
Abstract
Background
The flat periwinkles, Littorina fabalis and L. obtusata, are two sister species widely distributed throughout the Northern Atlantic shores with high potential to inform us about the process of ecological speciation in the intertidal. However, whether gene flow has occurred during their divergence is still a matter of debate. A comprehensive assessment of the genetic diversity of these species is also lacking and their main glacial refugia and dispersal barriers remain largely unknown. In order to fill these gaps, we sequenced two mitochondrial genes and two nuclear fragments to perform a phylogeographic analysis of flat periwinkles across their distribution range.
Results
We identified two main clades largely composed by species-specific haplotypes corresponding to L. obtusata and L. fabalis, with moderate to strong support, respectively. Importantly, a model of divergence with gene flow between the two species (from L. obtusata to L. fabalis) was better supported, both in Iberia and in northern-central Europe. Three mitochondrial clades were detected within L. fabalis and two within L. obtusata, with strong divergence between Iberia and the remaining populations. The largest component of the genetic variance within each species was explained by differences between geographic regions associated with these clades. Our data suggests that overall intraspecific genetic diversity is similar between the two flat periwinkle species and that populations from Iberia tend to be less diverse than populations from northern-central Europe.
Conclusions
The phylogeographic analysis of this sister-species pair supports divergence with gene flow. This system thus provides us with the opportunity to study the contribution of gene flow and natural selection during diversification. The distribution of the different clades suggests the existence of glacial refugia in Iberia and northern-central Europe for both species, with a main phylogeographic break between these regions. Although the genetic diversity results are not fully conclusive, the lower diversity observed in Iberia could reflect marginal conditions at the southern limit of their distribution range during the current interglacial period.
Similar content being viewed by others


Background
It is now widely recognized that reproductive isolation can progress in the face of gene flow [1]. However, examples where gene flow actually drives speciation (e.g. hybrid speciation or reinforcement) are less common [2]. Phylogeographic studies are an important source of information to understand the evolutionary history of a species [3]. Besides contributing to elucidate the demographic history of populations across the species distribution range, they provide crucial data to infer the geographic context of divergence between populations or species where specific hypotheses, such as the role of gene flow in speciation, can be further tested.
A well-established phylogeographic model for temperate regions of the Northern hemisphere claims that during Pleistocene glaciations many species were able to persist within refugia located at lower latitudes; and when the environmental conditions improved, i.e. during interglacial periods, populations expanded from southern refugia, in some cases meeting and hybridizing at higher latitudes [4,5,6,7]. These cyclic events of isolation followed by gene flow after secondary contact are known to have shaped the genetic diversity of many different species distributed across these regions [6, 8] and could even have contributed to speciation [9]. However, this phylogeographic model is probably an oversimplification particularly among marine taxa inhabiting the intertidal realm, as northern refugia during the last glacial maximum (LGM) have been suggested for several marine species, both on the western and eastern sides of the Atlantic [10, 11].
Intertidal species had to face both a reduction of suitable habitats during glacial periods due to ice-sheets covering most northern coastal areas and dramatic changes in the shoreline (sea level) within refugial areas [12,13,14]. These shifts not only had the potential to change the local selective pressures, in terms of wave exposure etc., but also to foster the contact and gene flow between populations and closely related species. Thus, in order to fully understand the phylogeographic patterns of intertidal species it is important to analyze the genetic diversity across the entire species range, without making assumptions about the location of refugia or the areas where hybridization and gene flow are likely to have happened.
Since the fossil record is often incomplete, the comparison of phylogeographic patterns among closely related species with a similar geographic distribution can be very useful to identify extrinsic barriers to gene flow impacting on both species or to disclose differences related to species-specific life history or ecological requisites [15,16,17,18,19]. For instance, closely related species with similar dispersal capacity and historical distribution range but showing distinct levels of genetic diversity when facing marginal habitat conditions can inform us about the ability of different taxa to resist environmental changes in different regions across their range [20,21,22,23]. This is fundamental to accurately predict the species responses to natural or human-mediated climate changes. Comparative phylogeographic studies between sister species can, in addition, reveal where and when introgressive hybridization may have occurred and its role in adaptation and speciation [2, 24].
The flat periwinkles comprise two sister species of the genus Littorina, L. fabalis and L. obtusata, that started to diverge about 1 million years (My) ago (see [25]). They have wide distribution ranges: both are present in the northern Atlantic coasts, from southwest Greenland to Iceland and across the Norwegian Sea to the White Sea; they are present in British Isles and show a rather continuous distribution from Scandinavia to southern Portugal [26]. In addition, L. obtusata is also present in the Atlantic coast of North America. The two species have internal fertilization, no free-swimming larvae and a generally low dispersal capacity, although they can occasionally raft longer distances attached to drifting macroalgae, which they also use as their main habitat [26]. Despite the wide range of overlap at the macrogeographic scale, the two species have relevant ecological differences. L. fabalis is generally present at the lower intertidal and L. obtusata is usually found in the mid-upper intertidal [27]. In non-tidal areas (e.g. Swedish coast) there is instead a division along the shoreline between moderately exposed and sheltered sites: L. obtusata is rare or absent in the former while both species are commonly found in the latter [27]. The two species are herbivores, but L. obtusata grazes on both Fucus spp. and Ascophyllum nodosum (the latter typical from sheltered habitats) whereas L. fabalis browses on epiphytes of the former. Predation, mainly exerted by crabs, is thought to have influenced the local distribution and life history of L. obtusata and L. fabalis, possibly contributing to their divergence [27]. Although the two species are morphologically very similar, adult L. obtusata tend to be larger than adult L. fabalis, presenting also a longer life-span [28]. There are no shell traits that completely distinguish the two species but male reproductive organs are diagnostic [26, 29, 30].
Gene flow between the two species has been suggested based on sharing of common mtDNA haplotypes throughout the Northeast Atlantic, although incomplete lineage sorting could not be ruled out [25]. In a recent study focussed in the Iberian Peninsula and based on microsatellites, hybrids between the two species were identified in one location in north Portugal [31]. Notably, since the effective population size of mtDNA is usually one quarter of that for nuclear markers, under neutrality, lineage sorting is expected to occur faster in mtDNA than for average nuclear markers [32, 33]. Thus, if shared haplotypes are only observed at mtDNA, introgressive hybridization is supported. Instead, if shared haplotypes are observed in both nuclear and mitochondrial markers, incomplete lineage sorting cannot be excluded. Therefore, the extension of the mtDNA analysis together with nuclear markers along the distribution range is important to clarify how widespread current and past hybridization is in this system.
At the intraspecific level, not much is known about their phylogeographic history apart from the fact that Western Atlantic populations of L. obtusata were colonized after the LGM from populations in Europe [34]. For each species, the location of glacial refugia, and whether populations persisted in northern areas during the LGM, as well as of putative phylogeographic barriers have not been previously investigated. Thus, it is currently unknown how population history and genetic diversity have contributed to the vast phenotypic variability observed in this system.
Several ecotypes have been described for L. fabalis. In northern Europe, a small (SS) and a large (LM) ecotype are predominantly found in sheltered and moderately-exposed sites in terms of wave action, respectively [35, 36]. In the Iberian Peninsula, three morphotypes/ecotypes are found in exposed (ME), intermediate (FI) and sheltered (ZS) shores dwelling in different macroalgae/seagrass (Mastocarpus spp., Fucus spp., and Zostera spp., respectively) [31, 37]. However, a phylogenetic/phylogeographic analysis of the relationship between these ecotypes is still lacking, and so is an assessment of the relative importance of ecotype evolution and geography in the genetic differentiation between flat periwinkles’ populations.
