respectively. We found that a large number of genes involved in key steps in the biosynthesis of endosperm storage compounds were upregulated after pollination in kernels, and that abnormal development and senescence appeared in unpollinated kernels (KUP). We also identified several genes with functions in the maintenance of silk structure that were highly expressed in silk. Further investigation suggested that the expression of autophagy-related genes and senescence-related genes is prevalent in maize kernels and silks. In addition, pollination significantly altered the expression levels of senescence-related and autophagy-related genes in maize kernels and silks. Notably, we identified some specific genes and transcription factors (TFs) that are highly expressed in single tissues.
Conclusions
Our results provide novel insights into the potential regulatory mechanisms of self-pollinated and unpollinated maize kernels and silks.
Similar content being viewed by others


Background
Maize (Z. mays L.), which has been domesticated over the past ~ 10,000 years, is one of the most important cereal crops worldwide and is grown for use as food, feed and raw material for industry [1, 2]. As a result of breeding progress and artificial selection, the yield of maize has increased extensively over the past hundred years, but further increases are needed to meet the demands imposed by the rapid development of industrialized economies [3]. Pollination, an important and complex process with significant effects on in vivo kernel setting and yield stability, is sensitive to the impacts of floral morphology [4] and water [5, 6], nitrogen [7] and other environmental conditions [8]. Nevertheless, the poor understanding of the regulatory mechanisms underlying pollination is an impediment to improving the pollination rate and thereby increasing yield in maize.
Maize is a monoecious plant with unisexual male and female flowers, and floral development in maize is illustrated using the ABCDE model [9, 10]. The male inflorescence (named tassel) is located in the apex of the plant and generates abundant pollen. The female inflorescence (named ear) is located within the axial vegetative leaves and produces silks [10,11,12]. In the maize pollination period, mature pollen grains are released from anthers in the tassel and drop onto the surface of silks. Then, compatible pollen grains hydrate, germinate, and produce pollen tubes that grow down to the ovules, with complete fertilization following [13]. During the pollination process, maize silks play a vital role in accepting pollen grains for the completion of fertilization, but there are few studies on this topic. Maize kernel filling plays an important role in maize yield determination and mainly involves the conversion of imported sucrose and amino acids into starch and storage proteins in the endosperm [14,15,16]. The developmental pattern of double fertilization has been extensively studied [4, 17]. After double fertilization, the zygote begins to undergo asymmetric cell division to form progenitors of the embryo and endosperm. Then, after further cell division, cell expansion and endoreduplication, the embryo and endosperm enlarge significantly. Genetic studies have identified a large number of genes involved in key steps of the regulation of embryogenesis and the biosynthesis of endosperm storage compounds, such as opaque2 (o2) [18], Shrunken2 [19], and knotted1 (kn1) [20]. However, little is known about the developmental pattern of uncompleted double-fertilized maize ovules, which are known as unpollinated kernels (KUP).
In high-throughput RNA sequencing (RNA-seq) experiments, the total mRNA of collected samples can be extracted and sequenced to determine expression levels of genes. Recently, high-throughput transcriptomic approaches have proven powerful for studying the regulatory networks of cereal kernel development [16, 17, 21]. With the development of RNA-seq technology, the detection of transcript expression levels and particular structures has become more precise, and several pipelines for RNA-seq analysis have been developed [22,23,24]. Meanwhile, the reference maize B73 genome has been improved to version 5 (https://www.maizegdb.org/, Zm-B73-reference-NAM-5.0), which is more complete and accurate than previous versions. The improved version has accelerated the application of genomics and transcriptomics in the genetics and molecular biology fields. In this study, we selected HISAT-StringTie-DESeq2 as an RNA-seq analysis pipeline and conducted RNA-seq to analyse transcript changes in self-pollinated and unpollinated maize ear tissues (kernels and silks) using the maize B73 version 5.0 genome as a reference. Our objective was to preliminarily explore the molecular mechanisms of maize kernel and silk development through the identification, exploration and annotation of differentially expressed transcripts (DETs) between self-pollinated and unpollinated maize kernels and silks, and the analysis of transcription factors (TFs). Ultimately, this work will provide new insights into the molecular mechanisms of maize kernel and silk development.
