
Abstract
Background
Phylogeographical approaches explain the genetic diversity of local organisms in the context of their geological and geographic environments. Thus, genetic diversity can be a proxy for geological history. Here we propose a genus of woodland isopod, Ligidium, as a marker of geological history in relation to orogeny and the Quaternary glacial cycle.
Results
Mitochondrial analysis of 721 individuals from 97 sites across Japan revealed phylogenetic divergence between the northeastern and southwestern Japan arcs. It also showed repeated population expansions in northeastern Japan in response to Quaternary glacial and interglacial cycles. Genome-wide analysis of 83 selected individuals revealed multiple genetic nuclear clusters. The genomic groupings were consistent with the local geographic distribution, indicating that the Ligidium phylogeny reflects its regional history.
Conclusion
Ligidium DNA sequence analysis can provide insight into the geological, geographical, and paleoenvironmental history of the studied region.
Similar content being viewed by others

Background
The geographic distribution of genetic diversity results from biological history and reflects fragmentation, bottlenecks, sudden population expansions, and migration routes. Studies of phylogeography explain such genetic diversity in the context of past events [1]. An early phylogeographic study identified the refugia and post-warming dispersal routes of various animals, from grasshoppers to bears, in the Quaternary ice ages [2]. The formation of islands following plate movements explains the dramatically different faunas bordering Wallace’s Line in the Indo-Australian Archipelago [3]. South American species divergence was affected by Pleistocene climate change and orogenic events in the Miocene and Pliocene [4]. These results suggest that phylogeographic analysis can provide insight into past geological and environmental events (e.g., [5, 6]). However, the usefulness of the approach has not been evaluated.
Conventional paleoenvironmental reconstruction using geological records can generate continuous time-series records at specific sites but is at a disadvantage when reconstructing the spatial distribution of the paleoenvironment. A phylogeographical approach would compensate for this weakness. Such an approach can increase the spatial resolution of the reconstructed paleoenvironment by increasing the spatial sampling density of the target species (Fig. 1a). We selected terrestrial isopods of the genus Ligidium as a case study for a phylogeographical analysis with the aim of assessing regional environmental changes in Japan’s paleohistory. Ligidium fulfills requirements for a marker organism, including high sensitivity to environmental disturbances (such as climate change or volcanic eruptions), low mobility, marked regional genetic differentiation, and ease of collecting populations.

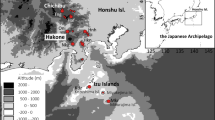
Conceptual diagram of this study and paleogeographic and sampling maps. a Conceptual diagram of the method used to analyze Ligidium as a paleogeological marker. b Paleogeographic map of the Japanese Archipelago. Colored areas are land. Map constructed based on a geological survey of Japan [7]. c Map of Ligidium sampling sites. Stars, locations where individuals used for restriction site-associated DNA sequencing (RAD-seq) were collected. Illustrations were created in Adobe Illustrator
The following six species have been reported in Japan: Ligidium japonicum Verhoeff, 1918; L. koreanum Flasarova, 1972; L. paulum Nunomura, 1976; L. ryukyuense Nunomura, 1983; L. kiyosumiense Nunomura, 1983; and L. iyoense Nunomura, 1983 [8]. All species except L. koreanum are endemic to Japan [8]. The selected genus Ligidium exhibits a strong forest preference [9, 10], dislikes dryness, and is not highly mobile [11]. Furthermore, it is easy to collect a sufficient number of samples. Because a large area in the Japanese Archipelago is forested, this genus has a wide distribution. Studies of Ligidium have been conducted in Greece, China, and Japan [12,13,14,15]. For example, in the Aegean Islands of Greece, Ligidium can be sorted into genetic clusters corresponding to their paleogeographic histories, suggesting that the islands’ isolation was a major determinant of their genetic diversity [12]. In China, the uplift of the Qinghai–Tibet Plateau likely affected Ligidium species divergence [13]. Our preliminary analysis showed that the Hokkaido population in northern Japan experienced a bottleneck caused by the Last Glacial Maximum (LGM) [14], suggesting that this genus is sensitive to glacial cycles. Furthermore, the phylogeography of independent lineages of L. japonicum reflects its dispersal history from the mountains to the plains and peninsulas in the Kanto Plain of Japan [15].
We analyzed the cytochrome oxidase I (COI) region of mitochondrial DNA (mtDNA) and performed genome-wide analysis via restriction site-associated DNA sequencing (RAD-seq) to examine the regional distribution of the genetic diversity of Ligidium throughout Japan (Fig. 1c, Table 1). The Honshu arc of the Japanese Archipelago was separated near the Fossa Magna 22–7 million years ago (Ma) (Fig. 1b). The southwestern Japan arc remained relatively stable and exposed, whereas the northeastern Japan arc was submerged in the sea with scattered islands until 3 Ma (Fig. 1b) [7, 16]. Northeastern Japan experienced repeated changes between glacial coniferous and interglacial broadleaf vegetation in response to glacial–interglacial cycles after 2.5 Ma [17]. Based on knowledge of the biology of Ligidium, we formulated the following two hypotheses. First, due to its low mobility, Ligidium may exhibit east–west divergence reflecting the formation history of the Japanese Archipelago, as seen in insects with low dispersal ability [18].