In this study we address the putative location and impact of glacial refugia, of geographic or ecological barriers to migration, and of hybridization in shaping the genetic diversity of flat periwinkles across their distribution range. To target these questions, we reconstruct the phylogeographic history of the two flat periwinkle species using both mitochondrial and nuclear markers, specifically aiming to understand: i) if introgressive hybridization (i.e. gene flow) occurred during the divergence of the two species; ii) whether northern refugia existed for these species during the LGM; iii) where are the main phylogeographic barriers for each species; iv) if genetic variability differs between L. fabalis and L. obtusata; and v) if genetic differentiation within L. fabalis is more influenced by ecology or geography.
Results
DNA sequence variation
The mitochondrial sequence alignments used for phylogenetic inferences were 615 and 585 bp long for COI and Cyt-b, respectively. These rendered a concatenated mtDNA dataset of 1200 bp sequenced for 344 individuals among the 462 initially available for this study (Table 1, Fig. 1), including 114 different haplotypes (GenBank a.n.: MN045635 - MN045748 and MN045750 - MN045863) defined by 136 variable positions (Additional file 3: Tables S1 and S2). Once the nuclear sequences were phased, the alignment of Thio consisted of 511 bp (491 bp after trimming indels as shown in Additional file 3: Table S3), with 19 different alleles (GenBank a.n.: MN061433 - MN061451) found among 225 individuals and defined by 24 variable positions (Additional file 3: Tables S4 and S5); while the alignment of Cal consisted of 550 bp (539 bp after trimming indels as shown in Additional file 3: Table S3), with 23 alleles (GenBank a.n.: MN061409 - MN061431) found among 133 individuals and also defined by 24 variable positions (Additional file 3: Tables S6 and S7). Due to their low variability, the nuclear fragments were combined in the nuDNA dataset to infer the interspecific relationships of flat periwinkles. A total of 1022 bp were retained after removing all indels, resulting in 66 different sequences over 102 individuals sequenced for both genes.

Map of flat periwinkle sampling sites characterized in this study. Littorina obtusata samples are represented by orange circles and L. fabalis samples by green squares. Symbols filled with both colors (i.e. half orange and half green) indicate populations where mitochondrial introgression was observed: orange circles half filled with green correspond to L. obtusata samples carrying typical mtDNA haplotypes from L. fabalis; green squares half filled with orange correspond to L. fabalis samples carrying typical mtDNA haplotypes from L. obtusata; the filling is not proportional to the percentage of introgression. A particular locality where hybrid individuals were previously identified (Cabo do Mundo, Portugal) is represented by a black star. a Overview of the sampling sites across the distribution range of the two species, highlighted by dotted shorelines: in white the region where only L. obtusata is found and in black the regions where both L. obtusata and L. fabalis occur along the North Atlantic coast (adapted from [26]). In the middle, L. obtusata and L. fabalis specimens (from Iberian Peninsula), in the standard position for morphometric analysis of the shell. b - e Zoom-in of the sampling sites in Iberian Peninsula, Wales, Norway and Sweden. Locality codes follow those in Table 1 with the letters referring to the country. Maps were obtained from DEMIS World Map Server (http://www2.demis.nl/worldmap/mapper.asp) and Google Maps (https://www.google.com) in 2018
Phylogenetic relationships between flat periwinkles
The two flat periwinkles grouped together with a posterior probability of 1 relative to the outgroup for all datasets (mtDNA, nuDNA and mt + nuDNA) (Fig. 2, Additional file 1: Figure S1 and Additional file 2: Figure S2). Two major clades, largely corresponding to the two species, were observed for the mitochondrial and nuclear genes altogether (mt + nuDNA) or the nuclear genes concatenated (nuDNA), but only the monophyly of L. fabalis is well supported, while the L. obtusata clade presents a moderate support (Fig. 2 and Additional file 2: Figure S2). A similar topology concerning the major clades but with lack of support for the monophyly of each species was obtained for the mtDNA alone (Additional file 1: Figure S1). A reliable divergence time estimate could not be obtained with IMa2 [38] due to lack of convergence. Nevertheless, a rough estimate was calculated assuming a mtDNA divergence rate of 1.76% per My (95% HPD: 1.26–3.14%). This rate was obtained by dividing the average divergence between L. saxatilis and flat periwinkles (Dxy = 0.044) by the split time between L. fabalis and L. saxatilis (2.5 My, 95% HPD: 1.4 My-3.5 My [11]). In this way, the pairwise divergence between flat periwinkles (Dxy = 0.015) for the mtDNA after removing potential introgressed individuals suggests that the time to the most recent common ancestor (TMRCA) of these two species is around 0.85 My ago (95% HPD: 0.48 My-1.19 My).

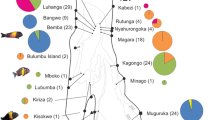
Maximum likelihood (ML) tree for all genes concatenated (mt + nuDNA; 2222 bp). Numbers on branches represent bootstrap values equal to or over 80% for ML (left) and posterior probabilities equal to or over 0.90 for BI (right). (*) Support value not provided by the ML method. The tree was rooted with L. saxatilis. Colors refer to lineages, roughly in agreement with geography. For L. fabalis, Iceland, Norway, Sweden, Shetlands are indicated in dark blue; UK, Ireland, France, in dark green; and Iberian Peninsula, in dark orange. For L. obusata, Iberian Peninsula is indicated in light orange and all remaining samples (USA, Iceland, Norway, Sweden, Shetlands, UK), in light blue
Introgressive hybridization between flat periwinkles
The nuDNA data showed that L. fabalis and L. obtusata do not share any nuclear alleles except for in one of the sampled populations, Cabo do Mundo (Portugal), where hybrids were previously found based on microsatellites [31]. Nine heterozygotes between species-specific alleles were here identified over the 24 individuals from Cabo do Mundo with information for at least one nuclear marker. This supports the former hypothesis of ongoing hybridization in this locality [31] (Fig. 3a and b).

Median-Joining (MJ) haplotype networks for each nuclear gene. a Thio. b Cal. Circle areas are proportional to allele/haplotype frequency. Numbers on branches indicate substitution steps between alleles/haplotypes (when more than one). Pie chart fillings correspond to species: solid colored circles – L. fabalis; low-transparency colored circles – L. obtusata. Individuals from Cabo do Mundo (admixed) are represented in white. The remaining colors refer to lineages, roughly in agreement with geography (North America – grey; Iceland, Norway, Sweden, Shetlands – blue; UK, Ireland, France – green; Iberian Peninsula – orange). Numbers within black squares indicate lineages
In contrast, the mtDNA data revealed a notable amount of haplotype sharing between species (Table 2, Fig. 4). Typical L. obtusata haplotypes were found in seven out of 13 L. fabalis populations (representing 26% of total L. fabalis individuals), with higher incidence in Scandinavia and in particular in the Swedish exposed site (fSW2) where a total replacement of L. fabalis haplotypes by those typically found in L. obtusata was observed. In contrast, within the 12 L. obtusata populations, typical L. fabalis haplotypes were only found in the two British populations (oWA1 and oWA2) (representing 3% of total L. obtusata individuals). The model-based analysis implemented in IMa2 detected significant migration between the two species (i.e. migration rate significantly different from zero) but only in one direction, from L. obtusata into L. fabalis. This occurred in the two main geographic regions that were independently analyzed, northern-central Europe and the Iberian Peninsula, while the estimated migration rate was higher in the first (m = 0.243 vs m = 0.061, respectively).