Results
Overview of RNA-seq data
In this study, self-pollinated and unpollinated maize ear tissues (kernel and silk; KSP, KUP, SSP and SUP) were selected for RNA sequencing with two biological replicates (Fig. 1). Initially, a total of 215.62 Gb of raw data were obtained after completing the paired-end sequencing protocol. We filtered the raw reads according to a quality score of less than 20 and removed adaptor sequences with fastp software. The Q-scores for more than 97.44% of the reads were Q20, and 36,731,405 ~ 43,427,959 paired reads were obtained for each group of samples (Table 1). We mapped these clean reads to maize reference genome sequences using the HISAT2 procedure. Among the mapped reads, divergent alignment rates were observed between the kernel and silk samples: the kernel samples showed rates of 78.48% ~ 81.79%, and the silk samples showed rates of 49.16% ~ 61.22%. In particular, the SSP alignment rate was 49.16% ~ 49.47%, which may have been the result of senescence in the silks; these results are consistent with previous reports [25,26,27]. After the completion of the HISAT-StringTie-Ballgown analysis pipeline, the FPKM value matrix was obtained with the default stringent criteria of Ballgown. As shown in Fig. 2A, the two biological replicates of each sample were strongly correlated, and only one transcript was detected for most genes (58.54%) (Fig. 2B). To validate the differential expression results from our transcriptome sequence data analysis, the expression of 10 randomly selected DETs with only one transcript was evaluated by qRT-PCR. The selected transcripts included WRKY transcription factor 74, a gibberellin receptor GID1L2 precursor, asparagine synthetase3, sweet4c and others (Fig. 3, Additional file 1). The expression levels determined by qRT-PCR were in agreement with the changes in transcript abundance determined by RNA-seq analysis, which suggested that our transcriptome profiling data were highly reliable.

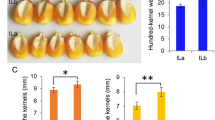
Phenotypes of self-pollinated and unpollinated ears. A Comparisons of self-pollinated and unpollinated ears, B self-pollinated and unpollinated kernels, and C self-pollinated and unpollinated silks

Summary of the RNA-seq data from this study. A The PCA plot for all samples. B Proportions of transcripts detected from different genes in all samples. C Venn diagram of exclusively detected transcripts. D Venn diagram of differentially expressed transcripts in kernels and silks

Quantitative RT-PCR (qRT-PCR) validation of differentially expressed genes (DETs) between self-pollinated and unpollinated maize ears. Error bars indicate standard error (SE) of three biological replicates
Transcriptional changes in self-pollinated and unpollinated maize ear tissues
To perform a more detailed analysis of the transcriptional changes in self-pollinated and unpollinated maize ear tissues, genes with low expression (FPKM ≤1) were filtered. In total, 24,030 transcripts (20.36% of all maize transcripts) were detected in all samples (Additional file 2), 13,406 transcripts were common to more than one group, and 1011, 1183, 916 and 381 transcripts were exclusively detected in KSP, KUP, SSP and SUP, respectively (Fig. 2C). Subsequently, we annotated the transcripts exclusively detected in KSP, KUP, SSP or SUP. Notably, we found that a large number of transcripts associated with starch biosynthetic and metabolic processes and carbohydrate metabolism showed enrichment in KSP, whereas in KUP, most of the transcripts detected exclusively in these kernels were related to plant-type cell wall organization. In silks, transcripts associated with the regulation of unidimensional cell growth and pollen tube growth were detected in SSP, but only xyloglucosyl transferase activity, which is important in maintaining cell homeostasis, was obtained for SUP (Additional file 3).
Functional characterization of DETs in self-pollinated and unpollinated maize ear tissues
To obtain a complete list of the DETs from self-pollinated and unpollinated maize ear tissues, we selected the prepDE Python script to obtain transcript count matrices. DESeq2 with stringent criteria (log2FC > 2 or log2FC < − 2 and p.adj < 0.05) was used to identify significant DETs. As a result, 5285 and 3225 DETs were determined to be significantly differentially expressed in the kernel (KSP vs KUP) and silk (SSP vs SUP) groups, respectively (Fig. 2D). Relative to KUP, there were 2331 upregulated DETs and 2954 downregulated DETs in KSP, and relative to SUP, there were 1782 upregulated DETs and 1443 downregulated DETs in SSP (Additional file 4). To explore which biological processes play key roles after pollination in kernels and silks, GO enrichment analysis of DETs was carried out. In the kernel groups (KSP vs KUP), a large number of upregulated KSP DETs were enriched in starch biosynthetic and metabolic processes, cellular glucan metabolic processes, and rRNA metabolic processes. More importantly, the most enriched cellular component categories of the upregulated KSP DETs were the cytosolic ribosome and amyloplast (Fig. 4A). However, a large number of downregulated DETs from KSP showed annotations related to the responses to jasmonic acid, salicylic acid and chitin, and the mainly enriched cellular component category of downregulated DETs from KSP was plant-type cell wall (Fig. 4B). Additionally, in the kernel group, we found a large number of DETs involved in metabolism, genetic information processing, environmental information processing and cellular processes, and more KEGG pathways related to metabolism were associated with the upregulated KSP DETs than with the downregulated KSP DETs; these pathways included the biosynthesis of amino acids, carbohydrate metabolism and fatty acid metabolism, among others (Fig. 5A). More strikingly, more KEGG pathways involved in transcription, translation, cell growth and death, DNA replication and repair were associated with the upregulated DETs in KSP. These results are consistent with the development of kernels.

