
Abstract
Background
In order to assess genetic diversity of a set of 41 Caricaceae accessions, this study used 34 primer pairs designed from the conserved domains of bacterial leaf blight resistance genes from rice, in a PCR based approach, to identify and analyse resistance gene analogues from various accessions of Carica papaya, Vasconcellea goudotiana, V. microcarpa, V. parviflora, V. pubescens, V. stipulata and, V. quercifolia and Jacaratia spinosa.
Results
Of the 34 primer pairs fourteen gave amplification products. A total of 115 alleles were identified from 41 accesions along with 12 rare and 11 null alleles. The number of alleles per primer pair ranged from 4 to 10 with an average of 8.21 alleles/ primer pair. The average polymorphism information content value was 0.75/primer. The primers for the gene Xa1 did not give any amplification product. As a group, the Indian Carica papaya accessions produced a total of 102 alleles from 27 accessions. The similarity among the 41 accessions ranged from 1% to 53%. The dendrogram made from Jaccard’s genetic similarity coefficient generated two major clusters showing that the alleles of Jacaratia spinosa and Vasconcellea accessions were distinctly different from those of Carica papaya accessions. All the alleles were sequenced and eleven of them were allotted accession numbers by NCBI. Homology searches identified similarity to rice BLB resistance genes and pseudogenes. Conserved domain searches identified gamma subunit of transcription initiation factor IIA (TFIIA), cytochrome P450, signaling domain of methyl-accepting chemotaxis protein (MCP), Nickel hydrogenase and leucine rich repeats (LRR) within the sequenced RGAs.
Conclusions
The RGA profiles produced by the 14 primer pairs generated high genetic diversity. The RGA profiles identified each of the 41 accessions clearly unequivocally. Most of the DNA sequences of the amplified RGAs from this set of 41 accessions showed significant homology to the conserved regions of rice bacterial leaf blight resistance genes. These information can be used in future for large scale investigation of tentative disease resistance genes of Carica papaya and other Caricaceae genus specially Vasconcellea. Inoculation studies will be necessary to link the identified sequences to disease resistance or susceptibility.
Similar content being viewed by others


Background
Papaya (Carica papaya L.), is one of the major fruit crops cultivated in tropical and sub-tropical zones. Over 6.8 million tonnes of this fruit are produced worldwide with India in the lead having an annual output of about 3 million tonnes [1]. Other leading producers are Brazil, Mexico, Nigeria, Indonesia, China, Peru, Thailand and Philippines. Papaya is eaten both fresh and cooked, and is processed into pickles, jams, candies, fruit drinks and juices. Papain, an enzyme purified from papaya latex, is extracted for export. The enzyme is used in medicine, breweries, textile and leather processing industries. Susceptibility to insect, pest and diseases are the major constraints limiting papaya production. Papaya ringspot virus (PRSV), Xanthomonas fruit rot, black spot, die back and root rot cause huge crop loss each year. The structural makeup and functional mechanisms of genes that confer disease resistance in Carica papaya is largely unknown and only a few genetic markers linked to resistance genes have been identified [2]-[5]. Although bio-engineering efforts have been successful in controlling PRSV [6] and improved agricultural practices like application of pesticides and nutritional supplements have been used in disease control of papaya; no durable solution is available due to the breakdown of resistance by high pathogenic variability. Vasconcellea, a related genus from the family Caricaceae, has the potential as a source of novel genes for quality traits and disease resistance especially against papaya ringspot virus [7],[8]. Resistances to several other diseases which affect Carica papaya have also been identified in the Vasconcellea genepool, including: resistance to black spot, (V. cundinamarcensis) [7]; die back, (V. parviflora) [7]; and root rot, (V. goudotiana) [7]. However hybridization between Carica papaya and Vasconcellea have been largely limited by post-zygotic instabilities, including embryo abortion and infertility of the hybrids [7],[8]; thus presenting a significant barrier for the successful introgression of desirable disease resistance traits into C. papaya.
The susceptibility of papaya to diseases coupled with the difficulty in producing viable intergeneric crosses has lead to the adoption of molecular biology tools, PCR-based strategies and in-silico genomic evaluation of defense gene homologs, as a means for crop improvement and search of naturally occurring resistance in existing genotypes of papaya and related species [9]. With the publication of the 372 Mb draft sequence of the papaya genome [10],[11], defense associated nucleotide-binding site (NBS)-encoding genes have been identified. Majority of the plant disease resistance proteins identified to date belong to a limited number of classes, of which those containing nucleotide-binding site (NBS) motifs are the most common. Amaral et al. [12] used the primer combination P1b and RNBS-D [13] to amplify RGAs in Carica papaya transgenic variety Embrapa PTP18 and Vasconcellea cauliflora. Forty eight clones were sequenced from each of the two species and the only RGA that was identified was from Carica papaya transgenic variety Embrapa PTP18. This RGA showed homology to the putative disease resistant protein RGA3 of Solanum bulbocastanum (gb|AAP45165.1|). Detailed in-silico analysis of the putative resistance genes (R-genes) identified by Ming et al. [11] have been done by Porter et al. [9]. They found that despite having a significantly larger genome than Arabidopsis thaliana, papaya has fewer NBS genes, belonging to both Toll/interleukin-1 receptor (TIR) and non-TIR subclasses. They also proposed that Papaya NBS gene family shares most similarity with Vitis vinifera homologs, but seven non-TIR members with distinct motif sequence represents a novel subgroup.
Although the order of plant disease resistance genes is not syntenic across taxa, majority of the defence related genes are structurally and functionally conserved across most plant species and the proteins coded have been grouped into various classes [14]-[16]. Synteny is the maintenance of the ordered sequence or the relative positions of the genes on the chromosome across species. With the increased availability of plant genome sequence information, syntenic relationships among the various taxa are being gradually elucidated. Studies have revealed that the gene families encoding transcription factors are syntenic throughout the angiosperm kingdom while others are subject to various aberrations [17]. Abrouk et al [18] analysed monocot synteny using rice as the reference genome and found that on the basis of short conserved sequence regions 77% of the genes were conserved among the five cereal genomes of rice, maize, wheat, Sorghum and Brachypodium. Similar analysis of eudicot synteny with grape as the reference genome showed 77% gene conservation between Arabidopsis, grape, poplar, soybean and papaya. Synteny has also been found between rice and Arabidopsis [19]. There are no reports of synteny between rice and papaya as of yet. However this experimentation has been based on the probable structural and functional conservation of disease resistance genes between rice and papaya.