Median-Joining (MJ) haplotype networks for the mtDNA genes concatenated (COI and Cyt-b). Circle areas are proportional to haplotype frequency. Numbers on branches indicate substitution steps between haplotypes (when more than one). Pie chart fillings correspond to species: solid colored circles – L. fabalis; low-transparency colored circles – L. obtusata. Individuals from Cabo do Mundo (admixed) are represented in white. The remaining colors refer to lineages, roughly in agreement with geography (North America – grey; Iceland, Norway, Sweden, Shetlands – blue; UK, Ireland, France – green; Iberian Peninsula – orange). Numbers within black squares indicate lineages. Letters within haplotypes refer to L. fabalis ecotypes/habitats
Phylogeographic patterns, genetic differentiation and diversity
Among the markers analyzed, mtDNA was the most informative at the intraspecific level revealing five main clades that largely correspond to major geographic regions (Fig. 4). Two clades were observed within L. obtusata, corresponding to a split between the Iberian Peninsula (clade 5) and the rest of the samples (northern-central Europe and North America – clade 4), with evidence for additional substructure within Iberia due to one very divergent mtDNA haplotype (HM64) present in two individuals from Rio de Moinhos (oPT1), Portugal (Fig. 4). Three clades were observed within L. fabalis, essentially corresponding to a split between northern Europe (clade 1) and central Europe (clade 2) apart from the Iberian Peninsula (clade 3). However, some individuals for northern Europe clustered within clade 2 (5 individuals of 39) and some individuals from central Europe within clade 1 (6 individuals of 16, all from France) (Fig. 4). These same clades were also recovered by the mt + nuDNA dataset, although most of them with strong support only under BI (Fig. 2).
The analysis of molecular variance (AMOVA) when populations were grouped by species revealed that most of the variance was explained by differences among species for the nuclear markers (64 and 71%) but it was very similar between species (40%) and among populations within species (43%) for mtDNA (Table 3). The AMOVA within each species separately, when grouping populations by geographic regions taking into account the genetic information described above (two groups for L. obtusata and three for L. fabalis) showed that most mtDNA variance was explained by differences between geographic regions (73 and 57%, respectively) (Table 4). The mtDNA divergence between the two L. obtusata geographic groups was higher than the mean divergence between the three L. fabalis groups (Dxy = 0.017 vs Dxy = 0.007 and Da = 0.013 vs Da = 0.006, respectively; Table 5), and slightly higher than between the two species (Dxy = 0.015 and Da = 0.007). Both the AMOVA and divergence estimates are in agreement with an early split between the Iberian clade and the one distributed northwards.
Within L. obtusata, a lower number of mtDNA haplotypes was observed in the Iberian Peninsula compared with the rest of the distribution of this species (Fig. 4), which rendered significantly lower haplotype and nucleotide diversity in Iberia (oIP) compared with northern-central Europe (oNE) (Tables 6 and 7). The star-like cluster of haplotypes represented by samples from northern-central Europe and North America (oUSA) suggests a recent demographic expansion, which was supported by significantly negative FS values in both regions as well as significantly negative Tajima’s D in North America (Table 6). Haplotype diversity was also significantly lower in Iberia (oIP) than in northern-central Europe (oNE) according to nuclear markers (Additional file 3: Table S8). Similarly, within L. fabalis all markers show a higher genetic diversity in northern-central Europe (fNE) than in Iberia (fIP) (Tables 6, 7 and Additional file 3: Table S8), with significant differences in haplotype and nucleotide diversity for the mtDNA and Thio data.
Mitochondrial haplotype and nucleotide diversity were, on average, higher in L. obtusata than in L. fabalis, for each of the two geographic regions where both species are present (northern-central Europe and Iberian Peninsula) (Tables 6 and 7). However, this tendency was not consistent over all localities (Table 6) and only the difference in haplotype diversity remained significant in both regions after Bonferroni correction for multiple testing (Table 7). In contrast, nuclear diversity was in general higher in L. fabalis than in L. obtusata for each region (NE and IP) (Additional file 3: Table S8), but the difference was only significant in the case of Thio for haplotype diversity.
Within L. fabalis in the Iberian Peninsula, the population sample of the exposed ecotype (ME, fPT2) had the lowest mtDNA diversity, whereas the population found in Fucus spp. (FI, fSP2) had the highest diversity, with the population sample of the ecotype confined to Zostera spp. (ZS, fSP1) showing intermediate levels (Table 6). In northern Europe, the number of haplotypes retained after removing putatively introgressed individuals did not allow to reliably compare genetic diversity between sheltered and moderately-exposed sites. Nevertheless, the AMOVA after grouping populations according to wave exposure in northern-central Europe (Norway, Sweden and Wales) revealed no significant differentiation between ecotypes/habitats, while most of the variation was explained by differences among populations within ecotypes/habitats (ΦSC = 0.610, p-value < 0.001) (Additional file 3: Table S9). In contrast, when populations were grouped according to geography (country), the highest percentage of variation was allocated among countries (57%, ΦCT = 0.574, p-value = 0.058) (Additional file 3: Table S9).
Discussion
Phylogenetic perspective of the diversification of flat periwinkles
The previously proposed monophyly of flat periwinkles [39] was confirmed here. Our results suggest that the TMRCA of the two species is around 0.85 My ago, in agreement with previous estimates based on the entire mitogenome (0.8 My [40]). However, we need to take into account that both estimates are derived from mtDNA data and assume a molecular clock. Different substitution rates and deviations from the neutral or nearly neutral model could render different inferences. Thus, a phylogenomics approach will contribute to improve divergence time estimates between the two species. Interestingly, L. obtusata started to diverge into two clades shortly after the divergence between the two species had started. Such relatively close split events could explain the lack of strong support for the monophyly of L. obtusata, although the morphology of the reproductive organs (among other traits [41]) supports the existence of two species of flat periwinkles.
Divergence of flat periwinkles in the face of gene flow
Past mtDNA introgression between the two species was proposed in previous studies [25]. Here we confirm that gene flow has occurred between L. obtusata and L. fabalis and we also find that it was asymmetric from the former into the latter. This is in line with previous research showing that males from both species prefer large females and mating attempts between small L. fabalis males and large L. obtusata females are common [42]. If this asymmetry is the result of demographic or selective factors (e.g. adaptive introgression) needs to be further evaluated. However, we cannot exclude that introgression in the opposite direction had also occurred. Three haplotypes present in L. obtusata individuals from central Europe clustered with the typical L. fabalis haplotypes from Iberia, supporting such introgressive hybridization from L. fabalis into L. obtusata. This could have happened either in UK, in Iberia before the northward migration, or even during migration as suggested for other species [43]. However, these individuals were not included in the IMa2 analyses as they would violate the model assumptions (see below), and the introgression from L. fabalis into L. obtusata could not be formally tested using these samples. Intriguingly, no single L. fabalis individual carrying typical Iberian haplotypes were observed outside Iberia.
Our results of the isolation with migration (IMa2) analysis also show that introgressive hybridization occurred both in the Iberian Peninsula and in northern-central Europe. Genetic exchange between the two species was higher in the latter region where the two species are more often sympatric, mainly in the sheltered areas of their distribution. However, these results need to be interpreted with caution, since our sampling sites in Iberia are composed mostly from a single species (i.e. allopatric), which could have contributed to a detection of lower introgression when compared with some northern-central European sites.
In contrast, for the nuclear markers, no shared alleles were observed between the two species except in Cabo do Mundo (Portugal) where hybridization had been previously detected [31]. The lack of shared nuclear alleles between species further support the mtDNA introgression scenario, as incomplete lineage sorting is expected if there is more haplotype sharing in nuclear markers on average than in mtDNA, which is the opposite to what we observed. Although the species specific nuclear alleles may be explained by divergent selection (except in Cabo do Mundo), Tajima’s D for these markers was not significant in any population or species, as would be expected under a local adaptation scenario. An assessment of the level of gene flow between L. fabalis and L. obtusata across the genome is a necessary next step to map barrier loci and to understand the extent of the genomic regions involved in reproductive isolation [44, 45].
Main phylogeographic barriers between populations
Concerning the distribution of intraspecific genetic diversity in this system, the most relevant (and significant) component of the mtDNA variation is due to differences among populations from different geographic regions. The genetic discontinuity between populations from the Iberian Peninsula and those from northern Europe supports the existence of a strong barrier between these regions, which corroborates the patterns observed for other marine taxa (e.g. [14, 19, 46,47,48,49] and references therein) including the closely related species L. saxatilis [11, 50]. Despite this phylogeographic break, the detection of three haplotypes in five individuals from Wales (UK) clustering into an otherwise Iberian clade suggests that migration, although rare, can have occurred between these biogeographic regions. Future studies aiming to identify putative hybrid zones between Iberian and northern European clades of both species would be useful to inspect the genomic regions involved in possible adaptation to different environmental conditions.