The most significantly enriched Gene Ontology (GO) annotations of DETs in self-pollinated and unpollinated maize ear tissues. A The most enriched GO terms of upregulated KSP DETs. B The most enriched GO terms of downregulated KSP DETs. C The most enriched GO terms of upregulated SSP DETs. D The most enriched GO terms of downregulated SSP DETs. The horizontal axis indicates the number of each GO term present in the DET dataset

Kyoto Encyclopedia of Genes and Genomes (KEGG) functional classification of DETs between self-pollinated and unpollinated maize ears. A Self-pollinated and unpollinated kernels. B Self-pollinated and unpollinated silks. Values beside the bars represent the numbers of components in each pathway present in the DET dataset
We performed GO and KEGG pathway enrichment analyses of the silk groups to explore the biological processes that play key roles after pollination. According to the GO enrichment analysis results, the regulation of cell growth and cell tip growth, pollen tube growth and the regulation of unidimensional cells were annotated among the upregulated SSP DETs, and the most enriched cellular component categories among the upregulated SSP DETs were pollen tubes and cell projections (Fig. 4C). However, photosynthesis, cutin biosynthetic process and plant-type cell wall organization were annotated categories among the downregulated SSP DETs (Fig. 4D). In addition, we found a large number of metabolic, genetic information processing, environmental information processing and cellular process KEGG pathways among the silk DETs. Pathways involved in protein folding, sorting and degradation were more enriched among the upregulated SSP DETs, whereas pathways involved in DNA replication and repair were more enriched among the downregulated SSP DETs (Fig. 5B).
Notably, the results of the enrichment analysis of DETs were similar to those obtained for transcripts exclusively detected in kernels and silks, with enrichment detected for categories such as starch biosynthetic and metabolic processes, the regulation of unidimensional cell growth, and pollen tube growth (Fig. 4, Additional file 3). This similarity may be explained by the fact that most of the DETs were derived from genes expressed specifically in different stages of kernels and silks.
Expression levels of senescence- and autophagy-related genes
Researchers have found that preventing pollination induces the initiation of senescence in maize leaves, which is always accompanied by cell death [28]. According to the enrichment analysis results, the DETs from kernels and silks were significantly enriched in cell growth and death pathways. Therefore, we focused on the expression of senescence-related genes in self-pollinated and unpollinated maize ear tissues. In the public leaf senescence database (https://bigd.big.ac.cn/lsd/), we observed 70 senescence genes with genomic sequences corresponding to 134 transcripts in maize. Among these genes, 60 genes (71 transcripts) were expressed in maize kernels and silks (Additional file 5). Moreover, the expression levels of senescence genes were clearly different between maize kernel and silk tissues, and more senescence genes were highly expressed in maize silks than in maize kernels.
Autophagy is a primary intracellular degradation pathway that contributes to nutrient recycling and is related to senescence [29]. To observe the expression levels of autophagy-related genes, we obtained 40 autophagy-related genes corresponding to 109 transcripts from the public database and determined the expression of these genes in maize ear tissues. As shown in Additional files 6, 35 autophagy-related genes (57 transcripts) were expressed in maize kernels and silks. Similar to the senescence-related genes, the majority of autophagy-related genes exhibited higher expression in maize silks than in kernels. Overall, these results indicate that the expression of autophagy-related genes and senescence-related genes is prevalent in maize kernels and silks and that pollination changes the expression levels of several senescence-related and autophagy-related genes in maize kernels and silks.
Identification of TFs associated with maize ear tissue development
The regulation of gene expression is essential for plant growth and development, governing the perception and responses of plant cells and tissues to different stimuli. TFs are key genomic regulatory elements that play crucial roles in regulating the expression of related genes. Here, we extracted TFs from the DETs to explore the expression of a common set of TF families in self-pollinated and unpollinated maize ear tissues. Strikingly, a total of 518 differentially expressed TFs from 46 families were identified in the four maize ear tissues (Additional file 7), and several small TF family genes were found to be upregulated in one of the tissues (Fig. 6), which implied that they may play a role in the development of only one of the four tissues examined in our study. Notably, the TF family members with high relative expression differed among different tissues. As shown in Fig. 6, most TF family members, such as bHLH, C2H2, ERF, GRAS, GRF, TCP, and WRKY TFs, were highly expressed in KUP tissues relative to the other studied tissues, but some TF family members, such as C3H, and GATA TFs, were highly expressed in KSP, SSP and SUP tissues.