Using degenerate PCR primers designed from the various classes of disease resistance, a number of workers like Leister et al. [20], Kanazin et al. [21] and Yu et al. [22] have developed a targeted technique for isolating homologous genes and DNA sequences. The term RGA (resistance gene analog) is used to denote such cloned homologous gene sequences for which no function has yet been assigned in the plant species [23]. Once found, the RGA can be used as probe to screen BAC or cDNA libraries, as a marker to be applied in marker assisted selection and to obtain resistance by their over expression in the plant genome.
Rice is the model monocot. Its genome has been sequenced and information regarding the structure and function of its disease resistance genes, including those against bacterial leaf blight (BLB) are publicly available [24]-[32]. BLB is caused by the vascular pathogen Xanthomonas oryzae pv. oryzae (Xoo), a gammaproteobacteria. It is one of the most serious diseases leading to crop failure in rice growing countries. Xoo enters rice leaves typically through the hydathodes at the leaf margin, multiplies in the intercellular spaces of the underlying epithelial tissue, and moves to the xylem vessels to cause systemic infection [25]. Rice Bacterial leaf blight (BLB) resistance genes Xa1 and Xa21 belongs to the CC/NBD/LRR (coiled coil/nucleotide binding domain/leucine rich repeat) [31] and extracellular LRR/kinase domain classes [27] respectively. The BLB resistance gene xa5 is a transcription factor and Xa26 codes for a receptor kinase like protein. A signal-anchor-like sequence is predicted at the amino (N)-terminal region of BLB resistance gene Xa27 and it localizes to the apoplast. The previous attempt to isolate and identify RGAs used degenerate primers designed by Bertioli [13] using a protein alignment of L6 rust R-gene (resistance gene) from Linum usitatissimum, R-gene N against tobacco mosaic virus from Nicotiana glutinosa, gene NL25 from Solanum tuberosum mRNA, gene RPS5 of A. thaliana for resistance to Pseudomonas syringae, R-gene Mi-1 against nematodes and aphids from Lycopersicon esculentum, and gene Rpp8 of A. thaliana; and by Kanazin [21] using the conserved P-loop sequence. However attempts to identify RGAs using primers developed from known resistance genes from rice was not done before and that is what we have tried to do in this study.
Most Genetic diversity studies use DNA primers that are from random genomic locations. While, genetic diversity studies using targeted genic sequences could be more informative, useful and valuable. Das et al. [33] had designed 34 pairs of primers from the conserved motifs of 6 bacterial leaf blight resistance genes of Oryza sativa – Xa1, xa5, Xa21, Xa21(A1), Xa26 and Xa27, for the assessment of genetic diversity amongst rice acccessions. In this study we have used those 34 primer pairs to identify RGAs in 41 accessions of Carica papaya, Vasconcellea sp and Jacaratia spinosa. The other objectives of this study were, to obtain the genetic relationship amongst the 41 Caricaceae accessions using the polymorphism of the amplified DNA bands using statistical methods, and to analyze the sequences of the obtained DNA bands for the presence of homology and conserved domains.
Method
Plant materials
The germplasm set in this study included 1 accession each from 27 Indian and 7 foreign commercially popular Carica papaya cultivars, 1 accession each of V. goudotiana, V. microcarpa, V. parviflora, V. pubescens, V. stipulata and V. quercifolia and 1 accession of South American tree species Jacaratia spinosa. The collection was maintained at the experimental farm of Acharya J.C. Bose Biotechnology Innovation Centre, Bose Institute at Madhyamgram, West Bengal, India. Fully expanded fourth leaf from the top was used as source material for genomic DNA isolation. The category, cultivar name, source and number of accessions used in this study for each accession are given in Table 1.
Designing primers for bacterial leaf blight resistance
Thirty four primer pairs were designed from publicly available sequences of six rice bacterial leaf blight resistance genes using the software BatchPrimer3 (http://probes.pw.usda.gov/batchprimer3). The forward and reverse primers for the markers were coded BDTG1 to BDTG34. The primers were designed to include only the exons and so as to amplify about 500 to 700 base pairs [33]. Details of the markers are given in Table 2.
Isolation of genomic DNA and PCR amplification
Genomic DNA isolation was done according to the method of Walbot [34]. PCR amplification of this DNA was performed with the designed markers. DNA amplification was carried out in 25 μl volumes using 200 μl thin-walled PCR tubes (Axygen, USA) in a MJR thermal cycler. Each reaction mixture contained 100 ng of genomic DNA, 1 μM of each of the two primers, 1× PCR buffer, 1.5mM MgCl2 solution, 1mM of dNTP mixture, 1 unit of Taq DNA polymerase and the volume was made up to 25 μl with PCR-grade water. The temperature profile used for PCR amplification comprised 97°C for 5 mins, followed by 35 cycles of 1 min at 95°C, 1 min at 59.5-61.8°C and 2 min at 72°C. The final extension was at 72°C for 10 min.
Polyacrylamide gel electrophoresis
The PCR products were resolved by native polyacrylamide gel electrophoresis (PAGE) following the protocol given by Sambrook et al. [35], in a 6% gel in vertical electrophoresis tank (gel size of 16 cm × 14 cm, Biotech, India) with Tris-Acetate-EDTA buffer at 150V. The gel, after electrophoresis, was stained with ethidium bromide (5μg of EtBr in 200ml of Tris-Borate-EDTA buffer) washed thoroughly with double distilled water and photographed using a Gel Documentation System (Biorad, USA).
Allele scoring
Under UV light a cluster of two to five discrete bands (stutter) was apparent in the stained gels for most of the markers. The size (in nucleotides) of the most intensely amplified band was determined using the software Quantity One (Biorad, USA), based on the migration of the band relative to molecular weight size markers (100bp DNA ladder SibEnzyme) included in the gel [36]. The band with the lowest molecular weight for each primer pair was assigned allele number 1 and the progressively heavier bands were assigned incrementally. For any individual primers pair, the presence of an allele in each of the accession was recorded as “1” and the absence of an allele was denoted as “0” [36]. Null alleles were assigned when no amplification product was generated [37]. When an allele was found in less than 5% of the germplasms under study, it was designated as rare [38].
Genetic relationship analysis using RGA profiles
A 1/0 matrix was constructed for each primer pair using the information of presence or absence of alleles and was used to calculate genetic similarities among the accessions according to Jaccard’s coefficient [39] using NTSYS-pc software package (version 2.02e) [40]. Using pairwise similarity matrix of Jaccard’s coefficient [39] a phylogenetic tree was made by unweighted pair-group method of arithmetic average (UPGMA) and neighbor-joining (NJoin) module of the NTSYS-pc. Support for clusters was evaluated by bootstrap analysis using WinBoot software [41] through generating 1,000 samples by re-sampling with replacement of characters within the 1/0 data matrix. The average polymorphism information content (PIC) was calculated for each primer pair in accordance with the method Anderson et al., [42].
