
Abstract
Background
The Pleistocene cyclic sea-level fluctuations are thought to have markedly affected the distribution and genetic architecture of Atlanto-Mediterranean biota. Despite the acknowledged key role played by these historical events in shaping population genetic structure of marine species, little is still known about the processes involved in shaping the spatial distribution of genetic variation within intertidal species. We intended in this study to reconstruct the phylogeography of a common and widely distributed coastal species across the East Atlantic and Mediterranean Sea (the warty crab Eriphia verrucosa), aiming to unravel potential microevolutionary processes likely involved in shaping its genetic polymorphism. For this purpose, a total of 155 specimens of E. verrucosa from 35 locations across the entire distribution range were analyzed by comparing a 453 basepairs region of the mitochondrial gene cytochrome oxidase subunit 1 (Cox1).
Results
Our results unveiled the prevalence of high genetic connectivity among East Atlantic and Mediterranean populations, with noticeable genetic distinctiveness of the peripheral population from the Azores. Spatio-temporal patterns of genetic diversification and demographic history allowed retrieving genetic imprints of late Pleistocene vicariant event across the Gibraltar Strait followed by subsequent postglacial expansion events for both the East Atlantic and Mediterranean regions. Integrative evidences from the outcomes of comparison of regional genetic diversification, as well as evolutionary and biogeographic histories reconstructions, support the existence of potential glacial refugia for E. verrucosa in the East Atlantic and western Mediterranean. Our results also revealed low levels of genetic variability along with recent demographic and spatial expansion events for eastern Mediterranean warty crabs, suggesting that the eastern areas within the distribution range of the species might have been recently colonized from putative glacial refugia.
Conclusions
These findings provide new insights into the phylogeography and evolutionary history of a common but poorly studied Atlanto-Mediterranean decapod species. Specifically, they contribute to the understanding of the impact of historical processes on shaping contemporary population genetic structure and diversity in intertidal marine species.
Similar content being viewed by others

Background
One of the main aims of evolutionary genetics is to discern the evolutionary processes responsible for driving and shaping geographic distribution of genetic variation of populations, as well as disentangling the origin of their genetic structure [1,2,3,4]. Integrative evidences from palaeogeographic, palaeoclimatic and phylogeographical investigations have brought in-depth knowledge to this issue, pointing out to the significant impact of Pleistocene climatic shifts on forging contemporary genetic polymorphisms [5,6,7,8].
The Pleistocene cyclic sea-level fluctuations are thought to have markedly affected the coastal environment and the evolutionary history of its biota [5] and regarded as one of the most important processes involved in shaping the contemporary geographic distribution of genetic variation [6, 9]. Noteworthy, these historic events have greatly influenced the distribution and evolutionary dynamics of populations [10, 11] following repeated habitat contractions and expansions of marine organisms. It has been postulated that habitat fragmentation induced by a lower sea level during glacial maxima could lead to a genetic bottleneck, with geographic isolates persisting in glacial refugia, and therefore to a heterogeneous population structure. Phylogeographic investigations across different parts of the globe have documented such patterns and allowed retrieving cryptic refugia in many marine species, such as in the seaweed Palmaria palmata across the English Channel [12] and the brown alga Sargassum polycystum across the southern Chinese coast [13]. The occurrence of phylogeographic breaks along with genetic imprints of glacial refugia have been also invoked in population genetic studies of East Atlantic and Mediterranean marine species such as in the gastropod Nassarius nitidus [14] and the green crab Carcinus aestuarii [8]. In contrast, range expansion primed by a rising sea level, following environmental warming during interglacials, could result in rapid population growth and consequent genetic homogeneity as a result of secondary contact between previously isolated evolutionary lineages [11]. The impact of these historical factors, intensified by the effects of contemporary environmental and oceanographic gradients, could have been potentially involved in shaping present day genetic variation and population structure in marine species [8, 15].
The Mediterranean Sea and the contiguous northEast Atlantic Ocean represent a suitable area to study biogeographical processes and investigate evolutionary patterns of diversification in marine species [2, 8, 16]. Indeed, the severe palaeogeographic and palaeoclimatic shifts that this region has undergone throughout its history, resulting in the onset of specific oceanographic features across its coastline, have set the stage for the impact of evolutionary and demographic processes on forging genetic variation of marine species [14, 17]. Recent phylogeographic investigations have provided evidence for the occurrence of pronounced genetic boundaries between the East Atlantic and the western Mediterranean, and between the Western and Eastern Mediterranean basins [2, 8, 15, 18,19,20,21,22]. Further pronounced genetic breaks have also been documented in the eastern Mediterranean, notably between the Adriatic-Ionian seas and the Aegean-Marmara-Black seas [1, 8, 22,23,24,25,26].