The observation of two clades in northern-central European populations of L. fabalis further points to some degree of isolation between UK/France and Scandinavia, which was also detected for L. saxatilis [11, 51, 52]. The presence of one divergent haplotype within Iberian populations of L. obtusata suggests that clades with deeper divergence exists in the region. Finally, the very shallow divergence between western and eastern Atlantic L. obtusata populations is consistent with a recent colonization of the former from northern European populations, probably after the LGM, as earlier proposed [34, 53].
Comparative genetic diversity and putative location of main glacial refugia
The two species present similar genetic diversity, although it was slightly higher (but significant) in L. obtusata than in L. fabalis for mtDNA (in terms of haplotype diversity) both in northern Europe and Iberia regions, while the reverse was observed for one of the nuclear markers (Thio). Whether these differences reflect a larger female effective population size in L. obtusata or simply stochasticity needs to be further investigated, including more nuclear markers and more even sample sizes.
Higher genetic diversity was observed in northern Europe when compared with the Iberian Peninsula both in L. fabalis and L. obtusata, consistent with the existence of separate refugia in these regions during the LGM. Although such higher diversity in the north could suggest a larger refugium in this region, it is important to emphasize that the larger geographic area covered by our sampling in northern Europe, mainly for L. fabalis, in respect to the Iberian Peninsula could have biased our estimates. At the same time, the fact that the Iberian populations are located near the limit of the species distribution ranges [26], where they probably face marginal environmental conditions, could have contributed to higher fluctuations in population sizes and consequently to reduced genetic diversity [54, 55]. The recent disappearance of various fucoid species from the southern limit of their distribution, including the Iberian Peninsula, due to increased temperatures (e.g. [56]; reviewed in [14]) suggests that flat periwinkles are at high risk of a southern contraction in the Iberian Peninsula due to the lack of suitable habitats.
The existence of northern glacial refugia for multiple marine taxa has been previously claimed [10, 57], in particular for the seaweed species inhabited by flat periwinkles ([58, 59]; reviewed in [14]), as well as for the rough periwinkle L. saxatilis [11]. Although the exact location of northern refugia for flat periwinkles is yet unknown, since they live and feed on these seaweeds, common refugia for seaweeds and snails seem reasonable.
Contribution of geography versus ecology to genetic differentiation
Within L. fabalis, the differences among the ecotypes/habitats described for northern Europe and UK correspond to a small component of the genetic variation when compared to differences among countries, with populations of each country grouped regardless of the wave-exposure level of the sites where the samples were collected. This suggests that the differentiation between sampling locations independently of the ecotype is more marked than between ecotypes/habitats. Grouping of populations according to geography rather than ecology, at both local or larger scales (within northern Europe and across the entire European shore, respectively) can be interpreted as support for parallel evolution between L. fabalis ecotypes. Although a single origin of the ecotypes followed by gene flow within countries can also originate the observed pattern [60], it resembles the genetic variation in mtDNA, nuclear introns and AFLP found in L. saxatilis ecotypes across the same geographic regions [51]. However, the future implementation of a demographic modeling approach using multiple markers will be necessary to formally test the hypotheses of parallel divergence of the ecotypes, as in L. saxatilis [51]. As well, the identification of markers under divergent selection between ecypes and their distribution across the genome will be crucial to understand the genetic architecture of ecotype divergence in L. fabalis.
Conclusions
Our study supports introgressive hybridization between L. obtusata and L. fabalis during their divergence. Whether the direction of gene flow is only from the former to the latter, as shown here, or it may also occur in the opposite direction remains an open question. Further studies are needed to assess the role of gene flow in the diversification of flat periwinkles, as well as to identify the genomic regions permeable between species. The distribution of the main genetic clades within each species suggests the existence of a northern refugium for both species besides the one identified in Iberia, as proposed for many intertidal taxa with a similar distribution range. Besides, the high genetic divergence between the Iberian clade and those present at higher latitude suggests that the different ecotypes described for L. fabalis in northern-central Europe and those in Iberia could have a different genetic basis. However, this hypothesis requires a proper assessment taking mainly into account the genetic variability involved in adaptive evolution. Finally, despite the different number of populations sampled in each region, the lower genetic diversity observed in Iberian populations, close to the southern distribution limit of both species, compared to northern-central European populations poses some conservation concerns that deserve future attention.
Methods
Sample collection
To characterize the phylogeographic patterns of flat periwinkles, samples were collected during 2012 from 19 sites within 15 localities along the northeast Atlantic coast covering most of the distribution range of L. fabalis and L. obtusata and different degrees of wave exposure (Fig. 1, Table 1). Although the initial aim was to sample sites where the two species do not currently co-exist (i.e. allopatric), this was not possible in some locations of northern and central Europe where neither of the species could be found alone (Fig. 1, Table 1).
To understand the impact of the habitat on the distribution of the genetic variability, representatives of the different ecotypes described for L. fabalis [31] were intentionally included. Hence, in northern and central Europe (Norway, Sweden and Wales) samples were collected from both sheltered (5, 7 and 9 in Fig. 1c, d and e) and moderately exposed (6, 8 and 10 in Fig. 1c, d and e) sites, where the SS and LM ecotypes predominate, respectively (Fig. 1c-e). The degree of wave exposure of sampling sites was determined by the abundance of Ascophyllum nodosum, following Tatarenkov and Johannesson [36]: this macroalgae is common in sheltered sites but generally absent from moderately-exposed sites. In the Iberian Peninsula (Spain and Portugal), the classification of sampling sites as sheltered, intermediate and exposed was based on the presence/prevalence of different macroalgae/seagrass, following Rolán and Templado [37]: Zostera spp., Fucus spp. And Mastocarpus spp., respectively (sites 14, 15 and 18 in Fig. 1b). However, the relative degree of exposure is only comparable within these regions. Furthermore, for consistency in the classification of sampling sites according to wave exposure within each region, only the sites sharing the same characteristics were classified, avoiding potential differences among locations (Table 1). Finally, samples from the Portuguese locality where hybrids between L. fabalis and L. obtusata were previously reported (Cabo do Mundo [31]) were also analyzed (site 19, Fig. 1b, Table 1).
Snails were transported alive to the lab, frozen and then transferred to 98–100% ethanol (or directly preserved in ethanol) for posterior analyses. Later on, soft tissues were removed from shells and sexed under a stereomicroscope (Olympus SZx16) with an attached camera (Olympus SDF PLAPO 1XPF). Morphological assignment of individuals to species was done as in Carvalho et al. [31]. Briefly, males were classified based on penis morphology, one of the most used diagnostic characters between L. fabalis and L. obtusata [26]. According to this criterion, the penis filament is about 30 to 60% of total penis length in the former and 10 to 25% in the latter species (see pages 232 and 206, respectively from [26]). Females were classified based on shell appearance. Since some overlap in shell morphology exists between species [26], whenever possible analyses were preferentially based on males to avoid any potential species misclassification. From all collected individuals, 402 were selected for this study: 211 L. fabalis, 161 L. obtusata and 30 individuals from Cabo do Mundo (a population mainly composed by admixed individuals, see [31]). In total, males represented 72% of the samples (76, 66 and 61%, respectively) (Table 1).