Sequencing and analysis of polymorphic DNA bands
All the alleles were sequenced. They were eluted using QIAquick Gel Extraction Kit following standard protocol. DNA sequences of the eluted products were determined according to Sanger et al. [43]. Sequencing was done using BioRad sequencer at Bose Institute with a BigDye Terminator v3.1 cycle sequencing kit according to the manufacturer’s manual (Applied Biosystems, Darmstadt, Germany).The sequences were submitted to the NCBI and were analyzed using publicly available software Basic Local Alignment Search Tool, [44] or BLAST, of NCBI (http://www.ncbi.nlm.nih.gov/BLAST/) to find homology. Conserved domains were identified in the sequences using the publicly available software of NCBI conserved domains (http://www.ncbi.nlm.nih.gov/BLAST/).
Results
Genetic diversity: number of alleles
The analysis of the PCR profiles of the 41 Caricaceae accessions generated using the 34 RGA primer pairs is summarized in Table 3. Fourteen out of 34 RGA primers used produced polymorphic profiles while the rest of the 20 primer pairs failed to generate amplification products. A total of 115 alleles were produced by the 14 RGA primer pairs; the number of alleles ranging from 4 (BDTG 21) to 10 (BDTG11, BDTG12, BDTG14, BDTG19, BDTG25 and BDTG31). The average number of alleles was 8.375 per locus.
As a group the total number of alleles for 6 Vasconcellea and 1 Jacaratia accessions was 50 with an average of 3.57 alleles /locus. The smallest number of alleles identified was 2, amplified by BDTG13, BDTG21 and BDTG34. The highest number of alleles in this category was 7, amplified by BDTG14. The total number of alleles from the 7 foreign Carica papaya accessions was also 50 with an average of 3.57 alleles/locus. The lowest number of alleles identified in this category was 1 (amplified by BDTG 22) and the highest was 5 (amplified by BDTG17, BDTG19, BDTG25 and BDTG30). The 27 Indian Carica papaya accessions produced 102 alleles with an average of 7.29 alleles/locus. The lowest and highest number of alleles identified in this category was 4 (by markers BDTG 21 and BDTG 25) and 13 (by marker BDTG14) respectively.
When grouped according to the category of motif, the average number of alleles produced by the 14 RGA primer pairs amplifying the LRR motif, the kinase motif, the charged domain and the TFIIA domain were 7.8, 8, 6 and 10 alleles/primer pair respectively.
Details of the amplification products obtained from the RGA primer pairs
BDTG11 and BDTG12, primer pairs designed from the TF IIA domain of the gene xa5, amplified 10 alleles each. The primer BDTG11 was developed from exon 1 and BDTG12 was designed from exon 2 of gene xa5. Four rare alleles were identified by BDTG12 and no null alleles were found. The primer pairs BDTG13, BDTG14 and BDTG17 designed from the receptor kinase domain of the Xa26 gene amplified 12 alleles while the rest of the primer pairs, BDTG15, BDTG16 and BDTG18 failed to amplify. The primers pairs BDTG13 and BDTG17 identified 1 rare and 1 null allele each while BDTG14 identified 2 rare and 2 null alleles. BDTG 19, the primer pair designed from the Xa27 gene produced 10 alleles and rare or null alleles were absent. Except for the signal sequence, the primer pairs developed from the other regions of the gene Xa21 amplified 36 alleles (Table 3). Those primer pairs were BDTG21, BDTG22, BDTG23, BDTG24 and BDTG25. BDTG21, designed from LRR domain, exon 2, of gene Xa21 produced 4 alleles. No rare or null alleles were identified. The primer pair BDTG22, designed from LRR domain, exon 2, of gene Xa21 produced 8 alleles and 1 null allele. BDTG23 designed from LRR domain, exon 2, of gene Xa21 produced 8 alleles and a null allele. The primer pair BDTG 24 designed from the charged domain, exon 3 of gene Xa21 produced 6 alleles and 1 null allele. The primer pair BDTG25 designed from kinase domain of gene Xa21 produced 10 alleles and 4 rare alleles and 1 null allele. The primer pairs BDTG30, BDTG31 and BDTG34 designed from gene Xa1(A1), produced amplification products, the rest i.e. BDTG 26, BDTG 27, BDTG 28, BDTG 29, BDTG32 and BDTG33 did not produce any amplification product. BDTG30 produced 9 alleles and one null allele. The primer pair BDTG31 produced 10 alleles. No null or rare alleles were produced. The kinase domain (exon 3) of Xa1(A1) was amplified by the primer pair BDTG34 and it produced 6 alleles and 1 null allele. The primer pairs for the gene Xa1 did not amplify any product.
PIC values
The PIC values, which denote allelic diversity and frequency among germplasms, had an average value of 0.763 per primer pair. The range of PIC value was 0.611 for primer pair BDTG13 to 0.852 for the primer pair BDTG14. That means the most diverse region as well as the region with minimum diversity lies within the same gene. Categorically average PIC value for the Vasconcellea accessions was 0.661 per primer pair with a range of 0.245 for primer pair BDTG21 and BDTG25 to 0.939 for primer pairs BDTG14 and BDTG31. For the foreign papaya accessions the average PIC value was 0.716 per primer pair and range of PIC value was 0.245 (BDTG30) to 0.939 (BDTG23). The Indian papaya accessions had an average PIC value of 0.92 per primer pair. The range of PIC value for them was 0.528 (BDTG 31) to 0.992 (BDTG14, BDTG25 and BDTG34). From the PIC values it is evident that allelic diversity is the highest among the Indian papaya accessions. An ANOVA test (Additional file 1: Table S1) was done with the PIC values of the different categories of germplasm. It was proved from that test that the PIC values of the three categories of papaya germplasms used in this study were significantly different from each other.
Rare and Null alleles
A total of 12 rare alleles were identified with an average of 0.86 rare alleles per loci. The highest number of rare alleles (4 rare alleles) was observed in the profile of the primer pairs BDTG12 and BDTG25. The accession of Jacaratia spinosa had 3 rare alleles, Vasconcellea microcarpa and V. parviflora had 2 while V. pubescens, V. quercifolia and V. stipulata each had one rare allele. The Carica papaya accessions Solo 109 and CO1 each had 1 rare allele. A total of 11 null alleles were detected. The primer pairs BDTG14 and BDTG34 each produced 2 null alleles while primer pairs BDTG13, BDTG17, BDTG22, BDTG23, BDTG24, BDTG25 and BDTG30 produced 1 null allele each. The accessions Orissa local had 2 while CO1 and Madhu had 1 null allele each. Seven null alleles were identified amongst the other Caricaceae accessions. Vasconcellea quercifolia and Jacaratia Spinosa had 2 while V. goudotiana, V. microcarpa and V. pubescens had 1 null allele each.