The above-mentioned biogeographic units could stem from the impact of historical, hydrographic and environmental processes. From a historical point of view, a marked drop of sea level during Pleistocene glaciation peaks [27] might have caused change in circulation patterns across main marine corridors, and consequently disrupted the biotic exchange across both the Gibraltar and Siculo-Tunisian Straits, leading to dramatic shifts in species range [19, 28, 29]. As a consequence, most of the current genetic patterns of marine coastal organisms could have been affected by past vicariance events, due to periodical closing of corridors that prevented larval interchange and gene flow [29]. This assumption has been consolidated by the outcome of recent phylogeographic investigations that have identified lower latitude Pleistocene marine refugia such as in the case of the seahorse Hippocampus hippocampus [30] and the intertidal gastropod Nassarius nitidus [14]. In addition to the potential impact of Quaternary climatic fluctuations (which might have left a strong footprint on the genetic structure of Atlanto-Mediterranean marine species), the contemporary oceanographic features (i.e., frontal systems and ocean currents) as well as gradual changes in abiotic factors (i.e., temperature and salinity) could also be involved in disrupting population connectivity [15, 18, 31]. Indeed, contemporary barriers to gene flow are mainly linked to the Almeria-Oran Oceanographic Front (accounting for the observed phylogeographic break between the East Atlantic and Mediterranean Sea) [19, 32] as well as to the particular hydrographic isolation patterns across the Siculo-Tunisian Strait [33] responsible for driving population genetic differentiation between Western and Eastern Mediterranean basins [4]. Besides, the potential interplay between oceanographic (involving mainly the isolating effects of Peloponnes anticyclonic front [34] and the Black Sea current) and environmental isolation of the Adriatic, Ionian and Aegean seas has been postulated to account for the genetic discontinuity recorded in the eastern Mediterranean [19, 23, 25].
Among Atlanto-Mediterranean marine biota, intertidal marine species are still less investigated in terms of their phylogeographic structure and evolutionary history. Furthermore, contemporary spatial distributions of their genetic diversity are thought to have been shaped by the impact of Pleistocene climate oscillations [35]. Hence, population genetic investigation of intertidal marine species would help not only with assessing the potential impact of postulated biogeographic boundaries within the East Atlantic and Mediterranean, but also unraveling the effect of microevolutionary processes (including glacial-induced fragmentation and postglacial-induced recolonisation) on forging patterns of genetic variability and structure.
The warty crab Eriphia verrucosa (Forskål, 1775) (Decapoda, Brachyura, Eriphiidae) represents a good model to address the above mentioned issues. This littoral decapod is one of the most ecologically important coastal species, playing a crucial role in structuring intertidal communities, as a highly predatory shore crab [36, 37]. It has a wide geographic distribution, stretching from the Mediterranean Sea (including the Black Sea) to the East Atlantic Ocean from Brittany to Mauritania and the Azores [38, 39]. The species inhabits stony coastal zones and occupies infralittoral crevices at shadow zones with a well-developed algal coverage. It mainly occurs in the lower intertidal zone (being more abundant in the infra and mesolittoral zones than in the supralittoral) [39] where it can be encountered among stones and seaweeds along rocky coastlines in shallow waters down to depths of 15 m [40]. The reproduction of E. verrucosa begins in May or June [41]. Spawning occurs from late July to the end of August [42], depending on water temperature. The warty crab is considered as very prolific, being characterized by high fecundity [41,42,43]. The dispersal potential of the species is probably high, since the complete larval development takes place in the ocean with four zoeal stages and one megalopa stage [44]. According to the experimental study held by Lumare and Gozzo [44], larval development of E. verrucosa can take 49 days at a temperature of 21 °C. Considering all of these eco-biological aspects, genetic homogeneity and panmixia could be expected among populations of this crustacean species. However, recent phylogeographic investigations, carried out in other Atlanto-Mediterranean decapod species (with similar life-history traits as E. verrucosa), revealed restricted patterns of gene flow, such as in the green crab Carcinus aestuarii [4, 8], the marbled crab Pachygrapsus marmoratus [21] and the littoral prawn Palaemon elegans [2, 15]. The outcome of these studies pointed out to the involvement of other factors, such historical isolating processes, contemporary oceanographic discontinuities as well as larval behavior, in shaping patterns of population genetic structure of littoral decapods.