Numbers and percentages of upregulated TFs in different tissues: we calculated the percentage of upregulated TFs in each tissue/all upregulated TFs
Discussion
In the present study, we performed RNA-seq to explore the molecular mechanisms underlying development in self-pollinated and unpollinated maize kernels and silks, and the most recent and accurate reference genome of maize B73 was used to map clean paired-end reads. Our results showed that at least 18,158 genes with 24,030 transcripts were required to program maize kernel and silk development. Over 40% of the genes produced more than two transcripts during this process. Global comparisons of gene expression highlighted significant changes in the gene expression patterns of kernels and silks after pollination. We also found some specific genes and TFs that were highly expressed in only one of the studied tissues.
Genes involved in the development of KSP and KUP
Large-scale gene expression analyses can be implemented through GO and KEGG pathway enrichment analyses, and the integration of these analyses can provide abundant information about the regulation of gene networks at the cellular level. In maize, upon the completion of pollination, the embryo and endosperm begin to form gradually in normal kernels, and sucrose and amino acids are quickly converted into starch and storage proteins in the endosperm, with the starch accumulation rate reaching a maximum at 20 DAP [16]. According to the results of GO and KEGG enrichment analyses, a large number of DETs involved in starch biosynthetic and metabolic processes and carbohydrate metabolism were highly expressed in KSP. Starch is the main component of mature maize kernels, and starch content is one of the main targets of maize breeding. Starch biosynthesis requires the proper execution of a series of coordinated enzymes [30], and the enzymes ADP-glucose pyrophosphorylase (AGPase), soluble starch synthase (SSS) and starch branching enzyme (SBE) are the major enzymes that catalyse starch biosynthetic substrate production and starch chain elongation and branching [30]. The starch synthase gene zSSIIa has been shown to encode granule-bound starch synthase I (also called Waxy protein) in the maize endosperm. Our results showed that zSSIIa was highly expressed in KSP, whereas it showed almost no expression in KUP, SSP and SUP (Additional file 8). Zmdull1 encodes a starch synthase, most likely starch synthase II, which plays a determinant role in the biosynthesis of endosperm starch; it was highly expressed in KSP, similar to zSSIIa. AGPase provides ADP-glucose as the glucosyl donor for starch synthesis, and Brittle2 (Bt2) encodes a characteristic AGPase in maize endosperm starch biosynthesis [7]. In this study, the expression of this gene was not detected in any sample, but another AGPase gene, Shrunken2, was highly expressed in KSP [31], whereas it showed almost no expression in KUP, SSP or SUP. In addition, there were other genes involved in the starch biosynthesis pathway that were highly expressed in KSP, such as Shrunken1, sucrose synthase-1 (Sus1), and starch branching enzyme1 (Sbe1) [17].
In maize, the seeds store most amino acids as proteins rather than as free amino acids in the endosperm. Zeins are the most important seed storage proteins in maize endosperm and can be divided into α, β, γ, and δ zeins [32]. Previous research has confirmed that there are 30 α, 1 β, 3 γ, and 1 δ zein genes in B73 bacterial artificial chromosomes (RefGen_v2) and that three-quarters of zein genes are highly expressed in endosperm [17]. Maize o2 makes a large contribution to the synthesis of a 22-kD α-zein protein and several other zein proteins in maize endosperm, and mutations of the o2 gene result in small, unexpanded protein bodies in maize endosperm [33]. In this study, we found that the maize o2 gene was highly expressed only in KSP and that zein genes were significantly upregulated in KSP. These results provide preliminary evidence that the expression pattern of zein genes might be regulated by o2. In KUP, genes involved in starch and zein biosynthesis showed low or no expression, whereas genes involved in senescence and autophagy showed relatively high expression. In maize B73 leaves, free glucose and starch accumulation, chlorophyll loss and senescence have been shown to be significantly triggered by the prevention of pollination [34]. In this study, the highly expressed DETs in KUP were enriched in GO categories related to the responses to jasmonic acid, salicylic acid and chitin, and a large number of TFs were highly expressed. Likewise, we found plant-specific TCP TF family members containing a basic helix-loop-helix (bHLH) TCP domain that were highly expressed in the KUP samples [35]. In plants, jasmonic acid, salicylic acid and chitin are related mainly to plant disease and insect resistance [36], and in maize leaves, jasmonic acid is known to promote senescence mainly through the degradation of chlorophyll [37]. TCP proteins mainly regulate plant proliferation and cell division, and the overexpression of several TCP genes can disturb plant tissue development [38, 39]. Considering these findings together, we speculate that abnormal development and senescence appeared in KUP and that a larger number of TFs were specifically expressed to regulate kernel development.