Clustering of the Caricaceae accessions
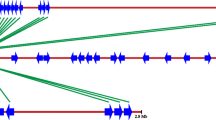
The dendrogram given in Figure 1 was made from genetic similarity values derived from the 1/0 matrix of the RGA profiles (Additional file 2: Table S2 1/0 matrix). The strength of the dendrogram nodes was estimated with a bootstrap analysis using 1000 permutations. The similarity among the Caricaceae accessions ranged from 1% to 53%. Two distinct clusters had separated at 1% level of similarity; “Cluster A”, consisted of 40 accessions and “Cluster B” consisting of just the one accession of Jacaratia spinosa. Cluster A was divided into 2 sub-clusters X and Y at 7.5% level of similarity. Both the clusters X and Y underwent further sub-divisions and segregated into 7 smaller clusters at various levels of similarity, as shown in Figure 1. The most significant segregation was at the 24.4% level of similarity at which point all the 6 accessions of Vasconcellea separated out from the rest of the accessions. There were two other significant clusters: the cluster separating at 15% similarity consisted of 5 accessions each of the Indian and the foreign caricas, while the cluster separating at 15.9% level of similarity consisted of 9 Indian Carica papaya accession and one foreign accession Hortus Gold. The maximum genetic similarity of 53%, was observed between the accessions Kapoho (foreign Carica papaya) and Madhu (Indian Carica papaya).

Dendrogram of 41 Caricaceae genotypes based on Jaccard's genetic similarity coefficient.
Sequence analysis
The information about the details of homology searches are given in Table 4. A total of 563 sequences were obtained, of which 394 showed significant homology with various sequences of Oryza sativa. Out of the 41 DNA sequences amplified by BDTG11 (gene xa5), 35 showed significant homology with Oryza sativa Indica Group cultivar IRGC 16339 xa5 gene, partial cds. Out of the 31 sequences amplified by BDTG12, ten were allotted accession numbers by NCBI. The sequences JM426511.1 (from Vasconcellea parviflora), JM426525 (from Vasconcellea stipulata), JM426506 (from Vasconcellea quercifolia), JM426460 (from CO5), JM426516 (from Bangalore Dwarf) were significantly homologous to Oryza nivara cultivar 106133 XA5 (xa5) gene, complete cds JM426495 (from Pusa Nanha) and HR614236 (from CO1) were significantly homologous to Oryza sativa Japonica Group Os05g0107700 (Os05g0107700) mRNA. The sequences JM170468 (from Pusa Giant), JM170470 (from Vasconcellea pubescens) and JM170472 (from Shillong) were significantly homologous to sequence of Oryza sativa Japonica Group Os08g0280600 (Os08g0280600) mRNA. The other 21 sequences derived from the PCR profiles of BDTG12 (gene xa5) were significantly homologous to the sequence Oryza sativa Indica Group cultivar IRGC 27045 xa5 gene. Among the sequences amplified by the BDTG12, JM426506 (from Vasconcellea quercifolia) and JM426460 (from CO5) showed significant homology with conserved domain of Gamma subunit of transcription initiation factor IIA. The sequence JM426495 (from Pusa Nanha) showed significant homology with conserved domain of Cytochrome P450. The sequence JM170472 (from Shillong) showed significant homology with the conserved domain of Methyl-accepting chemotaxis protein (MCP) signaling domain. Out of the sequences amplified by the primer BDTG13, 33 sequences were significantly homologous to the sequence Oryza sativa isolate BDTG13-Bhasa receptor kinase (Xa26) gene and one, HR614235.1 (from CO1) showed homology with the sequence Oryza sativa Japonica Group Os03g0579200 (Os03g0579200) mRNA, complete cds. The sequence HR614235.1 (from CO1) was also significantly homologous to the conserved domain of Nickel-dependent hydrogenase. The sequences amplified by the primer pair BDTG17 were significantly homologous to the sequence of Oryza sativa (japonica cultivar-group) bacterial blight resistance protein XA26 (Xa26 gene), complete cds. Some of the sequences were also homologous to the conserved domain of LRR receptor-like protein kinase. The sequences amplified by the primer pair BDTG21 were significantly homologous to the sequence of Oryza sativa Indica Group Xa21 gene for receptor kinase-like protein, complete cds, cultivar: Zheda8220. The conserved domains of LRR could be identified within these sequences. The sequences amplified by the primer pairs BDTG22, BDTG23, BDTG24 and BDTG25 respectively were significantly homologous to the sequences of Oryza rufipogon Xa21F pseudogene. The sequences amplified by the primer pairs BDTG30 and BDTG31 were significantly homologous to the sequence of Oryza sativa Japonica Group Os11g0559200 (Os11g0559200) mRNA. The sequences amplified by the marker BDTG30 were significantly homologous to the conserved domains of LRR receptor-like protein kinase. The sequences amplified by the primer pair BDTG34 were significantly homologous to Oryza longistaminata receptor kinase-like protein gene family. The sequences amplified by the primer pairs BDTG14 and BDTG19 did not show any significant homology.
Discussion
According to Nordborg and Weigel [45] genomic potential and its association with phenotypic variation of any plant species can be achieved by documentation of genomic polymorphism at specific loci controlling various traits using specific genomic region based primers. This variation then has to be coupled with association mapping, a method popularly known as Genome Wide Association mapping. In this study we have used 34 pairs of primers [33] developed from conserved domains of 6 BLB resistance genes of rice, to detect the presence of amplified DNA bands (RGAs) and their polymorphism in a set of 41 Caricaceae accessions. Of these 34 primer pairs, 14 gave amplification profiles in this set of accessions. Since the primers were originally designed to amplify conserved domains of rice BLB resistance genes, they are not expected to behave as random primers and will only amplify sequences with a certain degree of stringency. Apart from clear and consistent amplification profiles, stutter bands, i.e. minor PCR products of lower intensity and lacking or having extra repeat units than the main allele, [46] were also present in the profiles of most of the markers used. Null alleles were present probably due to mutations in the binding region of one or both of the primers, thereby inhibiting primer annealing [37].