In light of these considerations, in this study we investigated the following questions: (1) Can the main postulated barriers to gene flow spanning the East Atlantic and the Mediterranean Sea restrict gene flow among populations of E. verrucosa allowing for the occurrence of significant patterning of population genetic structure? (2) Did palaeoclimatic and palaeogeographical evolution of the surveyed geographic area during the Quaternary significantly impact contemporary spatial distribution of genetic polymorphisms and their variations through space and time, and what are the potential evolutionary processes likely involved in shaping genetic diversity and affecting spatial genetic structure within this decapod species? In order to answer these questions, we examined the mitochondrial phylogeography of the species and reconstruct its evolutionary and demographic histories. An intensive sampling of E. verrucosa was conducted from its entire distribution range, stretching from the East Atlantic to the Black Sea (Fig. 1 and Table 1), and sequences of the mitochondrial cytochrome oxidase subunit 1 (Cox1) gene were generated and compared. When amplifying this marker in the sampled specimens, we made sure that we sampled the region of the gene that has been shown to be variable enough for unveiling significant genetic differentiation in previous studies on marine decapods [8, 15, 16]. Furthermore, the use of this marker allowed us to incorporate all previously published Cox1 sequences into the analyses in order to increase the size of the examined dataset. However, given that only small portion of the mtDNA genome was analyzed, we combined several analytical tools in order to recover the maximum amount of information contained in the mtDNA at different hierarchical levels (genealogy, phylogeographic structure, calibrated phylogeny, ancestral area reconstruction, and historical demography). Although the exclusive use of uniparentally inherited markers can provide a distorted understanding of phylogeographic structure, the smaller effective population size of mtDNA as compared to nuclear loci makes mtDNA an appropriate molecular marker for reconstructing species evolutionary histories [45].

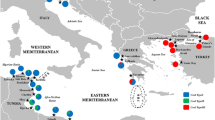
Sampling locations of Eriphia verrucosa across the East Atlantic and Mediterranean Sea. The main phylogeographic breaks are shown as dotted lines. G S: Gibraltar Strait; AOF: Almería-Oran Oceanographic Front; S-T S: Siculo-Tunisian Strait; PHB: Peloponnese Hydrographic Break (represented by the quasi-circular anti-cyclonic feature southwest of Peloponnese). The base map was constructed with the software DIVA-GIS 7.5.0 (http://www.diva-gis.org)
Results
Sequence variation, genetic diversity and assessment of regional genetic diversification
A total of 30 Cox1 haplotypes were identified in the 155 sampled individuals of E. verrucosa (Fig. 2 and Table 2). The newly generated sequences (147) allowed detecting 27 haplotypes; while the 12 sequences retrieved from GenBank contributed to the haplotype dataset by only 3 haplotypes (Additional file 1: Table S1). Details on the pattern of assignment of these latter sequences to the detected haplotypes in this study are given in Additional file 2: Table S2. The difference among the obtained haplotypes was due to 29 variable sites of which 12 were parsimony-informative and 17 were autapomorphic. The outcome of MEGA analysis retrieved unbalanced nucleotide composition of the analyzed Cox1 fragment (C = 20.46%; T = 34.96%; A = 27.36%; G = 17.22%), showing an A-T bias as expected for invertebrate mitochondrial DNA [46].

TCS parsimony network of Eriphia verrucosa, based on the alignment of 453 bp of the mitochondrial gene Cox1, showing the evolutionary relationships among the recorded haplotypes. Haplotype 2 corresponds to the ancestral haplotype. Small black circles correspond to missing (or hypothetical) haplotypes. Each connecting line indicates one mutational step between haplotypes. Circle sizes depict proportions of haplotypes; the smallest corresponds to 1 and the largest to 77 individuals. Cox1 lineages of E. verrucosa were defined based on the outcomes of both BEAST and RASP analyses (see Figs. 3 and 4)
An analysis of genetic diversity of the examined Cox1 dataset showed high total haplotype diversity (h = 0.733 ± 0.036) and low total nucleotide diversity (π = 0.0034 ± 0.0002). The total mean number of nucleotide differences (K) was 1.575. Details on these parameters of genetic variability in each examined population are reported in Table 2. At the regional scale, the East Atlantic specimens of E. verrucosa were shown to be genetically more variable than their Mediterranean counterparts (Table 2). Within the Mediterranean Sea, warty crabs from the Western Basin exhibited higher levels of genetic diversity than those from the Eastern Basin (Table 2). As nine out of the twelve GenBank sequences derived from the East Atlantic (eight from the Azores and one from Cádiz, Table 1), the recorded higher levels of genetic diversity in the East Atlantic than those observed in the Mediterranean could have been overestimated due to the inclusion of those sequences in the analysis. However, when re-analyzing the levels of three genetic diversity parameters (h, π, and K) in a dataset excluding GenBank sequences, we noticed that East Atlantic specimens of E. verrucosa remain genetically more diversified than their Mediterranean counterparts (Additional file 1: Table S1). This indicates that inclusion of GenBank sequences has no influence on the outcome of our genetic diversity analyses.