If tectonic history had influenced Ligidium, we would expect that populations would exhibit divergence in relatively stable southwestern Japan since ancient times. In northeastern Japan, submerged until approximately 7 Ma, Ligidium would exhibit increased genetic differentiation after the uplifting of land from 3 to 7 Ma [16]. Second, the Quaternary glacial cycles might have caused the recent population size increase of Ligidium in cold northeastern Japan, as on Hokkaido. We evaluated these hypotheses.
Results
Phylogeny based on mtDNA
The aligned COI sequences in the dataset were each 496 bases long. Phylogenetic trees for the mtDNA sequences were constructed using Bayesian analysis and maximum likelihood (ML) methods (Figs. 2 and 3). We identified 111 mtDNA-operational taxonomic units (OTUs) (Table 2) in the phylogenetic trees, which were divided into eight monophyletic groups (Clades I–VIII) supported by high posterior probabilities (> 0.99) and bootstrap values (> 85%). Individuals of Aomori at the northern tip of Honshu constituted Clade I. Clades II and III are distributed over a wide area from the Fossa Magna belt to northeastern Japan. Clades IV and V live in the Fossa Magna belt and eastern Japan, and Clade VI inhabits western and eastern Japan beyond the Fossa Magna. Clade VII is distributed from the west to the east of the Fossa Magna. Individuals from western Japan and the islands constitute Clade VIII. The monophyletic groups were consistent with four morphological species (Ligidium japonicum, L. koreanum, L. sp. NIIGATA1, and L. sp. EHIME1), excluding some lineages in the clade of L. koreanum (Fig. S1). Most of the specimens in northeastern Japan and the Fossa Magna areas constituted a clade (eastern Japan clade; Fig. 2b). However, some specimens from the eastern areas were closely related to the western samples (e.g., Clades VI and VII). By contrast, western samples included deeper-branching lineages compared to the eastern ones (Fig. 2b) and did not constitute one western clade. Interestingly, the phylogenetic analyses showed that the divergence of multiple lineages (Clades I–VIII) occurred around 7–3.5 Ma and after 3 Ma throughout Japan (Fig. 2, Table 3). However, depth differed between eastern and western Japan.

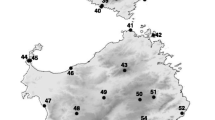
Bayesian phylogenetic tree of Japanese Ligidium with the geological history of the Japanese Islands and global climate change. a Paleogeographic map of the Japanese Archipelago. Colored areas are land. The map was constructed based on a geological survey of Japan [7]. b Bayesian phylogenetic tree. Colors for the operational taxonomic units (OTUs) match those of their sampling sites on the Japan map. See Table 2 for mitochondrial DNA (mtDNA)-OTU details. Colored borders and symbols indicate areas for which data were downloaded and downloaded data, respectively. We used the evolutionary rate of 1.68% per million years (3.36% divergence rate) for time estimation [19]. Clades I–VIII (dark boxes) have high posterior probabilities (> 0.99) and high bootstrap values (> 85%) in the maximum-likelihood (ML) tree (Fig. 3) and show characteristic branching. The numbers shown at tree nodes are ages. Horizontal lines are the node age ranges with 95% highest posterior density values. c Benthic δ18O records from 57 deep-sea sediment cores distributed worldwide over the past 5 million years [20]

mtDNA-based ML tree. Numbers at tree nodes are bootstrap values from 1000 pseudo-replicates (≥ 50%). Number on horizontal line is branch length measured as the number of substitutions per site. Colors correspond to the archipelago map and OTUs. Colored borders and symbols indicate areas for which data were downloaded and downloaded data, respectively. See Table 2 for details on the OTUs
In Japan, mtDNA-OTU 1 and 2 diverged recently, although samples were found across the northern (Hokkaido and Aomori) and central (Niigata) areas of Japan (Fig. 2b). We constructed a Bayesian tree and estimated divergence time to investigate the phylogenetic relationships of these samples (Fig. 4). Some southerly Niigata haplotypes were older than the Hokkaido and Aomori haplotypes. The oldest Niigata haplotype diverged ~ 1.5 Ma, whereas the haplotypes from Hokkaido and Aomori, located in the northernmost part of Japan, diverged ~ 0.4 Ma.

Bayesian tree of mtDNA-OTU 1 and 2 with divergence time estimation. Haplotype NJa is shared among Hokkaido, Aomori, and Niigata. Haplotypes Niigata2 and NJN1, 3, and 4 are from Niigata (Niigata inland area [site ID 32] and Echigo Plain [site ID 33]) We used an evolutionary rate of 1.68% per million years (3.36% divergence rate) for time estimation [19]. Horizontal lines are node age ranges with 95% highest posterior density values
Estimated historical demographic patterns inferred from mtDNA
We conducted population genetic analyses on 44 mtDNA-OTUs (northeastern Japan, 8; Fossa Magna belt, 19; western Japan, 17) based on the following criteria: close distribution, the presence of multiple haplotypes, and more than five individuals (Table 2). Mismatch distribution analysis detected population expansion and an expansion parameter (τ) was estimated to identify the generations in which the population size expanded (see Table S1 for calculation results). Smooth and unimodal frequency distributions indicated a recent, sudden population expansion in 17 mtDNA-OTUs, distributed as follows: northeastern Japan, 5; Fossa Magna belt, 5; western Japan, 7 (Fig. S2). Bimodal distributions were produced for 14 mtDNA-OTUs (northeastern Japan, 2; Fossa Magna belt, 7; western Japan, 5), showing that they might have experienced two expansions. Tajima’s D [21] and Fu’s Fs [22] neutrality tests assess the degree of deviation from neutral evolution and the presence or absence of natural selection. We found negative values for 35 mtDNA-OTUs, indicating population expansion (northeastern Japan, 7; Fossa Magna belt, 16; western Japan, 12), for both or either of the tests (Table 4). For the mtDNA-OTUs that exhibited expansion trends based on the mismatch distribution graph and neutrality tests, the expansion dates were 60 ~ 30 ka for nine mtDNA-OTUs (northeastern Japan, 3; Fossa Magna belt, 3; western Japan, 3), and 20 ~ 10 ka for another 10 mtDNA-OTUs (northeastern Japan, 2; Fossa Magna belt, 2; western Japan, 6) (Table 4).