In the dendrogram (Figure 1) the accessions of Vasconcellea species and Jacaratia spinosa had segregated from the Carica papaya accessions into different clusters. The Vasconcellea accessions had 7.5% similarity with the Carica papaya accessions whereas Jacaratia spinosa had only 1% similarity with either Carica papaya or Vasconcellea accessions. As indicated in a previous publication by Sengupta et al., [47] this finding was similar to that proposed by taxonomic descriptions of Badillo [48] and Amplified Fragment Length Polymorphism (AFLP) study of Van Droogenbroeck et al. [49]. Probably due to their similar lineage, the foreign Carica papaya accessions Sunrise Solo, Solo 109, Kapoho and Waimanalo had grouped into the same sub cluster (sub cluster 2). Such grouping was also obtained using the SSR profiles in a previous study [50]. The Indian Carica papaya accessions Pusa Dwarf, RT1, Ambasa local, Ranchi and Madhu were included in the same cluster as Sunrise Solo in the dendrogram of Figure 1. This indicates a similar genetic nature of the concerned loci amplified by the primers used in this study. Whether those Indian Carica papaya accessions share the same lineage with the foreign Carica papaya accessions is not known because their parentage has not been elucidated. The accessions of the Coimbatore varieties (CO1-CO7) are phenotypically distinct and were bred at Tamil Nadu Agricultural University by different workers [50]. Like the dendrogram obtained using SSR profiles [47], these accessions have segregated into different sub clusters in this case as well. These trends were also reiterated in a dendrogram derived from the combination of the SSR profiles and the RGA profiles (Additional file 3: Figure S1). In could be observed from that dendrogram (Figure 1) that Jacaratia spinosa had segregated out as a separate cluster all by itself and is only 2% similar with the rest of the Caricaceae accessions. In previous taxonomic classifications the genus Carica L. was divided into two sections, Carica and Vasconcellea. This segregation was based on the number of locules in the ovary as well as other morphological similarities between the two sections Based on genetic and morphological characteristics respectively Aradhya et al., [51] and Badillo [48] had separated the two sections into two different genera Vasconcellea Saint-Hilaire and Carica. According to the findings of Aradhya et al. [51], Olson [52],[53] and Kyndt et al [54] there is a possibility that Jacaratia shares a common ancestor with, or lies at the origin of Vasconcellea but not of Carica. In our dendrogram we see that Vasconcellea, Carica and Jacaratia have formed 3 distinct clusters. Moreover the similarity between Vasconcellea and Carica is more than the similarity between these two genus and Jacaratia spinosa. In our previous study of genetic diversity analysis with SSR [47], Vasconcellea and Jacartia were placed in the same cluster and Carica had segregated as a separate cluster. However in this case the alleles of the concerned loci were more similar between Vasconcellea and Carica hence they have been brought together and Jacaratia has separated as an outgroup.
In the same dendrogram of Additional file 3: Figure S1, accessions of Vasconcellea sp. along with Hortus Gold formed a separate sub cluster. The foreign Carica papaya accessions Solo109, Sunrise Solo, Kapoho and Waimanlo had grouped together in a single sub cluster. The accessions of the Coimbatore varieties (CO1 – CO7) and the Pusa Giant, Pusa Dwarf and Pusa Nanha have segregated into different sub clusters.
The conserved domains identified in the sequences were gamma subunit of transcription initiation factor IIA, Cytochrome P450, MCP, signaling domain, Nickel-dependent hydrogenase, LRR receptor-like protein kinase and LRRs. Out of these the LRR domain is present both in pathogen-associated molecular patterns (PAMP) receptors, and in majority of Resistance (R) proteins [55]. Some R proteins structurally resemble the PAMP receptor like kinases (RLKs), such as the rice Xa21 and Xa26 proteins [56]. LRR ribonuclease inhibitor (RI)-like subfamily are 20-29 residue sequence motifs present in many proteins that participate in protein-protein interactions and have different functions and cellular locations. A number of LRRs have been identified in this study, but the detailed structure, function and cellular location are not known and will be elucidated in future dissertations.
The sequences JM426506 and JM426460 amplified by the primer BDTG12 showed significant homology with the conserved domain of gamma subunit of transcription initiation factor IIA (TFIIAγ). The primer pair BDTG 12 was designed from the rice gene xa5. The mRNA transcribed by the gene xa5 translates to a protein which acts both as a transcription factor and a bacterial blight resistance protein in rice [57]. TFIIAγ is one of the general transcription factors for RNA polymerase II which increases the affinity of the TATA-binding protein (TBP) for DNA, in order to assemble the initiation complex. TFIIA also functions as an activator during development and differentiation, and is involved in transcription from TATA-less promoters (NCBI). The xa5 gene is unusual in that it is recessive and does not conform to one of the typical resistance gene structural classes [57]. Whether the xa5-like sequences identified in Caricaceae confers resistance to bacterial diseases or acts simply as a transcription factor is yet to be elucidated.
The sequence JM426495 amplified by the primer BDTG12 showed significant homology with the conserved domain of cytochrome P450 Among the cytochrome P450 enzymes, CYP51 sterol demethylases are one the most ancient and conserved [58]. Apart from its regular function in plants in the synthesis of essential sterols, CYP51 is used for the production of antimicrobial compounds (avenacins) that confer Fusarium rot resistance in oats [59]. Fusarium rot has previously been reported in papaya by Guevara et al. [60] and Correia et al. [61] and antifungal activity in leaves and seeds of Carica papaya L. cv. Maradol due to the presence of triterpenoid glycoside type saponins have already been proposed by Quintal et al. [62]. Perhaps the cytochrome P450 domain identified in our papaya samples also serves a similar function in the production of plant defense compounds.
The sequence JM170472 amplified by the marker BDTG12 was significantly homologous to Methyl-accepting chemotaxis protein (MCP), signaling domain. The cytokinin inducible genes IBC6 and IBC7, identified by Brandstatter and Kieber [63] from etiolated Arabidopsis. They encode proteins similar to Bacterial Response Regulators. The deduced amino acid sequence of IBC6 and IBC7 aligned significantly with the sequence of conserved regions of chemotaxis response regulators CheY from Escherichia coli. The CheY are commonly known as methyl-accepting chemotaxis proteins (MCPs), [64]. However no significant homology was observed between the sequence JM170472 and the sequences of IBC6 or IBC7 or CheY.
The sequence HR614235.1 amplified by the primer pair BDTG13 was significantly homologous with the conserved domain of Nickel-dependent hydrogenase. These enzymes indirectly influence plant productivity through its role in nitrogen-fixing symbionts [65]. A role for nickel in plant disease resistance has also been observed and has been attributed to a direct phyto-sanitary effect on pathogens, or to a role of nickel on plant disease resistance mechanisms [66],[67]. The presence of nickel in the bark of Carica papaya have already detected by Mishra et al., [68]. However the mechanism of this nickel in disease resistance is yet to be elucidated.