Genes involved in the regulation of SSP and SUP
Silks anchored to each ovule are a necessary component of maize ears, and the function of silks is equivalent to that of the stigma in typical flowering plants in supporting pollen hydration and germination. After pollen grain hydration, the silks form a pollen tube that penetrates the cortical parenchyma and reaches the ovule [40]. In this study, a large number of DETs involved in the regulation of cell growth and cell tip growth, pollen tube growth and the regulation of unidimensional cells were annotated in the group of upregulated SSP DETs. This implies that maize pollination is complete but that pollen tube growth is ongoing. The nutrient-rich, fluid-filled properties of maize silks facilitate their fertility-related functions but simultaneously make the tissue vulnerable to most maize fungal pathogens and insect pests [40]. A maize glycine-rich protein (ZmGRP5) has been identified to function in maintaining the structure of silks [40]. We found that this gene appeared to be relatively highly expressed in SSP and SUP, especially in SSP. Previous studies showed that two types of TFs function in maintaining the development of maize silks (ZmP1/ZmP2 and ZmbZIP25) [41, 42]. Our results indicated that these two types of TFs were highly expressed in SSP and SUP. Moreover, we found additional TFs that were highly expressed in SSP and SUP, such as MYBs and WRKYs; the functions of these TFs in maize silk development are unclear and require further study. Additionally, we found different genes related to senescence and autophagy that were highly expressed in SSP and SUP and even in kernels. The detailed mechanisms of senescence and autophagy during pollination need to be further investigated in the future.
Conclusions
In this study, RNA-seq analysis was conducted to investigate the development of self-pollinated and unpollinated maize kernels and silks. We found that a large number of genes involved in key steps of the biosynthesis of endosperm storage compounds were upregulated after pollination. Furthermore, abnormal development and senescence appeared in KUP. We also identified several genes with functions in maintaining the structure of silks that were highly expressed in silks. In addition, we identified various TFs expressed in self-pollinated and unpollinated maize kernels and silks, especially in KUP. This large collection of genes provides a rich resource for future maize kernel and silk development studies, which will greatly enhance our understanding of the genetic control of early seed development in maize.
Methods
Plant materials and growth conditions for field experiments
KA105, a superior female inbred line of two nationally approved commercial hybrids (SD650: No.20200261; SD620: No.20200264), was selected from the Shaan A group cultivated by Northwest A&F University [43]. The plants were cultivated at approximately 67,500 plants/ha in summer in Yangling, Shaanxi Province, China. Field experiments were carried out under normal field management without water or nutrient stress. The ears were bagged with Kraft paper bags before the silking stage. After silking, several plants were manually self-pollinated, whereas others were left bagged. Two biological replicates of self-pollinated and unpollinated ears were harvested 20 days after pollination (DAP). Then, kernels and silks were separated from maize ears, frozen in liquid nitrogen and stored at − 80 °C until RNA extraction. The examined samples were referred to as self-pollinated kernels (KSP), unpollinated kernels (KUP), self-pollinated silks (SSP) and unpollinated silks (SUP).
RNA isolation, cDNA library construction and RNA-seq
Total RNA from all samples was extracted with an RNA Sample Total RNA Kit (Tiangen, China), and a 2100 Bioanalyzer was then used to evaluate RNA quality. Qualified RNA samples were digested with DNase I (Takara, Japan) at 37 °C for 30 min. Dynabeads Oligo (dT) 25 (Life, USA) was used for mRNA purification. Sequencing libraries were constructed according to the manufacturer’s instructions of the employed NEBNext Ultra™ RNA Library Prep Kit for Illumina (NEB, USA). Then, the generated libraries were sequenced on the Illumina HiSeq 2500 platform to generate 125 bp short paired-end reads.
RNA-seq data analysis
Fastp software was used to obtain clean reads by trimming adaptor sequences and removing low-quality reads (quality score < 20) from the raw reads [44]. Then, clean reads were mapped to the maize genome sequence (B73_RefGen_v5, https://www.maizegdb.org/) with HISAT2, and SAM files were sorted and converted to BAM files using SAMtools. StringTie was utilized to assemble transcripts and to estimate the expression of the transcripts based on the BAM files. The fragments per kilobase of transcript per million fragments mapped reads (FPKM) value matrix of all samples was extracted with the R package Ballgown. For differential expression analysis, we used the prepDE Python script to obtain transcript count matrices, and DESeq2 with stringent criteria (log2FC > 2 or log2FC < − 2 and p.adj < 0.05) was used to confirm significant differences in transcript expression (DETs). The sequence data from this study can be found in the NCBI Sequence Read Archive under BioProject ID PRJNA745969.