Results of genome-wide analyses
Single nucleotide polymorphisms (SNPs) and loci calling
After trimming the raw reads, we used the denovo_map.pl pipeline and the populations program in Stacks for assembly, aligning, and SNP calling of paired-end reads [23]. No SNPs were shared by > 70% of all individuals. Therefore, we selected likely phylogenetically close individuals based on the mtDNA results and conducted repeated analyses to determine how to divide them to maximize the SNPs and members (Fig. S3, Table S3). We were able to define two groups: western and northeastern Japan. We selected 67 individuals in the northeastern group and 10 in the western group (Table S3). SNP calling for the outgroup (Ligia) was not successful. After complete filtering, we identified 67 individuals sharing 79 loci and 961 SNPs in northeastern Japan and 10 individuals sharing 301 loci and 1567 SNPs in western Japan (Table S3A). SNP calling remained unsuccessful for all individuals even when we used ipyrad [24]; therefore, grouping was conducted based on the Stacks results. More SNPs were called in ipyrad than in Stacks: 134 loci and 3319 SNPs in northeastern Japan, and 747 loci and 12,866 SNPs in western Japan (Table S3B). However, with only 24 loci and 644 SNPs, there were insufficient SNPs to conduct analyses between the eastern and western populations.
Nuclear DNA-based clusters
We performed Bayesian cluster analysis to assess population genetic structure for genome-wide data analyses. For the SNPs called by Stacks, the posterior probability values for the number of clusters (K) did not converge between K = 5 and 12, so the simulations were rerun 20 times for each K value from 5 to 12. After recalculating, because Ln(P) remained high for several values, the number of clusters was estimated based on the delta K method [25]. K was calculated to be 11, and the main nuclear clusters in each region were consistent with the geographical distribution in northeastern Japan (Fig. 5a). With the ipyrad SNPs, the nuclear clusters converged on 3 (Fig. 5b). Although the samples of mtDNA-OTU 1 exhibited slight genetic differences based on the mtDNA phylogenetic trees, they belonged to multiple nuclear clusters. Based on principal component analysis (PCA), the northeastern nuclear clusters were divided into three major clusters separated by distribution area. However, on Mt. Kakuda (Site ID 31) and in the Niigata inland area (Site ID 32), some clusters coexisted. This coexistence was also observed in the mtDNA-OTUs at the Niigata sites. We found only one nuclear cluster in Hokkaido, where there was only one mtDNA-OTU. In the western Japan populations, 10 samples were separated into two nuclear clusters based on their structure and the PCA results, which were consistent with the geographic distribution and mtDNA groups (Fig. 5c, d). However, this may be an effect of the small sample size.

Genome-wide analysis results. Genetic structure of the populations determined using Bayesian cluster analysis: a Stacks for northeastern populations, b ipyrad for northeastern populations, c Stacks for western populations, and d ipyrad for western populations. For each result of the genome-wide single-nucleotide polymorphism (SNP) cluster analysis, we depict the structure (top), structure map (left), and principal component analysis (PCA) plot (right). PCA is a dimensionality reduction technique that identifies correlations and patterns in a dataset. Same-colored circles indicate membership of the same cluster in Structure and the same sampling site in PCA. Each vertical line represents an individual, and the length is the posterior probability of membership in each cluster. The lists below the genetic structure data contain mtDNA and sampling site information (see Table 1)
Although all nuclear clusters were consistent with the geographic distributions, according to the mtDNA-OTU type, several individuals of the Niigata nuclear clusters possessed the same or genetically similar mtDNA haplotype as the Hokkaido individuals (Fig. 5a, b). Meanwhile, individuals from the Hokkaido–Aomori nuclear cluster did not have the Niigata mtDNA-OTU haplotype, indicating mtDNA introgression from the Hokkaido–Aomori population to the Niigata population in one direction. According to the morphological examination, the specimens from Hokkaido–Aomori were L. japonicum, but an undescribed species, Ligidium sp. NIIGATA1, was present in the Niigata specimens that had experienced introgression (Fig. S1). This suggests that intra-clade mtDNA introgression occurred within and between species.
Pairwise Fst
Pairwise Fst values for the genome-wide SNPs revealed a population structure in the nuclear clusters (Table 5). Fst, which represents the degree of genetic differentiation among populations, ranges from 0 to 1, with an Fst > 0.25 indicating high genetic differentiation [26]. We obtained high and significant Fst values for northeastern and western Japan including the mtDNA-OTU and nuclear clusters in which gene introgression had occurred (Table5, Fig. 5).