Information on disease resistance genes of papaya is scarce as compared to Arabidopsis and Oryza. Studies like this one pave way for the vast amount of work yet undone. According to existing reports [https://www.apsnet.org/publications/commonnames/Pages/Papaya.aspx] Xanthomonas oryzae is not pathogenic to Carica papaya or Vasconcellea sp. However Papaya fruits are frequently spoiled by soft rot caused by Xanthomonas campestris [69] under post harvest condition. There are no reports of pathogenicity of Xanthomonas campestris in Caricaceae under field conditions. Nevertheless the causal organisms of more destructive bacterial diseases of papaya like canker, leaf spot and internal yellowing, Erwinia sp., Pseudomonas carica-papayae and Enterobacter cloacae respectively are also gammaproteobacteria like Xanthomonas. Since the plant disease resistance genes are structurally and functionally conserved, there are possibilities that defence against the pathogenocity of Erwinia sp., Pseudomonas carica-papayae and Enterobacter cloacae are also mediated in a way similar to that against Xanthomonas oryzae in rice. Whether the identified DNA sequences from this study actually have any association with the soft rot disease or any other bacterial disease of papaya are yet to be unfolded. Such experiments were beyond the scope of this study and will be pursued by us in our future endeavors. Primers designed from known disease resistance genes from other plants should also be used to search for homologous DNA bands and sequences. There should be a large scale investigation on the LRR regions of Carica papaya and other Caricaceae genus specially Vasconcellea. Their uniqueness has already been shown in-silico by Porter et al., [9] and there are chances that DNA sequence analysis of LRR regions will bring forth some more special features. Cloning, characterization and expression analysis of the linked genes or DNA sequences should follow next.
Conclusion
Several researchers have proved that plant disease resistance genes are structurally and functionally conserved. Based on that principle this study has used 34 primer pairs designed from the conserved domains of 6 BLB resistance genes of rice to identify RGAs in accessions of Caricaceae. Several DNA bands were amplified by 14 primer pairs. The homology of the sequences of the amplified DNA bands with that of Oryza sativa clearly shows that some of the conserved regions of resistance genes are conserved across evolutionary distances between Caricaceae and Oryza while some others are not. The findings of this study should be informative for the elucidating the structure, function and genetic diversity of disease resistance genes of Carica papaya and other related species in future.
Availability of supporting data
The data set supporting the results of this article is included within the additionl file named Additional file 2: Table S2. 1/0 matrix.
Additional files
References
FAO: Statistical Databases of the Food and Agriculture Organization of the United Nations. 2011
Xu Y, Ouyang XX, Zhang HY, Kang GB, Wang YJ, Chen H: Identification of a RAPD marker linked to fusarium wilt resistant gene in wild watermelon germplasm (Citrullus lanatus var. citroides). Acta Bot Sin. 1999, 41: 952-955.
Wehner TC: Proc. IXth EUCARPA Meeting on Genetics and Breeding of Cucurbitaceae, L’institut National de la Recherche Agronomique (21–24 May 2008, Avignon, France). Cucurbitaceae. Overview of the Genes of Watermelon. Edited by: Pitrat M. 2008, 79-90.
Harris KR, Ling K, Wechter WP, Levi A: Identification and utility of markers linked to the zucchini yellow mosaic virus resistance gene in watermelon. J Amer Soc Hort Sci. 2009, 134: 1-6.
Ling KS, Harris KR, Meyer JDF, Levi A, Guner N, Wehner TC, Bendahmane A, Havey MJ: Non-synonymous single nucleotide polymorphisms in the watermelon eIF4E gene are closely associated with resistance to zucchini yellow mosaic virus. Theor Appl Genet. 2009, 120: 191-200. 10.1007/s00122-009-1169-0.
Fitch MM, Manshardt RM, Gonsalves D, Slightom JL, Sanford JC: Virus resistant papaya plants derived from tissues bombarded with the coat protein gene of papaya ringspot virus. BioTechnology. 1992, 10: 1466-1472. 10.1038/nbt1192-1466.
Drew RA, O’Brien CM, Magdalita PM: Development of interspecific Carica hybrids. Acta Horticulture. 1998, 461: 285-292.
Manshardt RM, Wenslaff TF: Interspecific hybridization of papaya with other species. J Amer Soc Hort Sci. 1989, 114: 689-694.
Porter BW, Paidi M, Ming R, Alam M, Wayne T, Nishijima WT, Yun J, Zhu YJ: Genome-wide analysis of Carica papaya reveals a small NBS resistance gene family. Mol Gen Genomics. 2009, 281 (6): 609-626. 10.1007/s00438-009-0434-x.
Arumuganathan K, Earle ED: Nuclear DNA content of some important plant species. Plant Mol Biol Rep. 1991, 9: 208-218. 10.1007/BF02672069.
Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH: The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature. 2008, 452: 991-996. 10.1038/nature06856.
Amaral PPR, Alves PCM, Martins NF, da Silva FR, de Capdeville G, Souza Júnior MT: Identification and characterization of a resistance gene analog (RGA) from the Caricaceae Dumort family. Rev Bras Frutic. 2006, 28 (3): 458-462. 10.1590/S0100-29452006000300026.
Bertioli DJ, Leal – Bertioli SC, Lion MB, Santos VL, Pappas JRG, Cannon SB, Guimaraes PM: A large scale analysis of resistance gene homologues in Arachis. Mol Gen Genomics. 2003, 270: 34-45. 10.1007/s00438-003-0893-4.
Hammond KE, Jones JG: Plant disease resistance genes. Annu RevPlant Physiol Plant Mol Biol. 1997, 48: 575-607. 10.1146/annurev.arplant.48.1.575.
Bent AF: Plant disease resistance genes: function meets structure. Plant Cell. 1996, 8: 1757-1771. 10.1105/tpc.8.10.1757.
Ellis J, Dodds P, Pryor T: Structure, function and evolution of plant disease resistance genes. Curr Opin Plant Biol. 2000, 3: 278-284. 10.1016/S1369-5266(00)00080-7.
Woodhouse MR, Tang H, Freeling M: Different gene families in Arabidopsis thaliana transposed in different epochs and at different frequencies throughout the rosids. Plant Cell. 2011, 23 (12): 4241-4253. 10.1105/tpc.111.093567.