Assessment and statistical comparison of seven genetic diversification parameters among three examined regions (Atlantic, western Mediterranean and eastern Mediterranean), after a rarefaction procedure involving 30 replicates, showed that the East Atlantic and western Mediterranean samples were significantly more diversified than their eastern Mediterranean counterparts for all parameters (except for haplotypic richness) (Table 3). Significant differences between the East Atlantic and western Mediterranean were also unveiled for all genetic diversity indices, with the former region being more variable than the latter (Table 3). Increase in the number of replicates did not influence the outcome of inter-regional comparison, as the same results were retrieved when re-carrying out the analyses with 60 replicates (Table 3).
Phylogenetic relationships among Cox1 haplotypes
The TCS statistical parsimony procedure yielded two resolved sub-networks centered around two main and common haplotypes (haplotype 2 and haplotype 11) (Fig. 2). These two clades or lineages were defined and delineated according to the outcome of the Bayesian phylogenetic analyses (Figs. 3 and 4). Both retrieved Cox1 lineages were separated by one mutational step. The two common haplotyes 11 (within lineage 1) and 2 (within lineage 2), separated by three mutational steps, were found in the East Atlantic as well as in the Western and Eastern Mediterranean basins (Fig. 2 and Additional file 3: Table S3). Such finding, showing no significant phylogeographic patterning, was supported by the outcome of PERMUT analysis. Indeed, the NST value (0.061) was not significantly higher than the GST value (0.048); P > 0.05, highlighting lack of a significant relationship between genealogy and geographic distribution of Cox1 haplotypes. Within lineage 1, a further separation by two mutational steps was noticed between two sets of haplotypes (or sub-clades), centered around haplotypes 1 and 11. The sub-clade around haplotype 1 was shown to be slightly prevailing in the East Atlantic. However, such observation should be interpreted with caution as this sub-clade is relatively common in a small subset of specimens from the Azores. Besides, the central haplotype 1 and the derived haplotype 3 have been also found in the eastern Mediterranean and may occur in the western Mediterranean but they probably went unnoticed because of the small sample size obtained for this region (only 45 specimens).

Bayesian phylogenetic analysis (as implemented in BEAST) exhibiting diversification pattern of the retrieved Cox1 haplotypes wihin Eriphia verrucosa through time. Node ages (mean values in black below the nodes) are highlighted in years before present (YBP) for the three main nodes. Values in grey (above the main nodes) correspond to the posterior probabilities (Bayesian inference) of the generated nodes. The specifically estimated Cox1 gene mutation rate, for Eriphia, of 4.87% per Myr was used to calibrate the genealogy and date tMRCA of Cox1 lineages

Biogeographic history of Eriphia verrucosa, highlighting ancestral area reconstruction based on Cox1 haplotypes. Couloured circles on each node (next to corresponding number) show the likelihood of occurrence of each ancestral haplotype at an inferred ancestral biogeographic region. Graphical results of ancestral distributions at each node of the phylogeny were obtained by S-DIVA (Statistical Dispersal-Vicariance Analysis) method, as implemented in RASP version 3.2. Biogeographic regions are shown in different colors and denoted with alphabetic letters (A, B, and AB). Green arrows indicate vicariance events at the corresponding nodes; while black arrows highlight possible dispersal events
Patterns of genetic differentiation and phylogeographic structure
Genetic differentiation among studied populations of E. verrucosa was found to be weak but significant based on both Tajima-Nei distances (ΦST = 0.043, df = 154, P = 0.026) and haplotype frequencies (FST = 0.045, df = 154, P = 0.008). Pairwise comparisons of genetic differentiation showed that only five (based on nucleotide divergence) and six (based on haplotype frequencies) out of the 105 pairwise genetic distances were significant after the B-Y FDR correction (Table 4). Most of the significant genetic distances, computed from nucleotide divergence and haplotype frequencies, were due to the genetic distinctiveness of the population of the Azores (Table 4). Re-analysis of pairwise genetic differentiation in a total dataset of 143 specimens (excluding the twelve sequences from GenBank) also unveiled marked genetic differentiation of the Azores population (Additional file 4: Table S4).