Discussion
Influence of the tectonic history of the Japanese Archipelago on the phylogenetic divergence of domestic Ligidium
We found high phylogenetic diversity in Ligidium in Japan, indicating that this genus is highly diversified. In particular, the phylogenetic divergences of several clades occurred around 7–3.5 Ma (Fig. 2, Table 3). Between 12 and 7 Ma, large areas of the northeastern part of the Japanese Archipelago and the Fossa Magna belt sank below sea level, but at 7–3 Ma, tectonic movement raised the land (Fig. 2a) [7, 27]. The northeastern and southwestern Japan arcs meet at the Fossa Magna region. Thus, the divergence seen at 7–3.5 Ma might have been triggered by uplift and exposure of the northeastern Japan arc.
Divergence of Clades II–VII (Fig. 2b) suggested that uplift and land exposure enabled the dispersal of Clades II–VII and their phylogenetic differentiation. Although Ligidium is distributed nationwide, only these six lineages spread on (Clades IV, V, and VII) or beyond (Clades II, III, and VI) the Fossa Magna and diverged at the time of the uplift and exposure. Clade I bifurcated in Aomori at the northern tip of Honshu, far from the Fossa Magna zone, but its bifurcation may also be attributed to the tectonic uplift of northeastern Japan. It is also possible that the phylogenetic divergence of Clades II, III, and V was due to the uplift of northeastern Japan and global cooling of the area 7 ~ 5.4 Ma. The divergence suggests a westward migration of Ligidium. Global ocean surface temperature records indicate that cooling was more marked at higher latitudes [28]. Thus, it is likely that migration occurred from high-latitude northeastern Japan to the Fossa Magna belt.
The nuclear and mtDNA results showed that Clade VI diverged from the western Japan lineage after 4.8 Ma (Table 3) and then dispersed to northeastern Japan with uplift of the northeastern Japan arc. Clade VI contained individuals from the nuclear cluster of the Niigata area (Site IDs 30, 31, and 32) in the nuclear ML tree (Fig. S1h). This nuclear cluster diverged from its ancestors earlier, but its internal clusters are immature, suggesting more recent dispersal to its current habitat.
Clade VIII, which encompassed Ligidium sp. CHUGOKU1, was found in an area far from the Fossa Magna zone (Figs. 2 and 3), suggesting that its divergence was not related to the uplift event. Clade VIII represents the divergence of Ligidium populations on Oki Island and Honshu. Oki Island repeatedly sank to the bottom of the sea but became terrestrial by 7–3 Ma, which corresponds to the time of this divergence [29].
Similarly, the formation history of the Japanese Archipelago may underlie why many species and genetic lineages in southwestern Japan diverged earlier from their ancestors than those in northwestern Japan. During the middle Miocene (16–10 Ma), the northeastern Japan arc comprised an archipelagic sea due to the expansion and large-scale subsidence of the Japan Sea [16, 29]. By contrast, the southwestern Japan arc west of the Fossa Magna was already elevated and had been mountainous for 15 million years [16, 29]. Older lineages are presumed to have inhabited southwestern Japan due to the ancient stable land areas. The clade belonging to the Chinese lineage might have differentiated when the Japanese Archipelago was connected to the continent and inhabited the southwestern Japan arc when it was separated from the Eurasian continent by the formation of the Japan Sea. This historical background may also explain the divergence depth similarities between the western Japanese lineage and continental Chinese Ligidium. According to genome-wide SNPs, genetic segregation of the western and eastern Japanese populations likely had the same cause. Similar segregations have been seen in other taxa [18, 30, 31].
Influence of quaternary glacial cycles on domestic Ligidium
The Northern Hemisphere ice sheets expanded and glacial–interglacial contrasts were enhanced after 2.5 Ma [20]. Our phylogenetic tree suggested that mtDNA-OTU 1 and 2 divergences between Hokkaido–Aomori and Niigata occurred repeatedly after the beginning of the Quaternary glacial cycle around 2.5 Ma (Fig. 4). Our data indicated a southward migration of L. japonicum from northeastern populations during the glacial cycle. Additionally, the conservation of the nuclear cluster indicated that the migration began recently.
Because warming is assumed to have increased the genetic diversity of L. japonicum in Hokkaido [14] after the LGM, it is possible that climate oscillations during the glacial cycle led to the diversification of Japanese Ligidium. Population genetic analyses of mtDNA sequences re-aligned according to their geographic distribution and mtDNA-OTU type showed that 34 of 45 OTUs underwent population expansions (Table 4). Although only nine groups were significant in neutrality tests, the mismatch distribution graphs and mtDNA phylogenetic tree results showed trends of population expansions (Fig. 2, Fig. S2, Table 4), so we discussed 34 groups. The northeastern Japanese lineages could be attributed to the Quaternary glacial cycle. In the northeastern populations, seven of eight groups had negative values in neutrality tests (Tajima’s D [21]; Fu’s Fs [22]). The estimated expansion dates of almost all these groups are within the last 50,000 years. The LGM period was the most recent major climatic event. As in the Hokkaido population [14], expansions between 20 and 10 ka might have been influenced by post-LGM warming. In addition, marine isotope stage (MIS) 4 (71–57 ka) was as cold as the LGM. The expansions between 30 and 60 ka correspond to MIS 3, the interval between the LGM and MIS 4. Analysis of pollen fossils in the sediments of Lake Nojiri, located in the middle of the Japanese Archipelago, indicated that MIS 3 was warmer than MIS 4 [32]. The mtDNA phylogenetic trees also suggested that Ligidium was affected by the glacial cycle. Multiple synchronous divergences in geographically distant regions occurred after 3 Ma (Fig. 2).