Sixty samples from four other localities (Iceland, Shetland Islands, Northwest Atlantic coast) that had been previously classified into one of the species (12 L. fabalis and 48 L. obtusata; Fig. 1, Table 1) were further included. Samples from Iceland and Shetland Islands had been analyzed for a 350 bp fragment of Cyt-b by Kemppainen et al. [25]; while samples from Massachusetts and Maine had not been genetically characterized before. All of them were sent as partial tissue samples in ethanol or RNAlater. Importantly, the species classification of these individuals was later confirmed by genetic data (see above). Finally, one sample of L. saxatilis was also collected in 2012 from one Portuguese locality (Praia do Carreço) to be used as outgroup.
Molecular methods
Genomic DNA was extracted from head-foot tissue using either a modified version of the standard high-salt protocol [61], where the lysis buffer was replaced by CTAB buffer [62], or the CTAB – chloroform protocol described in Galindo et al. [63]. DNA quality and quantity were assessed by 1% agarose gel electrophoresis. DNA concentration was standardized across samples by dilution in pure water. Two mitochondrial gene fragments (cytochrome oxidase I – COI, and cytochrome b – Cyt-b) and two nuclear exon-primed intron-crossing (EPIC) markers (thioredoxin peroxidase 2 – Thio, and calreticulin – Cal) were amplified using the following primer pairs: LCO1490 – HCO2198 [64], cytbF – cytbR [11], ThioPerF2 – ThioPerR and CalF – CalR2 [51], respectively.
After optimization, for mitochondrial fragments PCR was performed in 25 μL (final volume) containing 1 μL of template DNA, 2.5 μL of 10x reaction buffer, 1 μL of 50 mM MgCl2, 1 μL of 10 mM dNTPs (2.5 mM each), 1 μL of 10 μM forward and reverse primers and 0.15 μL of 5 U/μL of BioTaq polymerase (Bioline). PCR cycling conditions consisted of an initial denaturation at 95 °C for 3 min, followed by 35 cycles, each including denaturation at 95 °C for 20 s, annealing from 52 °C to 56 °C for 20 s, and extension at 72 °C for 30 s; and a final extension of 7 min at 72 °C. For nuclear fragments, PCR was performed in 20 μL (final volume) containing 1 μL of template DNA, 2 μL of 10x reaction buffer, 1 μL of 50 mM MgCl2, 0.5 μL of 10 mM dNTPs (2.5 mM each), 0.5 μL of 10 μM forward and reverse primers and 0.15 μL of 5 U/μL of BioTaq polymerase (Bioline). PCR cycling conditions consisted of an initial denaturation at 95 °C for 3 min, followed by 35 cycles, each including denaturation at 95 °C for 30 s, annealing from 52 °C to 56 °C for 30 s, and extension at 72 °C for 1 min; and a final extension of 7 min at 72 °C.
PCR products were visualized in 2% agarose gels and purified with Exo I and FastAP (Thermo Scientific). Sanger sequencing was performed at Macrogen Europe (Amsterdam, The Netherlands), using the corresponding primers (forward for mitochondrial and both forward and reverse for nuclear fragments).
Sequence data and haplotype determination
Chromatograms were visualized and manually checked using Geneious v6.0.5 (http://www.geneious.com), which was also used to align the corrected sequences. No stop codons were observed in the mitochondrial fragments. The two fragments were concatenated in a single dataset (mtDNA) retaining only individuals with available sequence for both genes. Several indels were identified in the nuclear fragments, seven from one to eight bp long in Thio and four from one to ten bp long in Cal (Additional file 3: Table S3). To assist haplotype reconstruction in individuals carrying alleles with different lengths, a subset of 18 samples (four PCR products for Thio and 14 for Cal) were cloned using TOPO TA Cloning kit (Invitrogen) according to the manufacturer’s protocol. Cloned fragments were amplified using the universal primers M13F and M13R-pUC and confirmed on agarose gel electrophoresis. After that three to eight clones per sample were sequenced in forward and reverse directions using the M13 primers at Macrogen Europe. Resulting sequences were assembled into contigs in Geneious. For the remaining samples, haplotype phases were manually solved by comparing forward and reverse sequences and using known haplotypes as reference. Indels were reduced to single positions for subsequent analyses (Additional file 3: Table S3). Afterwards, in individuals carrying alleles with the same length but with nucleotide differences at two or more positions, International Union of Pure and Applied Chemistry (IUPAC) ambiguity codes were manually added to those sites. Haplotypes were then inferred with the Bayesian method implemented in PHASE v2.1 [65], incorporating information from all known phases and considering models with (MR0) and without (MS) recombination, while the rest of parameters were set to default. Input files for PHASE were obtained with SeqPHASE [66]. Both models rendered the same result, all phases were unambiguously solved (p = 1) except for two individuals from the Cal dataset (p = 0.5 in one position), which were excluded from subsequent analyses.
Two additional datasets were built by combining the two nuclear fragments alone (nuDNA) and the mitochondrial and nuclear altogether (mt + nuDNA). For the nuDNA dataset, only individuals with sequence available for both nuclear fragments were retained. In this case, indel positions were completely removed from each alignment. Since the phase of the combined haplotypes from the two genes was not known, the two alleles of each locus were also summarized in a consensus sequence assigning back ambiguity codes to heterozygous sites. The mtDNA and nuDNA datasets were subsequently concatenated excluding individuals from Cabo do Mundo (because of their putative hybrid origin) as well as potential introgressed individuals from the remaining localities (i.e. individuals from one species carrying mtDNA haplotypes that clustered within those of the other species) and retaining only individuals with sequence available for both sets. The different alleles/haplotypes in each dataset were identified with FaBox v1.41 [67].
Phylogenetic inference and phylogeographic analysis
For all datasets (mtDNA, Thio, Cal, nuDNA and mt + nuDNA), phylogenetic trees were inferred using Maximum Likelihood (ML) and Bayesian Inference (BI) with L. saxatilis as outgroup (GenBank a.n.: MN045749, MN045864; MN061452, MN061453; and MN061432), using the best substitution model for each set selected with jModeltest v0.1.1 [68] according to the Bayesian information criterion (TPM3uf + I + G for mtDNA, JC for Thio, F81 + G for Cal, F81 + I for nuDNA, and HKY + I + G for mt + nuDNA). The ML analyses were done with PhyML v3.1 [69] using the nearest neighbor interchange (NNI) and the subtree pruning and regrafting (SPR) searching algorithms and 1000 bootstrap replicates. Bootstrap values ranging from 80 to 89% were considered moderate clade support, while those equal to or over 90% as strong support. The BI analyses were done with MrBayes v3.2.5 [70], and each consisted of two simultaneous runs starting with random trees, each run with four chains under default heating parameters and 20,000,000 generations sampled every 2000 steps. The standard deviation of split frequencies was used as a convergence index, considering values under 0.01 as indicative of run convergence. The first 25% of the samples were discarded as burn-in and the remaining summarized in a majority rule (50%) consensus tree. Posterior probabilities ranging from 0.90 to 0.94 were considered moderate support, while those equal to or over 0.95 as strong support. ML and BI resulting trees were visualized with FigTree v1.4.0 [71]. For mtDNA, Thio and Cal datasets, evolutionary relationships between haplotypes/alleles were further depicted by constructing Median-joining (MJ) networks [72] using the software Network 4.6.1.3 (http:// fluxus-engineering.com).
Divergence between populations and species
Average (Dxy) and net (Da) divergence between flat periwinkle species as well as between and within intraspecific geographic groups (two for L. obtusata: oN – L. obtusata from North America and from northern-central Europe, oIP – L. obtusata from Iberian Peninsula; and three for L. fabalis fNE1 – L. fabalis from northern Europe, fNE2 – L. fabalis from central Europe, and fIP – L. fabalis from Iberian Peninsula; taking into account the phylogeographic information presented in Fig. 4) were further estimated based on p-distance from mtDNA data using MEGA X [73]. Divergence (Dxy) was also calculated between flat periwinkles and L. saxatilis (the outgroup), as the average of five different replicates, each consisting of the single L. saxatilis individual sequenced here and four randomly chosen flat periwinkles (two from each species, excluding potential introgressed individuals). The aim was to convert nucleotide divergence into time given that the split between L. fabalis and L. saxatilis has been dated around 2.5 My (95% HPD: 1.4 My-3.5 My) based on partial Cyt-b sequences and fossil calibrations [11].