Examination of phylogeographic structure across potential barriers to gene flow, encompassing the East Atlantic and Mediterranean Sea, revealed none significant genetic subdivision within E. verrucosa across the Gibraltar Strait, the Almería-Oran Oceanographic Front, the Siculo-Tunisian Stait, and the Peloponnese hydrographic break (Table 5). The lack of genetic structure across these biogeographic boundaries was recorded according to both nucleotide divergence (Tajima and Nei distance) and haplotype frequencies (Table 5).
Evolutionary history and historical biogeography
BEAST analysis allowed unravelling a Cox1 subtitution rate of 4.87% per million years (Myr) with a 95% high posterior density interval (HPD) of 3.83–5.89%, which is within the range of substitution rates of 1.11–6.58% per Myr, estimated so far for the Cox1 gene in crustaceans [15, 47,48,49,50].
The yielded pattern of Cox1 haplotypes diversification clearly hints at the separation between two lineages or haplogroups (Fig. 3). Bayesian analyses of Cox1 sequences (posterior probability [PP] = 1.00) confirmed the mtDNA monophyly of E. verrucosa. The first mitochondrial clade (lineage 1) was found to be statistically supported (PP = 0.65); while the more genetically diversified clade (lineage 2) was poorly resolved (PP = 0.42) (Fig. 3). Within this latter clade, some derived nodes were shown to seemingly pre-date their ancestral nodes despite the strong convergence of the data. Referring to our earlier Bayesian phylogenetic reconstructions for intraspecific data [4, 8], we assume that such pattern could stem from the high frequency of diversification in relatively short period of time followed by longer period of relative stasis. When calibrating the haplotype phylogeny with the already determined Cox1 mutation rate, divergence among both retrieved lineages of E. verrucosa was found to occur approximately 66,270 years before present (YBP) (95% HPD: 37,100–114,194 YBP). Subsequent diversification within both retrieved mitochondrial lineages started nearly at the same time (lineage 2: 52,338 YBP [95% HPD: 31,719–78,900 YBP]; lineage 1: 50,066 YBP [95% HPD: 29,272–75,223 YBP]; Fig. 3).
Reconstruction of possible past geographic distributions of Cox1 haplotypes (Fig. 4) suggests that the origin of E. verrucosa was in a wide ancestral region encompassing the East Atlantic and Mediterranean regions (biogeographic region AB). The S-DIVA analysis allowed detecting a marked vicariance signal at the most ancestral node (basal node 59; Fig. 4) between the East Atlantic (regarded as the most likely ancestral range for lineage 1; biogeographic region A at node 38) and the Mediterranean Sea (considered a favored ancestral range for lineage 2; biogeographic region B at node 58). Re-analysis of the historical biogeography of E. verrucosa based on a total dataset of 143 sequences (excluding the GenBank sequences) allowed detecting the same patterns at the three most ancestral nodes (Additional file 5: Figure S1). Within lineage 1, several dispersal events were recorded at both the basal node (node 38) and the derived ones (nodes 32, 33, 35, 36, and 37) (Fig. 4). Within lineage 2, distinct dispersal waves (recorded at nodes 40, 42, 46, 49, and 50) were occasionally followed by vicariance events (noticed at nodes 39 and 45) (Fig. 4). The retrieved dispersal pathways within the two lineages suggest not only the occurrence of bidirectional dispersal events between the East Atlantic and Mediterranean Sea, but also further dispersal within the same region (Fig. 4). However, such interpretation should be cautiously considered owing to the small sample size of the East Atlantic in relation to the Mediterranean.