TF and transcript function analysis
The maize TF list was retrieved from PlantTFDB (http://planttfdb.cbi.pku.edu.cn). To analyse the potential functions of the proteins corresponding to the obtained transcripts, we first reannotated all maize proteins. Briefly, all maize (B73_RefGen_v5) proteins were functionally annotated according to an updated list of Gene Ontology (GO) terms, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, Pfam/SMART domains and Clusters of Orthologous Groups (COG) functional categories by using eggnog-mapper to build org.Zmays.eg.db [45]. The R package clusterProfiler was used to identify enriched GO terms with a cut-off of P-value < 0.05 [46]. For KEGG pathway analysis, the amino acid sequences of downregulated and upregulated transcripts were uploaded to BlastKOALA (https://www.kegg.jp/blastkoala/), and BLASTP analysis was then performed to obtain potential KEGG pathways.
Quantitative reverse transcription RT-PCR analysis (qRT-PCR)
Approximately 10 DETs identified among the two groups of samples were verified by qRT-PCR performed on a QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher, America) with a SuperReal PreMix Plus kit (SYBR Green) (Tiangen, China). Maize tubulin was used as an internal reference gene to normalize the relative expression of randomly selected DETs. Gene-specific primers were designed based on maize gene nucleotide sequences using Primer 5.0 (Additional file 1). All qRT-PCR experiments were performed using three biological replicates. The relative expression levels of each gene were calculated using the 2-∆∆CT method in comparison with the control.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the NCBI Sequence Read Archive (RNA sequencing data: BioProject PRJNA745969, https://dataview.ncbi.nlm.nih.gov/object/PRJNA745969).
Abbreviations
- DETs:
-
Differentially expressed transcripts
- TFs:
-
Transcription factors
- RNA-seq:
-
RNA sequencing
- KUP:
-
Unpollinated kernels
- KSP:
-
Self-pollinated kernels
- SUP:
-
Unpollinated silks
- SSP:
-
Self-pollinated silks
- 20 DAP:
-
20 days after pollination
- FPKM:
-
Fragments per kilobase of transcript per million fragments mapped reads
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- COG:
-
Clusters of Orthologous Groups
- qRT-PCR:
-
quantitative reverse transcription PCR
References
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science (New York, NY). 2009;326(5956):1112–5.
Xiao Y, Liu H, Wu L, Warburton M, Yan J. Genome-wide association studies in maize: praise and stargaze. Mol Plant. 2017;10(3):359–74. https://doi.org/10.1016/j.molp.2016.12.008.
van Heerwaarden J, Hufford MB, Ross-Ibarra J. Historical genomics of north American maize. Proc Natl Acad Sci U S A. 2012;109(31):12420–5. https://doi.org/10.1073/pnas.1209275109.
Faure JE, Rusche ML, Thomas A, Keim P, Dumas C, Mogensen HL, et al. Double fertilization in maize: the two male gametes from a pollen grain have the ability to fuse with egg cells. Plant J. 2003;33(6):1051–62. https://doi.org/10.1046/j.1365-313X.2003.01692.x.
Fuad-Hassan A, Tardieu F, Turc O. Drought-induced changes in anthesis-silking interval are related to silk expansion: a spatio-temporal growth analysis in maize plants subjected to soil water deficit. Plant Cell Environ. 2008;31(9):1349–60. https://doi.org/10.1111/j.1365-3040.2008.01839.x.
Bassetti P, Westgate ME. Water deficit affects receptivity of maize silks. Crop Sci. 1993;33(2):279–82. https://doi.org/10.2135/cropsci1993.0011183X003300020013x.
Ballicora MA, Iglesias AA, Preiss J. ADP-glucose Pyrophosphorylase: a regulatory enzyme for plant starch synthesis. Photosynth Res. 2004;79(1):1–24. https://doi.org/10.1023/B:PRES.0000011916.67519.58.
Alam MA, Seetharam K, Zaidi PH, Dinesh A, Vinayan MT, Nath UK. Dissecting heat stress tolerance in tropical maize (Zea mays L.). Field Crop Res. 2017;204:110–9. https://doi.org/10.1016/j.fcr.2017.01.006.
Chuck G, Meeley R, Hake S. Floral meristem initiation and meristem cell fate are regulated by the maize AP2 genes ids1 and sid1. Development. 2008;135(18):3013–9. https://doi.org/10.1242/dev.024273.
Thompson BE, Bartling L, Whipple C, Hall DH, Sakai H, Schmidt R, et al. Bearded-ear encodes a MADS box transcription factor critical for maize floral development. Plant Cell. 2009;21(9):2578–90. https://doi.org/10.1105/tpc.109.067751.
Cheng PC, Greyson RI, Walden DB. Organ initiation and the development of unisexual flowers in the tassel and ear of Zea Mays. Am J Bot. 1983;70(3):450–62. https://doi.org/10.1002/j.1537-2197.1983.tb06411.x.