In contrast to northeastern Japan, many western mtDNA-OTUs, such as L. koreanum and Ligidium sp. CHUGOKU1, yielded positive values in neutrality tests. However, the following groups had negative values and expansions within the last 100 ka: OTUs from islands (mtDNA-OTU 94, 95, and 96), two OTUs from the Chugoku region of western Honshu (mtDNA-OTU 4 and 6), one OTU from the southernmost of the four main islands of Japan, Kyushu (mtDNA-OTU 104), and two OTUs from the smallest of the four main islands of Japan, Shikoku (mtDNA-OTU 65 and 103).
The LGM and MIS 4 glacial periods may have affected the Chugoku groups as in northeastern Japan. These Chugoku groups of L. japonicum (Fig. S1, Table S4) suggest past migrations from the Fossa Magna. L. japonicum was affected by the LGM cooling in Hokkaido [14]. Moreover, the Hokkaido–Aomori population moved southward with the glacial cycle (Fig. 4). L. japonicum populations in northeastern Japan, including Hokkaido, were influenced by the glacial periods and might have been sensitive to climate change during those periods.
A population expansion of the mtDNA-OTU 4 clade occurred 10–7 ka. This population might have escaped from the sea level rise caused by the Jomon transgression. The Jomon transgression is associated with the combination of rapid glacial retreat and the slow uplift of the continental margin by hydro isostasy after the LGM, which peaked ca. 6 ka [33]. The mtDNA-OTU 103 population expansion corresponded to this time. The mtDNA-OTU 65 expansion date was slightly earlier than that of the Jomon transgression but coincided with the postglacial period. The population expansion in Kyushu occurred after MIS 4, suggesting the influence of warming after a cold period. Alternatively, the population might have been affected by the arrival of humans—archaeological evidence indicates that paleolithic people arrived in Japan from the continent 40–30 ka, after which their population expanded [34]. Kyushu faces Tsushima Island, a route of human expansion.
Discrepancies between the mtDNA and genome-wide analysis results
Classification often uses mtDNA barcodes as molecular tools. However, we identified mtDNA introgressions in individuals belonging to different nuclear clusters. Our results suggest that in Ligidium, hybridization could occur among individuals with a high degree of genetic differentiation (Fst > 0.88; Fig. 5, Fig. S1, Table 5). Therefore, species identification based on mtDNA may lead to misidentification. However, the mtDNA-OTU and nuclear clusters in which no gene introgression had occurred were consistent (Fig. S1). Also, genetic differences between the Ligidium sp. EHIME1 and the northeastern Japan populations were so large that SNP sampling was unsuccessful, again agreeing with the molecular data and morphological observations.
Conclusions
The modern phylogeography of Japanese Ligidium was determined by archipelago formation and climate change, and indicated highly endemic genomic evolution in each habitat. Although genetic structures affected by old events were conserved in the nuclear genome, mtDNA recorded changes due to relatively recent events such as glacial cycles. Thus, different timescale reconstructions may be possible depending on the molecular markers selected. Slowly evolving gene regions may enable us to trace back thousands of years of history. Because Ligidium lives throughout the Northern Hemisphere [8], its nuclear genome may provide insight into the history of Northern Hemispheric tectonic dispersal. Although the oldest fossil of Ligidium is Paleogenic [35], its DNA sequence may enable evaluation of older eras. Fossils of Oniscidea are rarely preserved because of the biochemical properties of the exoskeleton. Thus, they have likely lived since more ancient times than indicated by the fossil record [35]. Furthermore, as demonstrated in this study, a rapidly evolving genetic region may signal much younger geological events, such as volcanic eruptions.
Our results revealed high genetic diversity in domestic Ligidium. The intrageneric diversity of this taxon may exceed the number of species currently reported, and further work is needed to combine molecular data and morphological characters. Finally, evolution of the genome in the populations examined in this study highlights the importance of considering genetic structure in biological conservation taking also into account that most of the populations represent endemic taxa.
Materials and methods
Sample collection
We surveyed Ligidium populations in Japan. We collected 828 Ligidium specimens from 97 sites and sequenced 721 samples (Fig. 1, Table 1). Samples were preserved in 70–99.5% ethanol in 2-mL microtubes at 4 °C or room temperature.
DNA extraction, amplification, and sequencing
Genomic DNA was isolated from the muscles of the abdomen and legs with a DNA Mini Kit (Qiagen, Germantown, MD, USA). PCR amplification was conducted using the primer pair LCO-1490 and HCO-2198 [36]. Amplification and cycling conditions were as in a previous work [14]. Sequencing was conducted with an ABI 3130 Genetic Analyzer (Applied Biosystems, Waltham, MA, USA). Sequences were checked and assembled using MEGA7 [37]. Mitochondrial gene locus (CO1) sequences for 721 individuals were determined and aligned using ClustalW [38].