Demographic history reconstruction
The analysis of the Tajima’s D, Fu’s FS, and Ramos-Onsins and Rozas’s R2 tests in the defined populations of E. verrucosa showed significant deviations from mutation-drift equilibrium for eight populations (Table 2). With the R2 test being more reliable to infer past demographic fluctuations in small sample size, the occurrence of a potential expansion event can be roughly highlighted for six populations of the warty crab (Table 2). The significant output of the three examined neutrality tests, together with the small and non-significant value of Harpending’s raggedness index rg, consolidate the scenario of a sudden demographic expansion for the whole dataset as well as for the Mediterranean Sea and its Western and Eastern basins (Table 2). A signature of past demographic expansion was also highlighted for the East Atlantic, mainly inferred from the significant Fu’s FS along with the non-significant demographic index (rg) (Table 2).
Both demographic and spatial expansion models were accepted for the total dataset as well as for the East Atlantic and Mediterranean regions (Table 6). Only, the null hypothesis of expectation under a sudden demographic expansion model has not been statistically supported for the East Atlantic (Table 6). Both expansion scenarios were also validated for the Western and Eastern Mediterranean basins. Noteworthy, time since expansion, measured in mutational time units, allowed inferring marked older demographic and spatial expansion events recorded for the western Mediterranean than those retrieved for the eastern Mediterranean (Table 6).
The generated BSP plots, for the total dataset of E. verrucosa as well as for the East Atlantic and Mediterranean regions, showed a recent and sudden increase in effective population size, following a long stationary period (Fig. 5). The sudden expansion event occurred roughly at about 16,500 years ago (CI: 11,000–30,500 years ago) for the whole dataset (Fig. 5c). At the regional scale, East Atlantic specimens of E. verrucosa started expanding approximately at about 17,000 years ago (CI: 13,500–26,600 years ago) (Fig. 5a); while their Mediterranean counterparts underwent a marked significant increase in their effective size around 17,500 years ago (CI: 12,500–29,000 years ago) (Fig. 5b). All underlined expansion events markedly followed the Last Glacial Maximum period (between 26,500–20,000 years before present; [51]).

Bayesian skyline plots (BSP) for specimens of Eriphia verrucosa corresponding to the Atlantic Ocean (a), Mediterranean Sea (b) as well as to the whole dataset (c), showing changes in effective population size (Ne multiplied per generation time) over time (measured in years before present). The thick solid line depicts the median estimate, and the margins of the blue area represent the highest 95% posterior density intervals. The enclosed grey-shaded box corresponds to the Last Glacial Maximum (LGM) priod (between 26,500–20,000 years before present; [51])
Discussion
The present investigation is the first report on mitochondrial phylogeography and evolutionary history of Eriphia verrucosa across its entire distribution range. The main results of the study unveiled a lack of population genetic structure across biogeographic boundaries within the East Atlantic and Mediterranean, with noticeable genetic distinctiveness of the peripheral population from the Azores. They also showed that the late Pleistocene climate oscillations might have had a significant impact on shaping the genetic variability and structure of the examined warty crab species.
Lack of phylogeographic structure across postulated barriers to gene flow
The outcomes of various population genetic analyses concur in unveiling lack of phylogeograhic structure across postulated barriers to gene flow. Such finding clearly contrasts with previous ones from other phylogeographic investigations of marine species [2, 8, 19, 21, 22, 32] and suggests that the biogeographic boundaries encompassing the surveyed region do not seem to restrict gene flow within E. verrucosa. Given its supposedly high larval dispersal potential [44] and continuous distribution across the intertidal belt of the East Atlantic and Mediterranean, we hypothesize that the observed pattern of genetic homogeneity could be attributed to the fact that long drifting larvae of the warty crab can reach very distant locations along the East Atlantic-Mediterranean littoral and assure genetic connectivity owing to the unidirectional surface current, called the Atlantic Current, originating from the East Atlantic, moving eastwards along the western Mediterranean coast and reaching into the eastern Mediterranean [52]. This assumption could be supported by the outcome of the TCS Cox1 genealogy, in which a wide distribution of the common haplotype H2 (Fig. 2), being shared by all populations, can be noticed.
Nevertheless, given that highly dispersive intertidal species, occurring in sympatry with E. verrucosa, have been shown to display a significant differentiation across boundaries [2, 4, 8, 15, 20, 21], we can assume that other factors (including larval behaviour and wide tolerance toward gradual shift in abiotic features) could be interacting with the high larval dispersal potential of the species and account for such recorded genetic homogeneity. In this context, we advance the hypothesis that widely tolerant larvae of E. verrucosa towards disparate environmental features of the Atlantic and the Mediterranean (i.e., salinity and temperature [53]) could overcome the wide variety of physical, chemical and biological processes that could potentially affect the probability of their transition from one stage to another [54, 55]. This would deprive the occurrence of local genetic structuring, potentially triggered by the effect of natural selection. Further experimental investigations on eco-biological properties of the warty crab larvae are required to test this hypothesis.