Isolation with migration model
To test whether the observed sharing of mtDNA haplotypes between L. fabalis and L. obtusata could be explained to some extent by gene flow during divergence of the two lineages, the isolation with migration model implemented in IMa2 [38] was applied. As the flat periwinkle populations from the Iberian Peninsula were very different from the populations from northern Europe, and as the mtDNA sharing was mainly confined to within regions (see Results), the IMa2 analysis was performed separately for each region. Therefore, a two-populations model was employed in each case: L. fabalis vs. L. obtusata in the Iberian Peninsula, and L. fabalis vs. L. obtusata in northern Europe. All individuals from Cabo do Mundo were excluded from the first pairwise comparison because of their hybrid origin. Five L. obtusata individuals from Wales were further excluded from the second set because their mtDNA haplotypes clustered with L. fabalis haplotypes from the Iberian Peninsula (see Results), violating one of the assumptions of the model: the absence of migration from a third/unknown population. The analysis was based on mtDNA, Thio and Cal data.
For each analysis, six parameters were estimated: time since population split (t), migration rates between current populations (m1 > 2 and m2 > 1), and effective size of current and ancestral populations (θ1, θ2, θA). The HKY model was specified for each locus, and upper prior bounds (splitting time of 10, population size of 25 and 1 for migration) were optimized in preliminary runs. Each final run consisted of 30 chains with geometric heating and 10,000,000 steps after discarding 1000,000 as burn-in. Effective sample sizes (ESS) and trend plots were examined with Tracer v1.6 [74] to assure proper mixing and convergence of each run and consistence across runs. The significance of migration rates (i.e. significantly different from zero) was evaluated through the likelihood ratio test of Nielsen and Wakeley [75].
Genetic diversity and differentiation between and within species
Hierarchical analyses of molecular variance (3-level AMOVA) were performed to infer the main partition of variance at the species level grouping populations by species, and within each species grouping populations by geographic region but taking also into consideration phylogeographic information (Fig. 4), as described above. Additionally, an AMOVA was performed within L. fabalis at the ecotype level, including only sites where two ecotypes/wave-exposure levels were sampled in the same location (i.e. Norway, Sweden and Wales): populations were first grouped by wave-exposure and second by country (regardless of the ecotype), for comparison. The AMOVAs within each species were based on mtDNA data (higher power for intraspecific analyses, see above) after excluding putatively introgressed individuals, while for the species-level AMOVA the nuclear markers were also used.
Genetic diversity was also assessed at various levels: by species, by geographic region within species (North America, northern-central Europe and Iberian Peninsula), and by population (see Table 7 and Additional file 3: Table S8). These analyses were performed for each genetic marker, separately: mtDNA after removing potential introgressed individuals, Thio and Cal. Molecular diversity was calculated as the number of segregating sites (S), number of haplotypes (h), haplotype diversity (hd) and nucleotide diversity (π). Demographic stability was evaluated by means of Fu’s FS [76] and Tajima’s D [77] tests, assuming that the genomic regions analyzed have not been influenced by selection, and their significance was based on 1000 bootstrap replicates and subsequent Bonferroni correction [78] to account for multiple tests. The significance of the difference in haplotype and nucleotide diversity between geographic groups was calculated with the script genetic_diversity_diffs v1.0.3 [79, 80]. All other analyses in this section were performed in Arlequin v3.5 [81].
Availability of data and materials
Haplotype sequences were deposited in GenBank under the accession numbers MN045635 – MN045864 and MN061409 – MN061453. All other data are available in supplementary material files.
Abbreviations
- °C:
-
Centigrade degree
- a.n.:
-
accession numbers
- AFLP:
-
Amplified Fragment Length Polymorphism
- AMOVA:
-
Analysis of Molecular Variance
- BI:
-
Bayesian Inference
- bp:
-
base pairs
- Cal:
-
Calreticulin
- COI:
-
Cytochrome oxidase I
- CTAB:
-
cetyl trimethylammonium bromide
- Cyt-b:
-
Cytochrome b
- DNA:
-
deoxyribonucleic acid
- dNTPs:
-
deoxynucleotide solution mix
- EPIC:
-
Exon-Primed Intron-Crossing markers
- ESS:
-
Effective Sample Sizes
- Exo I:
-
Exonuclease I
- F1:
-
first generation hybrid
- F2:
-
second generation hybrid resulting from the cross between two F1s
- FastAP:
-
Thermosensitive Alkaline Phosphatase
- FI:
-
Fucus-Intermediate - Littorina fabalis individuals of the intermediately exposed ecotype from the Iberian Peninsula
- fIP:
-
individuals of L. fabalis from the Iberian Peninsula
- fNE:
-
individuals of L. fabalis from northern-central Europe
- h:
-
number of haplotypes
- hd:
-
haplotype diversity
- HPD:
-
Highest Posterior Density
- IUPAC:
-
International Union of Pure and Applied Chemistry
- LGM:
-
Last Glacial Maximum
- LM:
-
Large-Moderately exposed - L. fabalis individuals of the moderately exposed ecotype from northern Europe
- m:
-
migration rate per generation
- ME:
-
Mastocarpus-Exposed - L. fabalis individuals of the exposed ecotype from the Iberian Peninsula
- MgCl2 :
-
Magnesium chloride
- min:
-
minutes
- MJ:
-
Median-Joining
- ML:
-
Maximum Likelihood
- mM:
-
milimolar
- mtDNA:
-
mitochondrial DNA
- My:
-
million years
- NNI:
-
Nearest Neighbor Interchange
- nuDNA:
-
nuclear DNA
- oIP:
-
indivuduals of L. obtusata from the Iberian Peninsula
- oNE:
-
individuals of L. obtusata from northern-central Europe
- oUSA:
-
individuals of L. obtusata from the United States of America
- PCR:
-
Polymerase Chain Reaction
- S:
-
number of segregating sites
- s:
-
seconds
- SPR:
-
Subtree Pruning and Regrafting
- SS:
-
Small-Sheltered - L. fabalis individuals of the sheltered ecotype from northern Europe
- t:
-
time since population split
- Thio:
-
Thioredoxin peroxidase 2
- TMRCA:
-
Time to the Most Recent Common Ancestor
- U/μL:
-
enzyme units per microliter
- UK:
-
United Kingdom
- ZS:
-
Zostera-Sheltered - L. fabalis individuals of sheltered ecotype from the Iberian Peninsula
- Θ:
-
effective population size
- μL:
-
microliter
- π:
-
nucleotide diversity
References
Pinho C, Hey. Divergence with gene flow: models and data. Annu Rev Ecol Evol. 2010;41:215–30.
Abbott RJ, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N, et al. Hybridization and speciation. J Evol Biol. 2013;26(2):229–46.
Avise JC. Phylogeography: the history and formation of species. Cambridge: Harvard University Press; 2000.
Taberlet P, Fumagalli L, Wust-Saucy A-G, Cosson J-F. Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol. 1998;7(4):453–64.
Hewitt GM. Post-glacial re-colonization of European biota. Biol J Linn Soc. 1999;68(1–2):87–112.
Hewitt GM. Speciation, hybrid zones and phylogeography - or seeing genes in space and time. Mol Ecol. 2001;10(3):537–49.
Provan J, Bennett KD. Phylogeographic insights into cryptic glacial refugia. Trends Ecol Evol. 2008;23(10):564–71.
Hewitt GM. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc. 1996;58(3):247–76.
Feder JL, Gejji R, Powell THQ, Nosil P. Adaptive chromosomal divergence driven by mixed geographic mode of evolution. Evolution. 2011;65(8):2157–70.