Phylogenetic analyses using mtDNA
Phylogenetic trees were constructed with the Bayesian analysis tool BEAST2 [39] and the ML method in MEGA7 [37]. The Bayesian tree analysis conditions were the strict clock model and a constant population size for 50 million step runs with sampling every 100 steps. The results were checked with Tracer v1.7.1 [40] after discarding a 10% burn-in, summarized with TreeAnnotator v1.10.4 (http://beast.community/index.html), and visualized with FigTree v1. 4.4 [41]. Because we focused on the events of the exposure of the northeastern Japan arc and convergence of eastern and western Japanese islands uniting until 3 Ma, we used the evolutionary rate calibrated by the events before this date. Previous studies using such events as calibration points showed that arthropods have a divergence rate of 3.0% [6, 19]. Thus, we used the rate of tenebrionid beetles (divergence rate 3.36%, clock rate 1.68%) used in many phylogenetic studies. Its rate estimation was performed with a calibration point of the formation of the mid-Aegean trench at 9–12 Ma. We selected the Tamura 3-parameter [42] + G + I evolutionary model in MEGA 7 for the ML tree. The ML tree’s nodal support values were obtained with 1000 bootstrap pseudo-replicates. We used sequences of subgenus Ligidium (Austria, Greece, and Japan) and, as an outgroup, relatively closely related Ligia spp. (Japan, Hawaii, unknown area) belonging to the family Ligiidae of the order Isopoda in which the genus Ligidium was also assigned until recently when the family was found to be polyphyletic [43]. Samples of the subgenus Ligidium (Ligidium) spp. came from Kagawa, Shikoku, Japan. We also downloaded data for the following Ligidium species: L. ryukyuense (AB626261; this specimen was misidentified and closely related to L. koreanum, based on reexamination in this study) collected on Okinawa Island, Ligidium sp. SHIZU2 (LC496507) and Ligidium sp. SHIZU1 (LC496506) collected in Shizuoka, a prefecture central to the Pacific Ocean side of Honshu, 213 sequences of individuals sampled mainly in the regions around Tokyo, Japan, that were previously published by Yoshino and Kubota [15], Ligidium species from Europe, and Oniscidea sp. collected in Shanghai, China. Table S4 lists the accession numbers for the downloaded data. Data from Yoshino and Kubota [15] were also used in the population genetics analyses.
Historical demographic patterns were examined by mismatch distribution analysis [44] and neutrality tests with Arlequin 3.5.1.2 [45]. Mismatch distribution analysis was conducted with spatial and sudden expansion models to detect population expansions and estimate the expansion parameter (τ) representing the generation marking the start of the population expansion. In the analyses, smooth and unimodal frequency distributions indicate an expanding population [45]. The time since the expansion in generations (t) can be estimated with the formula t = τ/2uk, where u is the evolutionary rate per generation and k is the number of sequence base-pairs [46]. We estimated the time since the demographic expansion in millions of years (T) with the formula T = τ/2μk, where μ is the evolutionary rate per million years. We used an evolutionary rate of 0.087 for Ligidium [14]. We performed Tajima’s D [21] and Fu’s Fs [22] as neutrality tests to calculate the degree of deviation from neutral evolution and determine the presence or absence of natural selection. Negative values for Tajima’s D and Fu’s Fs statistics for a population indicate that the population experienced expansion. Statistics and 95% confidence intervals were computed with 10,000 bootstrap pseudo-replicates.
RAD-seq
We performed RAD-seq analysis to search for SNPs in individuals from Niigata and Hokkaido obtained in previous studies [14], two regions in northern Japan (Aomori and Sendai), and two regions in western Japan (Shizuoka and Sendai). Tables 1 and S1 list the samples used, and the sampling sites are shown in Fig. 1c. Genomic DNA was isolated from almost whole-body tissues with a DNA Mini Kit (Qiagen). Libraries for RAD-seq were prepared with EcoRI and BglII restriction enzymes [47, 48]. The library was sequenced with 150 + 150 bp paired-end reads in one lane of an Illumina HiSeqX instrument (Illumina, San Diego, CA, USA) by Macrogen (Seoul, South Korea). Raw reads were trimmed using Trimmomatic-0.39 [49] with the following parameters: ILLUMINACLIP: adapter.fasta:2:30:10:keepBothReads, SLIDINGWINDOW: 4:15, CROP: 132, HEADCROP: 2, and MINLEN: 130. Sequences are available at the DNA Data Bank of Japan (DDBJ) Sequence Read Archive (DRA014204). We used two pipeline programs to call the SNPs: denovo_map.pl provided by Stacks [23] and ipyrad [24]. Following a previous work [50], we varied the combinations of the denovo_map.pl parameters as follows: (n, M) = (2, 1), (3, 2), (4, 3), (5, 6), and selected (n, M) = (2, 1), which called the most SNPs. We used Stacks’ populations program to analyze populations of individual samples, calculate population genetics statistics, and export data in various output formats for analysis. PLINK v1.90b6.18 [51] was used for data handling. Alleles with a frequency of < 1% and sites with > 50% heterozygosity were removed. Only SNPs shared by more than 80% of the individuals were retained. With ipyrad, loci with frequencies of > 50% heterozygosity were removed, and SNPs shared by more than 70% of the local populations were retained. We retained SNPs shared by at least two individuals and filtered out individuals that did not have 80% of all SNPs using TASSEL 5 [52]. After filtering with TASSEL 5, we used PGDSpider to convert the vcf files for other analyses [53].
Genetic structure analysis
We tested the ability of Structure v. 2.3.4 [54] to determine the genetic structure of populations using Bayesian cluster analysis. Ten simulations were run, with the burn-in period and Markov chain Monte Carlo iterations set to 105 and 106, respectively. The maximum value of K was determined based on the mtDNA results and geographical distribution. For the Structure analysis, one SNP was randomly sampled from each locus to avoid the effect of linkage disequilibrium. The python script vcf_single_snp.py (radseq/vcf_single_snp.py at master · pimbongaerts/radseq · GitHub) was used to obtain the one SNP datum from ipyrad, and drawings were created using the R package pophelper [55]. In addition, PCA was performed to visualize the genetic differences among populations using the adegenet package in R [56, 57]. We obtained pairwise Fst values for the RAD-seq dataset using Arlequin 3.5.1.2. Fst values were used to test population structure, supported by cluster analysis, and statistical significance was based on 1000 restored extractions.