Alternatively, we can propose that high gene flow and a particular larval behaviour may not be the only causes of the lack of phylogeographic structure discerned within E. verrucosa, as other factors such as large effective population size could also be involved [5]. Indeed, given the high fecundity potential in this decapod species [41, 42], it is highly likely that the impact of genetic drift may be counteracted by large populations of the warty crab. Hence, the lack of population genetic structure over the surveyed distribution range could potentially stem from recent spatial and demographic expansions, as clearly observed in our study. In that case it may not necessarily reflect high levels of contemporary gene flow in this species, potentially induced by high larval dispersal potential and homogenizing oceanographic features [56].
While the above mentioned scenarios may provide possible explanations to the high genetic connectivity within E. verrucosa, they have to be considered with caution as the increase in sample size may yield new insights into the phylogeographic structure of the species across the postulated biogeographic boundaries. The recent study by Fratini et al. [21] showed that exhaustive increase in sample size allowed unraveling population genetic structuring (with mtDNA) in the marbled crab P. marmoratus, previously regarded as panmictic due to geographically and numerically limited datasets.
Genetic differentiation of the Azores population
Opposed to a background of high regional and and sub-regional genetic connectivity, the East Atlantic peripheral population of Azores was shown to be genetically distinct. Such finding has been already reported for other marine invertebrate and vertebrate species with high dispersal potential [57,58,59]. This pattern of differentiation may hint at the crucial role played by contemporary isolating factors and/or historical processes in shaping this noticeable genetic isolation. Owing to the marginal position of the Azores Islands, we hypothesize that the impact of the countercurrents, roaming the Azores [60, 61], might render larval dispersal to more eastern locations difficult. This could prevent gene flow and account for the observed pattern of genetic differentiation. Alternatively, the recorded genetic distinctiveness of the Azores population could be attributed to the impact of historical processes linked to the palaeogeographic and palaeoclimatic evolution of the surveyed region. Given the particular genetic polymorphism (high haplotype diversity) recorded at these isolated islands at the westernmost edge of the species range, we rather suggest that this region might have served as a historical repository for genetic diversity during glacial low sea levels of the Pleistocene [62]. The impact of these historical events might have been maintained and intensified by the effect of particular oceanographic circulation, leading to the genetic differentiation of the Azores, as described in our study.
Evolutionary history
In addition to the likely involvement of the above-discussed contemporary factors in shaping population genetic structure of E. verrucosa, historical processes might also have exerted significant impact on geographic distribution of genetic variability. This assumption is likely supported by the outcomes of regional genetic diversity, spatio-temporal patterns of genetic diversification, and demographic history. Thereby, a complex series of micro-evolutionary forces encompassing allopatric differentiation, followed by range expansion and secondary contact and admixture among refugial lineages, might have contributed to shaping genetic diversity and structure in this decapod species. Hence, we discuss the evolutionary history of E. verrucosa in the context of the following issues:
Potential glacial refugia within the East Atlantic and western Mediterranean
As far as can be inferred from the outcome of this study, several lines of evidence support the existence of potential marine glacial refugia in the East Atlantic and western Mediterranean. First, assessment and comparison of regional genetic diversification showed that East Atlantic and western Mediterranean samples of E. verrucosa were significantly more diversified than their eastern Mediterranean counterparts for most of the examined parameters. Second, BEAST mitochondrial genealogy highlights potential genetic signatures of late Pleistocene genetic divergence between two lineages. Third, reconstruction of possible past geographic distributions of Cox1 haplotypes, using the S-DIVA method, identified the East Atlantic and the Mediterranean Sea as the most likely ancestral ranges for both retrieved lineages within E. verrucosa. Such integrative evidences suggest that suitable habitats within the East Atlantic and western Mediterranean (although in need to be consolidated and specified by further palaeoenvironmental and palaeogeographic evidences) might have been important marine biodiversity refugia to retain ancestral relict populations of E. verrucosa during Pleistocene climate changes. This assumption is likely supported by the outcome of recent phylogeographic studies on other Atlanto-Mediterranean marine species, which have also identified potential glacial refugia within the East Atlantic (namely across the Iberian Peninsula and Macaronesia) and the western Mediterranean [14, 17, 30, 63,64,65]. However, based on the analysis of only single mtDNA marker in limited sample dataset, this finding has to be considered as tentative and need to be confirmed by the analysis of another variable nuclear marker along with numerically and geographically extended datasets. Furthermore, detailed investigation, involving fine-tuned ecological niche modeling, is required to specify the exact locations of the postulated refugia.