Maggs CA, Castilho R, Foltz D, Henzler C, Jolly MT, Kelly J, et al. Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology. 2008;89(Suppl 11):108–22.
Panova M, Blakeslee AMH, Miller AW, Makinen T, Ruiz GM, Johannesson K, et al. Glacial history of the North Atlantic marine snail, Littorina saxatilis, inferred from distribution of mitochondrial DNA lineages. PLoS One. 2011;6(3):e17511.
Voris HK. Maps of Pleistocene Sea levels in Southeast Asia: shorelines, river systems and time durations. J Biogeogr. 2000;27(5):1153–67.
Li JJ, Hu ZM, Duan DL. Survival in glacial refugia vs. postglacial dispersal in the North Atlantic: the cases of red seaweeds. In: Hu ZM, Fraser CI, editors. Seaweed: adaptation and evolution of seaweeds under environmental change. Heidelberg: Springer; 2016. p. 309–30.
Neiva J, Serrão EA, Assis J, Pearson GA, Coyer JA, Olsen JL, et al. Climate oscillations, and phylogeographic patterns of North Atlantic Fucaceae. In: Hu ZM, Fraser CI, editors. Seaweed phylogeography: adaptation and evolution of seaweeds under environmental change. Heidelberg: Springer; 2016. p. 279–308.
Marko PB. “What's larvae got to do with it?” disparate patterns of post-glacial population structure in two benthic marine gastropods with identical dispersal potential. Mol Ecol. 2004;13(3):597–611.
Reid DG, Kalpana L, Mackenzie-Dodds J, Kaligis F, Littlewood DTJ, Williams S. Comparative phylogeography and species boundaries in Echinolittorina snails in the central indo-West Pacific. J Biogeogr. 2006;33(6):990–1006.
Pelc RA, Warner RR, Gaines SD. Geographical patterns of genetic structure in marine species with contrasting life histories. J Biogeogr. 2009;36(10):1881–90.
Ayre DJ, Minchinton TE, Perrin C. Does life history predict past and current connectivity for rocky intertidal invertebrates across a marine biogeographic barrier? Mol Ecol. 2009;18(9):1887–903.
Tarnowska K, Krakau M, Jacobsen S, Wołowicz M, Féral JP, Chenuil A. Comparative phylogeography of two sister (congeneric) species of cardiid bivalve: strong influence of habitat, life history and post-glacial history. Estuar Coast Shelf S. 2012;107:150–8.
Faria R, Weiss S, Alexandrino P. Comparative phylogeography and demographic history of European shads (Alosa alosa and A. fallax) inferred from mitochondrial DNA. BMC Evol Biol. 2012;12:194.
Bulgarella M, Trewick SA, Minards NA, Jacobson MJ, Morgan-Richards M. Shifting ranges of two tree weta species (Hemieina spp.): competitive exclusion and changing climate. J Biogeogr. 2014;41(3):524–35.
Gemmell MR, Trewick SA, Crampton JS, Vaux F, Hills SFK, Daly EE, et al. Genetic structure and shell shape variation within a rocky shore whelk suggest both diverging and constraining selection with gene flow. Biol J Linn Soc. 2018;125(4):827–43.
Sivyer L, Morgan-Richards M, Koot E, Trewick SA. Anthropogenic cause of range shifts and gene flow between two grasshopper species revealed by environmental modelling, geometric morphometrics and population genetics. Insect Conserv Diver. 2018;11(5):415–34.
Hedrick PW. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol Ecol. 2013;22:4606–18.
Kemppainen P, Panova M, Hollander J, Johannesson K. Complete lack of mitochondrial divergence between two species of NE Atlantic marine intertidal gastropods. J Evol Biol. 2009;22(10):2000–11.
Reid D. Systematics and evolution of Littorina. London: Ray Society; 1996.
Williams GA. The comparative ecology of the flat periwinkles, Littorina obtusata (L.) and L. mariae Sacchi et Rastelli. Field Stud. 1990;7(3):469–82.
Williams GA. The effects of predation on the life histories of Littorina obtusata and Littorina mariae. J Mar Biol Assoc UK. 1992;72(2):403–16.
Sacchi CF. Sur le dimorphisme du penis chez Littorina obtusata (L.) (Gastr. Pros.). Compte rendu hebdomadaire des Se ances de l’academie des Sciences. Paris Serie D. 1966;262:2370–2.
Sacchi CF, Rastelli M. Littorina mariae, nov. sp.: les differences morphologiques entre ‘nains’ et ‘normaux’ chez l’espece L. obtusata (L.) (Gastr. Prosobr.) et leur signification adaptative et evolutive. Attidella Societa Italiana di Scienze Naturali e del Museo Civico di Storia Naturale di Milano. 1966;105:351–69.
Carvalho J, Sotelo G, Galindo G, Faria R. Genetic characterization of flat periwinkles (Littorinidae) from the Iberian Peninsula reveals interspecific hybridization and different degrees of differentiation. Biol J Linn Soc. 2016;118(3):503–19.
Neigel JE, Avise JC. Phylogenetic relationships of mitochondrial DNA under various demographic models of speciation. In: Nevo E, Karlin S, editors. Evolutionary processes and theory. New York: Academic Press; 1986. p. 515–34.
Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Mol Ecol. 2004;13:729–44.
Wares JP, Cunningham CW. Phylogeography and historical ecology of the North Atlantic intertidal. Evolution. 2001;55(12):2455–69.
Reimchen TE. Microgeographical variation in Littorina mariae Sacchi & Rastelli and a taxonomic consideration. J Conchol. 1981;30:341–50.
Tatarenkov A, Johannesson K. Habitat related allozyme variation on a microgeographic scale in the marine snail Littorina mariae (Prosobranchia: Littorinacea). Biol J Linn Soc. 1994;53:105–25.
Rolán E, Templado J. Consideraciones sobre el complejo Littorina obtusata-mariae (Mollusca, Gastropoda, Littorinidae) en el Noroeste de la penınsula Iberica. Thalassas. 1987;5:71–85.
Hey J. Isolation with migration models for more than two populations. Mol Biol Evol. 2010;27(4):905–20.
Reid DG, Dyal P, Williams ST. A global molecular phylogeny of 147 periwinkle species (Gastropoda, Littorininae). Zool Scri. 2012;41(2):125-136.
Marques JP, Sotelo G, Larsson T, Johannesson K, Panova M, Faria R. Comparative mitogenomic analysis of three species of periwinkles: Littorina fabalis, L. obtusata and L. saxatilis. Mar Genomics. 2017;32:41–7.
Reid DG, Rumbak E, Thomas RH. DNA, morphology and fossils: phylogeny and evolutionary rates of the gastropod genus Littorina. Philos Trans R Soc Lond B. 1996;351(1342):877–95.
Hintz-Saltin S. Mate choice and its evolutionary consequences in intertidal snails (Littorina spp.). Göteborg: University of Gothenburg; 2013. PhD Thesis
Neiva J, Pearson GA, Valero M, Serrão EA. Surfing the wave on a borrowed board: range expansion and spread of introgressed organellar genomes in the seaweed Fucus ceranoides L. Mol Ecol. 2010;19(21):4581–829.
Ravinet M, Faria R, Butlin RK, Galindo J, Bierne N, Rafajlović M, et al. Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. J Evol Biol. 2017;30(8):1450–77.
Martin SH, Davey JW, Salazar C, Jiggins CD. Recombination rate variation shapes barriers to introgression across butterfly genomes. PLoS Biol. 2019;17(2):e2006288.
Chevolot M, Hoarau G, Rijnsdorp AD, Stam WT, Olsen JL. Phylogeography and population structure of thornback rays (Raja clavata L., Rajidae). Mol Ecol. 2006;15(12):3693–705.
Fontaine MC, Baird SJE, Piry S, Ray N, Tolley KA, Duke S, et al. Rise of oceanographic barriers in continuous populations of a cetacean: the genetic structure of harbour porpoises in Old World waters. BMC Biol. 2007;5(1):30.