Ambrose BA, Lerner DR, Ciceri P, Padilla CM, Yanofsky MF, Schmidt RJ. Molecular and genetic analyses of the silky1 gene reveal conservation in floral organ specification between eudicots and monocots. Mol Cell. 2000;5(3):569–79. https://doi.org/10.1016/S1097-2765(00)80450-5.
Hiscock SJ, Allen AM. Diverse cell signalling pathways regulate pollen-stigma interactions: the search for consensus. New Phytol. 2008;179(2):286–317. https://doi.org/10.1111/j.1469-8137.2008.02457.x.
Spielbauer G, Margl L, Hannah LC, Romisch W, Ettenhuber C, Bacher A, et al. Robustness of central carbohydrate metabolism in developing maize kernels. Phytochemistry. 2006;67(14):1460–75. https://doi.org/10.1016/j.phytochem.2006.05.035.
Schultz JA, Juvik JA. Current models for starch synthesis and the sugary enhancer1 (se1) mutation in Zea mays. Plant Physiol Biochem. 2004;42(6):457–64. https://doi.org/10.1016/j.plaphy.2004.05.008.
Prioul JL, Mechin V, Lessard P, Thevenot C, Grimmer M, Chateau-Joubert S, et al. A joint transcriptomic, proteomic and metabolic analysis of maize endosperm development and starch filling. Plant Biotechnol J. 2008;6(9):855–69. https://doi.org/10.1111/j.1467-7652.2008.00368.x.
Chen J, Zeng B, Zhang M, Xie S, Lai J. Dynamic Transcriptome Landscape of Maize Embryo and Endosperm Development. Plant Physiol. 2014;166(1):252–64.
Schmidt RJ, Ketudat M, Aukerman MJ, Hoschek G. Opaque-2 is a transcriptional activator that recognizes a specific target site in 22-kD zein genes. Plant Cell. 1992;4(6):689–700. https://doi.org/10.1105/tpc.4.6.689.
Hannah LC, Tuschall DM, Mans RJ. Multiple forms of maize endosperm Adp-glucose Pyrophosphorylase and their control by Shrunken-2 and Brittle-2. Genetics. 1980;95(4):961–70. https://doi.org/10.1093/genetics/95.4.961.
Smith LG, Jackson D, Hake S. Expression of Knotted1 Marks shoot meristem formation during maize embryogenesis. Dev Genet. 1995;16(4):344–8. https://doi.org/10.1002/dvg.1020160407.
Fu J, Cheng Y, Linghu J, Yang X, Kang L, Zhang Z, et al. RNA sequencing reveals the complex regulatory network in the maize kernel. Nat Commun. 2013;4(1):2832. https://doi.org/10.1038/ncomms3832.
Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–67. https://doi.org/10.1038/nprot.2016.095.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. https://doi.org/10.1186/gb-2013-14-4-r36.
Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. https://doi.org/10.1038/nbt.2450.
Bassetti P, Westgate ME. Senescence and receptivity of maize silks. Crop Sci. 1993;33(2):275–8. https://doi.org/10.2135/cropsci1993.0011183X003300020012x.
Kessler SA, Grossniklaus U. She’s the boss: signaling in pollen tube reception. Curr Opin Plant Biol. 2011;14(5):622–7. https://doi.org/10.1016/j.pbi.2011.07.012.
Dresselhaus T, Franklin-Tong N. Male–female crosstalk during pollen germination, tube growth and guidance, and double fertilization. Mol Plant. 2013;6(4):1018–36. https://doi.org/10.1093/mp/sst061.
Koyama T. The roles of ethylene and transcription factors in the regulation of onset of leaf senescence. Front Plant Sci. 2014;5:8.
Cui X, Zheng J, Zheng J, Gong Q. Study of autophagy in plant senescence. Methods Mol Biol. 2018;1744:299–306.
Jeon JS, Ryoo N, Hahn TR, Walia H, Nakamura Y. Starch biosynthesis in cereal endosperm. Plant Physiol Biochem. 2010;48(6):383–92. https://doi.org/10.1016/j.plaphy.2010.03.006.
Greene TW. Maize endosperm ADP-glucose pyrophosphorylase Shrunken2 and Brittle2 subunit interactions. Plant Cell. 1998;10(8):1295–306.
Xu JH, Messing J. Organization of the prolamin gene family provides insight into the evolution of the maize genome and gene duplications in grass species. Proc Natl Acad Sci U S A. 2008;105(38):14330–5. https://doi.org/10.1073/pnas.0807026105.
Geetha KB, Lending CR, Lopes MA, Wallace JC, Larkins BA. Opaque-2 modifiers increase gamma-zein synthesis and alter its spatial distribution in maize endosperm. Plant Cell. 1991;3(11):1207–19. https://doi.org/10.1105/tpc.3.11.1207.