Abrouk M, Murat F, Pont C, Messing J, Jackson S, Faraut T, Tannier E, Plomion C, Richard Cooke R, Feuillet C, Salse J: Palaeogenomics of plants: syntenybased modelling of extinct ancestors. Trends Plant Sci. 2010, 15: 479-487. 10.1016/j.tplants.2010.06.001.
Salse J, Piegu B, Cooke R, Delseny M: Synteny between Arabidopsis thaliana and rice at the genome level: a tool to identify conservation in the ongoing rice genome sequencing project. Nucleic Acid Res. 2002, 30 (11): 2316-2328. 10.1093/nar/30.11.2316.
Leister D, Ballvora A, Salamini S, Gebhardt CA: A PCR based approach for isolating pathogen resistance genes from potato with potential for wide application in plants. Nat Genet. 1996, 14 (4): 421-429. 10.1038/ng1296-421.
Kanazin V, Frederick ML, Shoemaker RC: Resistance gene analogs are conserved and clustered in soybean. Proc Acad Natl Sci USA. 1996, 93 (21): 11746-11750. 10.1073/pnas.93.21.11746.
Yu YG, Buss GR, Saghai Maroof MA: Isolation of a super family of candidate disease-resistance genes in soybean based on a conserved nucleotide-binding site. Proc Acad Natl Sci USA. 1996, 93 (21): 11751-11756. 10.1073/pnas.93.21.11751.
Sakaguchi S: Linkage studies on the resistance to bacterial leaf blight, Xanthomonas oryzae (Uyeda et Ishiyama) Doeson, in rice. Bull. Natl. Inst. Agr. Sci. Ser. D. 1967, 16: 1-18. Japanese/English
Petpisit V, Khush GS, Kauffman HE: Inheritance to bacterial blight in rice. Crop Sci. 1977, 17: 551-554. 10.2135/cropsci1977.0011183X001700040018x.
Ronald PC, Albano B, Tabien R, Abenes L, Wu K: Genetic and physical analysis of the rice bacterial blight disease resistance locus, Xa21. Mol Gen Genet. 1992, 236: 113-120.
Song WY, Wang GL, Chen LL, Kim HS, Pi LY, Holsten T, Gardner J, Wang B, Zhai WX, Zhu LH, Fauquet C, Ronald P: A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science. 1995, 270: 1804-1806. 10.1126/science.270.5243.1804.
Song WY, Pi LY, Wang GL, Gardner J, Holsten T, Ronald PC: Evolution of the rice Xa21 disease resistance gene family. Plant Cell. 1997, 9: 1279-1287. 10.1105/tpc.9.8.1279.
Chen S, Huang ZH, Zeng LX, Yang JY, Liu QG, Zhu XY: High resolution mapping and gene prediction of Xanthomonas oryzae pv. oryzae resistance gene Xa7. Mol Breed. 2008, 22: 433-441. 10.1007/s11032-008-9187-1.
Garris AJ, McCouch SR, Kresovich S: Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics. 2003, 165: 759-769.
Lee KS, Rasabandith S, Angeles ER, Khush GS: Inheritance of resistance to bacterial blight in 21 cultivars of rice. Phytopathology. 2003, 93: 147-152. 10.1094/PHYTO.2003.93.2.147.
Yoshimura S, Yamanouchi U, Katayose Y, Toki S, Wang ZX, Kono I, Kuruta N, Yano M, Iwata N, Sasaki T: Expression of Xa1, a bacterial blight resistance gene in rice, is induced by bacterial inoculation. Proc Acad Natl Sci USA. 1998, 95: 1663-1668. 10.1073/pnas.95.4.1663.
Gu K, Yang B, Tian D, Wu L, Wang D, Sreekala C, Yang F, Chu Z, Wang GL, White FF, Yin Z: R gene expression induced by a type-III effector triggers disease resistance in rice. Nature. 2005, 435: 1122-1125. 10.1038/nature03630.
Das B, Sengupta S, Prasad M, Ghose TK: Genetic diversity of the conserved motifs of six bacterial leaf blight resistance genes in a set of rice landraces. BMC Genet. 2014, 15: 82-10.1186/1471-2156-15-82.
Walbot V: Preparation of DNA from single rice seedling. Rice Genet Newsl. 1988, 5: 149-151.
Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. 1989, Cold Spring Harbour Laboratory Press, New York, 2
Cho YG, Ishii T, Temnykh S, Chen X, Lipovich L, Park WD, Ayres N, Cartinhour S, McCouch SR: Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativa L.). Theor Appl Genet. 2000, 100: 713-10.1007/s001220051343.
Callen DF, Thompson AD, Shen Y, Phillips HA, Richards RI, Mulley JC, Sutherland GR: Incidence and origin of “null” alleles in the (AC) n microsatellite markers. Am J HumGenet. 1993, 52: 922-927.
Jain S, Jain RK, McCouch SR: Genetic analysis of Indian aromatic and quality rice (Oryza sativa L.) germplasm using panels of fluorescently-labeled microsatellite markers. Theor Appl Genet. 2004, 109: 965-977. 10.1007/s00122-004-1700-2.
Jaccard P: Nouvelle recherches sur la distribution florale. Bulletin de la Socie’te’ Vaudoise des Sciences Naturelles. 1908, 44: 223-270.
Rohlf FJ: NTSYSpc. Numerical Taxonomy and Multivariate Analysis System, 2. 1997, Exeter Software, Setauket, New York
Yap IP, Nelson R: Win Boot: A Programm for Performing Boot Strap Analysis of Binary Data to Determine the Confidence Limits of UPGMA-Based Dendrograms. 1995
Anderson PA, Lawrence GJ, Morrish BC, Ayliffe MA, Finnegan EJ, Ellis JG: Inactivation of the Flax rust resistance gene M associated with loss of a repeated unit within the leucine rich repeat coding region. Plant Cell. 1997, 9: 641-651. 10.1105/tpc.9.4.641.
Sanger F, Nicklen S, Coulson AR: DNA sequencing with chain-terminating inhibitors. PNAS. 1977, 74: 5463-5467. 10.1073/pnas.74.12.5463.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Nordborg M, Weigel D: Next-generation genetics in plants. Nature. 2008, 456: 11-10.1038/nature07629.
Walsh PS, Fildes NJ, Reynolds R: Sequence analysis and characterization of stutter products at the tetranucleotide repeat locus vWA. Nucleic Acids Res. 1996, 24: 2807-2812. 10.1093/nar/24.14.2807.
Sengupta S, Das B, Prasad M, Acharyya P, Ghose TK: A comparative survey of genetic diversity among a set of Caricaceae accessions using microsatellite markers. SpringerPlus. 2013, 2: 345-10.1186/2193-1801-2-345.