Raupach MJ, Bininda-Emonds ORP, Knebelsberger T, Laakmann S, Pfaender J, Leese F. Phylogeographical analysis of Ligia oceanica (Crustacea: isopoda) reveals two deeply divergent mitochondrial lineages. Biol J Linn Soc. 2014;112:16–30.
Jenkins TL, Castilho R, Stevens JR. Meta-analysis of Northeast Atlantic marine taxa shows contrasting phylogeographic patterns following post-LGM expansions. PeerJ. 2018;6:e5684.
Doellman MM, Trussell GC, Grahame JW, Vollmer SV. Phylogeographic analysis reveals a deep lineage split within North Atlantic Littorina saxatilis. Proc R Soc B. 2011;278(1722):3175–83.
Butlin R, Saura M, Charrier G, Jackson B, Andre C, Caballero A, et al. Parallel evolution of local adaptation and reproductive isolation in the face of gene flow. Evolution. 2014;68(4):935–49.
Morales HE, Faria R, Johannesson K, Larsson T, Panova M, Westram AM, Butlin JK. Genomic architecture of parallel ecological divergence: beyond a single environmental contrast. Sci. Adv. 2019;5:eaav9963.
Ilves KL, Huang W, Wares JP, Hickerson MJ. Colonization and/or mitochondrial selective sweeps across the North Atlantic intertidal assemblage revealed by multi-taxa approximate Bayesian computation. Mol Ecol. 2010;19:4505–19.
Antunes A, Faria R, Johnson WE, Guyomard R, Alexandrino P. Life on the edge: the long-term persistence and contrasting spatial genetic structure of distinct brown trout life histories at their ecological limits. J Hered. 2006;97(3):193–205.
Eckert CG, Samis KE, Lougheed SC. Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol Ecol. 2008;17:1170–88.
Nicastro KR, Gerardo IZ, Teixeira S, Neiva J, Serrão EA, Pearson GA. Shift happens: trailing edge contraction associated with recent warming trends threatens a distinct genetic lineage in the marine macroalga Fucus vesiculosus. BMC Biol. 2013;11:6.
Kettle AJ, Morales-Muñiz A, Roselló-Izquierdo E, Heinrich D, Vøllestad LA. Refugia of marine fish in the Northeast Atlantic during the last glacial maximum: concordant assessment from archaeozoology and palaeotemperature reconstructions. Clim Past. 2011;7(1):181–201.
Hoarau G, Coyer JA, Veldsink JH, Stam WT, Olsen JL. Glacial refugia and recolonization pathways in the brown seaweed Fucus serratus. Mol Ecol. 2007;16(17):3606–16.
Olsen JL, Zechman FW, Hoarau G, Coyer JA, Stam WT, Valero M, et al. The phylogeographic architecture of the fucoid seaweed Ascophyllum nodosum: an intertidal ‘marine tree’ and survivor of more than one glacial–interglacial cycle. J Biogeogr. 2010;37(5):842–56.
Faria R, Renaut S, Galindo J, Pinho C, MeloFerreira J, Melo M, et al. Advances in ecological speciation: an integrative approach. Mol Ecol. 2014;23(3):513–21.
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Press; 1989.
Winnepenninckx B, Backeljau T, De Wachter R. Extraction of high molecular weight DNA from mollusks. Trends Genet. 1993;9(12):407.
Galindo J, Morán P, Rolán-Alvarez E. Comparing geographical genetic differentiation between candidate and noncandidate loci for adaptation strengthens support for parallel ecological divergence in the marine snail Littorina saxatilis. Mol Ecol. 2009;18(5):919–30.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3(5):294–9.
Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68(4):978–89.
Flot JF. SeqPHASE: a web tool for interconverting phase input/output files and fasta sequence alignments. Mol Ecol Resour. 2010;10(1):162–6.
Villesen P. FaBox: an online toolbox for FASTA sequences. Mol Ecol Notes. 2007;7(6):965–8.
Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25(7):1253–6.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A. Höhna set al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42.
Rambaut A. 2012. FigTree version v1.4.0. Available at: http://tree.bio.ed.ac.uk/software/figtree
Bandelt H-J, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Rambaut A, Suchard MA, Xie D, Drummond AJ. 2014. Tracer v1.6. Available at http://beast.bio.ed.ac.uk/Tracer.
Nielsen R, Wakeley J. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics. 2001;158(2):885–96.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–25.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95.
Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43(1):223–5.
Alexander A, Steel D, Hoekzema K, Mesnick SL, Engelhaupt D, Kerr I, et al. What influences the worldwide genetic structure of sperm whales (Physeter macrocephalus)? Mol Ecol. 2016;25(12):2754–72.
Alexander A. 2017. genetic_diversity_diffs v1.0.3. Available at https://github.com/laninsky/genetic_diversity_diffs.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10(3):564–7.
Acknowledgements
We would like to thank Petri Kemppainen, Fernando Lima and Rui Seabra for some of the samples used in this study. We would also like to thank Emilio Rolán-Alvarez and Juan Galindo for suggestions of sampling sites in Spain; and João Carvalho, Carolina Pereira and Juan Galindo for their help in processing and extracting DNA of some samples.
Funding
This study was supported by: European Regional Development Fund (FCOMP-01-0124-FEDER-014272), FCT – Foundation for Science an d Technology (PTDC/BIA-EVF/113805/2009), ASSEMBLE (grant number 227799), the Centre of Marine Evolutionary Biology and a Linnaeus grant from Swedish Research Councils VR and Formas. RF was financed by FCT (SFRH/BPD/89313/2012) and is currently funded by the European Union’s Horizon 2020 research and innovation programme, under the Marie Sklodowska-Curie grant agreement number 706376. GS was financed by Volkswagen Stiftung (50500776). The funding bodies did not have any role in the design of the study, collection, analysis and interpretation of data, or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
GS, MD, MP, KJ and RF designed the study and planned the experiments. GS, MD, DC, KJ and RF sampled the individuals. GS performed the experiments and analyzed the genetic data; and DC analyzed morphological data for species distinction. GS and RF wrote the first draft. All the authors contributed to the final version. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The species sampled are not endangered or protected.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Maximum likelihood (ML) tree for the mitochondrial genes (COI and Cyt-b) concatenated (mtDNA dataset; 1200 bp). Numbers on branches represent bootstrap values equal to or over 80% for ML (left) and posterior probabilities equal to or over 0.90 for BI (right). The tree was rooted with L. saxatilis. Colors refer to lineages, roughly in agreement with geography. For L. fabalis, Iceland, Norway, Sweden, Shetlands are indicated in dark blue; UK, Ireland, France, in dark green; and Iberian Peninsula, in dark orange. For L. obusata, Iberian Peninsula is indicated in light orange and all remaining samples (USA, Iceland, Norway, Sweden, Shetlands, UK), in light blue. Putatively introgressed haplotypes between flat periwinkles are shown in black.
Additional file 2: Figure S2.
Maximum likelihood (ML) tree for the nuclear genes (Thio and Cal) concatenated (nuDNA dataset; 1022 bp). Numbers over branches represent bootstrap values equal to or over 80% for ML (left) and posterior probabilities equal to or over 0.90 for BI (right). The tree was rooted with L. saxatilis. Black - L. fabalis, grey - L. obtusata. (*) Support value not provided by the ML method.
Additional file 3:
Nine tables (one per sheet) containing information about polymorphic sites and distribution of alleles/haplotypes for the different fragments here analyzed; position of the indels and summary statistics for the nuclear fragments; and AMOVA results for the L. fabalis ecotypes.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Sotelo, G., Duvetorp, M., Costa, D. et al. Phylogeographic history of flat periwinkles, Littorina fabalis and L. obtusata. BMC Evol Biol 20, 23 (2020). https://doi.org/10.1186/s12862-019-1561-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-019-1561-6