Sekhon RS, Childs KL, Santoro N, Foster CE, Buell CR, de Leon N, et al. Transcriptional and metabolic analysis of senescence induced by preventing pollination in maize. Plant Physiol. 2012;159(4):1730–44. https://doi.org/10.1104/pp.112.199224.
Martin-Trillo M, Cubas P. TCP genes: a family snapshot ten years later. Trends Plant Sci. 2010;15(1):31–9. https://doi.org/10.1016/j.tplants.2009.11.003.
Clarke JD, Volko SM, Ledford H, Ausubel FM, Dong X. Roles of salicylic acid, jasmonic acid, and ethylene in cpr-induced resistance in arabidopsis. Plant Cell. 2000;12(11):2175–90. https://doi.org/10.1105/tpc.12.11.2175.
Hung KT, Kao CH. Promotive effect of jasmonates on the senescence of detached maize leaves. Plant Growth Regul. 1996;19(1):77–83. https://doi.org/10.1007/BF00024405.
Broholm SK, Tahtiharju S, Laitinen RA, Albert VA, Teeri TH, Elomaa P. A TCP domain transcription factor controls flower type specification along the radial axis of the Gerbera (Asteraceae) inflorescence. Proc Natl Acad Sci U S A. 2008;105(26):9117–22. https://doi.org/10.1073/pnas.0801359105.
Lucero LE, Uberti-Manassero NG, Arce AL, Colombatti F, Alemano SG, Gonzalez DH. TCP15 modulates cytokinin and auxin responses during gynoecium development in Arabidopsis. Plant J. 2015;84(2):267–82. https://doi.org/10.1111/tpj.12992.
Tao TY, Ouellet T, Dadej K, Miller SS, Johnson DA, Singh J. Characterization of a novel glycine-rich protein from the cell wall of maize silk tissues. Plant Cell Rep. 2006;25(8):848–58. https://doi.org/10.1007/s00299-006-0128-3.
Li W, Yu D, Yu J, Zhu D, Zhao Q. Functional Analysis of Maize Silk-Specific ZmbZIP25 Promoter. Int J Mol Sci. 2018;19(3):822.
Zhang P, Wang Y, Zhang J, Maddock S, Snook M, Peterson T. A maize QTL for silk maysin levels contains duplicated Myb-homologous genes which jointly regulate flavone biosynthesis. Plant Mol Biol. 2003;52(1):1–15. https://doi.org/10.1023/A:1023942819106.
Li T, Qu J, Tian X, Lao Y, Wei N, Wang Y, et al. Identification of Ear Morphology Genes in Maize ( L.) Using Selective Sweeps and Association Mapping. Front Genet. 2020;11:747.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. https://doi.org/10.1093/bioinformatics/bty560.
Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, et al. Fast genome-wide functional annotation through Orthology assignment by eggNOG-mapper. Mol Biol Evol. 2017;34(8):2115–22. https://doi.org/10.1093/molbev/msx148.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7. https://doi.org/10.1089/omi.2011.0118.
Acknowledgements
We thank the Young Scientists Fund of the National Natural Science Foundation of China (Grant No. 31701438) for project support.
Funding
This work was supported by the Young Scientists Fund of the National Natural Science Foundation of China (Grant No. 31701438). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
SX, XZ and JX conceived and designed the experiments. BY and YS performed the experiments. XG and JQ collected and processed the data. TL and YW analysed the data and wrote the paper and prepared figures and/or tables. All authors improved, read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primers used for qRT-PCR in this study.
Additional file 2: Table S2.
Matrix of filtered transcript expression.
Additional file 3: Table S3.
GO annotations of transcripts exclusively detected in KSP, KUP, SSP and SUP.
Additional file 4: Table S4.
Analysis of differentially expressed transcripts between kernels and silks.
Additional file 5: Figure S1.
Expression levels of senescence-related genes in KSP, KUP, SSP and SUP. The color scale represents the normalized FPKM values (blue indicates lower expression, red indicates higher expression).
Additional file 6: Figure S2.
Expression levels of autophagy-related genes in KSP, KUP, SSP and SUP. The color scale represents the normalized FPKM values (blue indicates lower expression, red indicates higher expression).
Additional file 7: Table S5.
Average expression of differentially expressed TFs.
Additional file 8: Table S6.
Genes involved in the development of self-pollinated and unpollinated kernels and silks.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, T., Wang, Y., Shi, Y. et al. Transcriptome profiling provides insights into the molecular mechanisms of maize kernel and silk development. BMC Genom Data 22, 28 (2021). https://doi.org/10.1186/s12863-021-00981-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-021-00981-4