Badillo VM: Carica L. vs. Vasconcellea St.-Hil. (Caricaceae) con la rehabilitación de este último. Ernstia. 2000, 10: 74-79.
Van Droogenbroeck B, Breyne P, Goetghebeur P, Romeijn-Peeters E, Kyndt T, Gheysen G: AFLP analysis of genetic relationships among papaya and its wild relatives (Caricaceae) from Ecuador. Theor Appl Genet. 2002, 105: 289-297. 10.1007/s00122-002-0983-4.
Ram M: Papaya. 2005, Indian Council of Agricultural Research, New Delhi India
Aradhya MK, Manshardt RM, Zee F, Morden CW: A phylogenetic analysis of the genus Carica L. (Caricaceae) based on restriction fragment length variation in a cpDNA intergenic spacer region. Genet Res Crop Evol. 1999, 46: 579-586. 10.1023/A:1008786531609.
Olson ME: Intergeneric relationships within the Caricaceae– Moringaceae clade (Brassicales) and potential morphological synapomorphies of the clade and its families. Int J Plant Sci. 2002, 163: 51-65. 10.1086/324046.
Olson ME: Combining data from DNA sequences and morphology for a phylogeny of Moringaceae (Brassicales). Syst Bot. 2002, 27: 55-73.
Kyndt T, Van Droogenbroeck B, Haegeman A, Roldán-Ruiz I, Gheysen G: Cross-species microsatellite amplification in Vasconcellea and related genera and their use in germplasm classification. Genome. 2006, 49: 786-798. 10.1139/G06-035.
Nürnberger T, Kemmerling B: Receptor protein kinases - pattern recognition receptors in plant immunity. Trends Plant Sci. 2006, 11: 519-522. 10.1016/j.tplants.2006.09.005.
Sun X, Cao Y, Yang Z, Xu C, Li X, Wang S, Zhang Q: Xa26, a gene conferring resistance to Xanthomonas oryzae pv. oryzae in rice, encodes an LRR receptor kinase-like protein. Plant J. 2004, 37: 517-527. 10.1046/j.1365-313X.2003.01976.x.
Iyer AS, McCouch SR: The rice bacterial blight resistance gene xa5 encodes a novel form of disease resistance. MPMI. 2004, 17 (12): 1348-1354. 10.1094/MPMI.2004.17.12.1348.
Qi X, Bakht S, Qin B, Leggett M, Hemmings A, Mellon F, Eagle J, Werck-Reichhart D, Schaller H, Leso A, Melton R, Osbourn A: A different function for a member of an ancient and highly conserved cytochrome P450 family: From essential sterols to plant defense. Proc Natl Acad Sci U S A. 2006, 103 (49): 18848-18853. 10.1073/pnas.0607849103.
Crombie WM, Crombie L: Pathogenicity of the take-all fungus to oats: its relationship to the concentration and detoxification of the four avenacins. Phytochemistry. 1986, 25: 2075-2083. 10.1016/0031-9422(86)80069-3.
Guevara BPE, Mejia ZE, Leon CMT, Romero LI, Gutierrez AJG: Control biologico de Colletotrichum gloeosporioides [(Penz.) Penz. y Sacc.] en papaya maradol roja (Carica papaya L.) y fisiologia postcosecha de frutos infectados. Rev Mex Fitopatol. 2004, 22: 198-205.
Correia KC, Souza BO, Câmara MPS, Michereff SJ: Departamento de Agronomia. 2013, Universidade Federal Rural de Pernambuco, Recife, 52171-900, Pernambuco, Brazil
Quintal PC, Flores TG, Buenfil IR, Tintore SG: Antifungal activity in ethanolic extracts of carica papaya L. cv. Maradol leaves and seeds. Indian J Microbiol. 2011, 51 (1): 54-60. 10.1007/s12088-011-0086-5.
Brandstatter I, Kieber JJ: Two genes with similarity to bacterial response regulators are rapidly and specifically induced by cytokinin in Arabidopsis. Plant Cell. 1998, 10: 1009-1020. 10.1105/tpc.10.6.1009.
Mutoh N, Simon MI: Nucleotide sequence corresponding to five chemotaxis genes in Escherichia coli. J Bacteriol. 1986, 165 (1): 161-166.
Brown PH, Welch RM, Cary EE: Nickel: a micronutrient essential for higher plants. Plant Physiol. 1987, 85: 801-803. 10.1104/pp.85.3.801.
Mishra B, Kushwaha R, Pandey FK: Determination and comparative study of mineral elements and nutritive value of some common fruit plants. J Pure Appl Sci Tech. 2012, 2 (2): 64-72.
Graham RM, Welch C, Walker D: A Role of Nickel in the Resistance of Plants to Rust. 1985, Proceedings of the third Australian Agronomic Conference, Hobart Tasmania, Australia
Mishra D, Kar M: Nickel in plant growth and metabolism. Bot Rev. 1974, 40: 395-452. 10.1007/BF02860020.
Liao CH, Wells JM: Association of pectolytic strains of Xanthomonas campestris with soft rots of fruits and vegetables at retail markets. Phytopathology. 1987, 77: 418-422. 10.1094/Phyto-77-418.
Acknowledgement
The authors wish to thank United States Department of Agriculture, Indian Council of Agricultural Research, Indian Institute of Horticultural Research, Orissa University of Agriculture and Technology and Tamil Nadu Agriculture University for contributing the Caricaceae accessions. They also wish to thank the Department of Science and Technology for providing the research funding through Bose Institute and for providing the fellowship to Basabdatta Das. Thanks are also due to the University of Calcutta for providing fellowship to Samik Sengupta.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
SS procured all the Caricaceae accessions and did all the experiments pertaining to DNA extraction, PCR, PAGE and collected data. BD was involved in data analysis and drafting of the manuscript and helped with data collection. MP did the bootstrap analysis and constructed the dendrogram. PA was involved with the conception of the work. TKG was involved with the conception of the work and gave the final approval to the version of the manuscript that is being sent for consideration for publication. All authors read and approved the final manuscript.
Electronic supplementary material
12863_2014_137_MOESM3_ESM.jpeg
Additional file 3: Figure S1.: Dendrogram of 41 Caricaceae accessions using SSR and RGA profiles based on Jaccard’s genetic similarity coefficient. (JPEG 94 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Sengupta, S., Das, B., Acharyya, P. et al. Genetic diversity analysis in a set of Caricaceae accessions using resistance gene analogues. BMC Genet 15, 137 (2014). https://doi.org/10.1186/s12863-014-0137-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-014-0137-0