Statistics and reproducibility
Tables 1, 2, and S1 list summaries of the sample size. Statistical analyses were conducted for distribution analysis, neutrality tests, phylogenetic tree constructions, structure analysis, and calculations of Fst. The methods used for reproducibility checks are described in the corresponding methods section. A P-value < 0.05 was considered indicative of statistical significance, and the results were reproducible through access to the genetic data (see “Availability of data and materials”).
Availability of data and materials
mtDNA sequences were deposited in the DDBJ. RAD-seq sequences are available at the DDBJ Sequence Read Archive (DRA014204). The result outputs, scripts of SNP data (Software: https://doi.org/10.5281/zenodo.7393599), and a list of mtDNA accession numbers (Supplemental information: https://doi.org/10.5281/zenodo.7393601) can be downloaded from DRYAD (https://doi.org/10.5061/dryad.zkh1893bw) without any permissions.
Abbreviations
- ka:
-
Thousand years ago
- LGM:
-
Last glacial maximum
- Ma:
-
Million years ago
- MIS:
-
Marine isotope stage
- ML:
-
Maximum likelihood
- mtDNA:
-
Mitochondrial DNA
- OTU:
-
Operational taxonomic unit
- PCA:
-
Principal component analysis
- RAD-seq:
-
Restriction-site-associated DNA sequencing
- SNP:
-
Single-nucleotide polymorphism
References
Avise JC. Phylogeography: the history and formation of species. Cambridge: Harvard Univ press; 2000.
Hewitt G. The genetic legacy of the Quaternary ice ages. Nature. 2000;405:907–13.
Lohman DJ, de Bruyn M, Page T, von Rintelen K, Hall R, Ng PK, von Rintelen T. Biogeography of the Indo-Australian archipelago. Annu Rev Ecol Evol Syst. 2011;42:205–26.
Turchetto-Zolet AC, Pinheiro F, Salgueiro F, Palma-Silva C. Phylogeographical patterns shed light on evolutionary process in South America. Mol Ecol. 2013;22:1193–213.
Collins GE, Hogg ID, Convey P, Sancho LG, Cowan DA, Lyons WB, et al. Genetic diversity of soil invertebrates corroborates timing estimates for past collapses of the West Antarctic Ice Sheet. Proc Natl Acad Sci. 2020;117(36):22293–302.
Ota H. Geographic patterns of endemism and speciation in amphibians and reptiles of the Ryukyu Archipelago, Japan, with special reference to their paleogeographical implications. Res Popul Ecol (Kyoto). 1998;40(2):189–204.
Kano K, Kato H, Yanagisawa Y, Yoshida F. Stratigraphy and geologic history of the Cenozoic of Japan (in Japanese). Tsukuba: Geol Surv; 1991.
Schmalfuss H. World catalog of terrestrial isopods (Isopoda: Oniscidea). – Stuttgarter Beiträge zur Naturkunde, Serie A, Nr. Stuttgart: Staatl Mus fur Nat Stuttg; 2003.
Fujita Y, Watanabe H. Terrestrial isopods and their habitats: A case study in Kyoto city (in Japanese). For Res. 1999;71:1–7.
Nasu T, Kitagawa K, Karasawa S. Species compositions of terrestrial isopods in public parks of a commuter town in Japan. ZooKeys. 2018;801:389.
Kamihira Y. The influence of highway construction in forests on soil macrofauna (in Japanese). Res Bull Coll Exp For Hokkaido Univ. 1987;44:789–96.
Klossa-Kilia E, Kilias G, Tryfonopoulos G, Koukou K, Sfenthourakis S, Parmakelis A. Molecular phylogeny of the Greek populations of the genus Ligidium (Isopoda, Oniscidea) using three mtDNA gene segments. Zool Scr. 2006;35(5):459–72.
Wang J, Yang J, Zeng X, Li W. Integrative taxonomy on the rare sky-island Ligidium species from southwest China (Isopoda, Oniscidea, Ligiidae). BMC Zool. 2022;7(1):1–21.
Harigai W, Saito A, Suzuki H, Yamamoto M. Genetic Diversity of Ligidium Isopods in Hokkaido and Niigata, Northern Japan. Based on Mitochondrial DNA Analysis Zool. 2020;37(5):1–12.
Yoshino H, Kubota K. Phylogeographic analysis of Ligidium japonicum (Isopoda: Ligiidae) and its allied species reveals high biodiversity and genetic differentiation in the Kanto region. Japan Entomol Sci. 2022;25(2):e12501. https://doi.org/10.1111/ens.12501.
Nakajima T. Tectonics of sedimentary basins in and around Japan since the opening of the Sea of Japan. Jour Geol Soc Japan. 2018;124:693–722.
Igarashi Y, Oba T. Fluctuations of monsoons and insolation in the northwest Pacific during the last 144 kyr from a high-resolution pollen analysis of the IMAGES core MD01-2421. Quat Sci Rev. 2006;25:1447–59.
Tojo K, Sekiné K, Takenaka M, Isaka Y, Komaki S, Suzuki T, Schoville SD. Species diversity of insects in Japan: their origins and diversification processes. Entomol Sci. 2017;20(1):357–81.
Papadopoulou A, Anastasiou I, Vogler AP. Revisiting the insect mitochondrial molecular clock: the mid-Aegean trench calibration. Mol Biol Evol. 2010;27(7):1659–72.
Lisiecki LE, Raymo ME. A Pliocene-Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanography. 2005. https://doi.org/10.1029/2004PA001071.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–25.
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Eco. 2013;22(11):3124–40.
Eaton DA, Overcast I. ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics. 2020;36(8):2592–4.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Hartl DL, Clark AG, Clark AG. Principles of population genetics. Sunderland: MA, Sinauer associates; 1997.
Matsuzaki KM, Itaki T, Tada R, Kamikuri SI. Paleoceanographic history of the Japan Sea over the last 9.5 million years inferred from radiolarian assemblages (IODP Expedition 346 Sites U1425 and U1430). Prog Earth Planet Sci. 2018;5(1):1–33.
Herbert TD, Lawrence KT, Tzanova A, Peterson LC, Caballero-Gill R, Kelly CS. Late Miocene global cooling and the rise of modern ecosystems. Nat Geosci. 2016;9(11):843–7.
Kano K. Cenozoic History of the Japanese lslands (in Japanese). HGI Report. 1993;3:33–50.
Okamiya H, Sugawara H, Nagano M, Poyarkov NA. An integrative taxonomic analysis reveals a new species of lotic Hynobius salamander from Japan. PeerJ. 2018. https://doi.org/10.7717/peerj.5084.
Nakagawa H, Seki S, Ishikawa T, Watanabe K. Genetic population structure of the Japanese torrent catfish Liobagrus reinii (Amblycipitidae) inferred from mitochondrial cytochrome b variations. Ichthyol Res. 2016;63(3):333–46.
Kumon F, Kawai Y, Kigoshi T. Late Quaternary Climate Changes around the Japanese Alps, Central Japan (in Japanese). Jour Geogr. 2013;122(4):571–90.
Yasuhara M, Irizuki T, Yoshikawa S, Nanayama S. Holocene sea-level changes in Osaka Bay, western Japan: ostracode evidence in a drilling core from the southern Osaka Plain. Jour Geol Soc Japan. 2002;108(10):633–43.
Kaifu Y. Origin of modern humans: The Present State and prospects for the future research (in Japanese). Anthropol sci Jpn ser. 2005;113(1):5–16.
Broly P, Deville P, Maillet S. The origin of terrestrial isopods (Crustacea: Isopoda: Oniscidea). Evol Biol. 2013;27(3):461–76.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3(5):294–9.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, et al. BEAST 2: a software platform for bayesian evolutionary analysis. PLoS Comput Biol. 2014. https://doi.org/10.1371/journal.pcbi.1003537.
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol. 2018;67(5):901–4.
Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007. https://doi.org/10.1186/1471-2148-7-214.
Tamura K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+ C-content biases. Mol Biol Evol. 1992;9(4):678–87.
Dimitriou AC, Taiti S, Sfenthourakis S. Genetic evidence against monophyly of Oniscidea implies a need to revise scenarios for the origin of terrestrial isopods. Sci rep. 2019;9(1):1–10.
Schneider S, Excoffier L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics. 1999;152(3):1079–89.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7.
Rogers AR. Genetic evidence for a Pleistocene population explosion. Evolution. 1995;49(4):608–15.
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE. 2012. https://doi.org/10.1371/journal.pone.0037135.
Sakaguchi S, Sugino T, Tsumura Y, Ito M, Crisp MD, Bowman DM, et al. High-throughput linkage mapping of Australian white cypress pine (Callitris glaucophylla) and map transferability to related species. Tree Genet Genomes. 2015;11(6):1–12.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Paris JR, Stevens JR, Catchen JM. Lost in parameter space: a road map for stacks. Methods Ecol Evol. 2017;8(10):1360–73.
Purcell S, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:559–75.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–5.
Lischer HEL, Excoffier L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics. 2012;28(2):298–9.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59.
Francis RM. pophelper: an R package and web app to analyse and visualize population structure. Mol Ecol Resour. 2017;17(1):27–32.
Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24(11):1403–5.
Jombart T, Ahmed I. adegenet 1.3–1: new tools for the analysis of genome-wide SNP data. Bioinformatics. 2011;27(21):3070–1.
Acknowledgements
We thank Gohta Kinoshita and Takashi Hayakawa for their kind guidance on the experiment. We thank Yudai Segawa, Sohei Kikuchi, Fangxian Wang, Masaya Inagaki, Isami Nakai, Kaho Harigai and Toru Harigai for their help with sampling. Renata Szarek contributed to the polishing manuscript.
Funding
This study was conducted with the support of Grants-in-aid for Scientific Research (C) from the Japan Society for the Promotion of Science (JSPS) KAKENHI (20K06093 to SK and Kiban S 19H05595 to MY). The funding body played no role in the design of the study and collection, analysis, interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
WH wrote the manuscript and created the illustrations. WH, CZ, AS, and TY analyzed DNA. SK and TY carried out morphological observations. AN conducted the RAD-seq analysis. HS offered the use of his laboratory and was involved in the writing and interpretation of the paper. MY designed the study.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
In Japan, collecting Ligidium specimens is permitted by the current laws, and we did not conduct sampling in protection areas. All surveys were also performed in accordance with the current regulations and laws in Japan.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Harigai, W., Saito, A., Zemmoto, C. et al. History of the terrestrial isopod genus Ligidium in Japan based on phylogeographic analysis. BMC Ecol Evo 23, 38 (2023). https://doi.org/10.1186/s12862-023-02144-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-023-02144-8