Late Pleistocene vicariance across the Gibraltar Srait followed by postglacial expansion and admixture among refugial lineages
Integrative outcomes of BEAST and RASP analyses allowed retrieving genetic imprints of a late Pleistocene vicariant event across the Gibraltar Strait, which occurred approximately around 66,300 years before present. This historical event corresponds to the glacial period of the late Pleistocene (Würm glaciation: 110,000–12,000 years before present; [66]). It is known that cyclic shifts in sea levels during the Pleistocene glacial and interglacial periods could have potentially affected palaeogeography as well as palaeoenvironment of the East Atlantic-Mediterranean region [28, 29]. Specifically, glacial cycles of the Pleistocene might have provided the opportunity for allopatric differentiation in intertidal marine invertebrate species with planktonic larval dispersal, not only due to potential loss of habitat and subsequent genetic drift, but also following the restriction of connectivity between the East Atlantic and Mediterranean across the Gibraltar Strait. Accordingly, we hypothesize that the two Cox1 lineages might have differentiated in distinct glacial refugia during the harsh climate of the Würm glacial cycle. During the Pleistocene glacial periods, marine regression (dropping in sea level of about 150 m) might have not only narrowed the Gibraltar Strait following the emergence of set of islands [67], but also led to the restriction of marine circulation between the East Atlantic and Mediterranean and subsequent interruption of gene flow across the Gibraltar Strait [19].
Contrary to what has been found for other Atlanto-Mediterranean decapod species [2, 16, 68], genetic discontinuity triggered by this historical isolation within E. verrucosa has not been maintained until the present, as gene flow has been intensively restored between allopatric populations. This pattern is clearly evidenced in the TCS mitochondrial genealogy and can be mainly explained by the outcome of demographic history reconstruction. In particular, the BSP analysis showed that all estimated expansion events for the East Atlantic and Mediterranean regions markedly followed the Last Glacial Maximum period (LGM, between 26,500–20,000 years before present; [51]). It has been postulated that the warming of the East Atlantic and Mediterranean Sea following the LGM might have led to the onset of favourable environmental conditions for the rapid growth and expansion of marine populations [17]. The simultaneous occurrence of demographic expansion of both regional groups supports this assumption and indicates that Atlanto-Mediterranean specimens of E. verrucosa might have responded similarly to the climatic change and new abiotic conditions. Hence, it is more likely that the evidenced postglacial demographic and range expansions for this species might have promoted secondary contact and admixture among potential refugial lineages.
Recent colonization of the eastern Mediterranean
Analysis of regional genetic diversification revealed significantly reduced levels of genetic variability in eastern Mediterranean specimens of E. verrucosa. This pattern was shown for all examined diversity parameters except for haplotypic richness. Based on these insights, and referring to the markedly younger demographic and spatial expansion events recorded for the eastern Mediterranean than those retrieved for the western Mediterranean, we hypothesize that areas located to the east of the distribution range of the species might have been recently and rapidly colonized from the nearest refugia, when new habitats became available again.
However, given the fact that the eastern Mediterranean was shown to be genetically more diversified than the western Mediterranean based on haplotypic richness, the validity of this assumption can be seriously questioned. Two possible explanations can be advanced in order to clarify this discrepancy. First, it has been shown that genetic diversity in re-colonized areas can be either higher or lower depending on the estimator [69]. Second, given the significantly lowest levels of private haplotypes recorded in the eastern Mediterranean against a background of higher haplotypic richness (after a rarefaction procedure), we can assume that unexpected increase in the latter parameter could be likely due to the fact that the eastern Mediterranean may correspond to a contact zone between founding haplotypes from potential refugial zones [62, 70]. According to the latter explanation, we hypothesize that the eastern Mediterranean might have been recently colonized from putative refugia within both the western Mediterranean and the East Atlantic. Nevertheless, it should be mentioned that owing to the limited examined samples in this study, these insights have to considered as tentative and need to be confirmed by analysis of another nuclear marker in exhaustive numerically and geographically datasets.
Evolutionary history scenario for E. verrucosa
Overall, based on the obtained preliminary results, we propose the following evolutionary history scenario for E. verrucosa: The harsh climatic changes, mainly mediated by significant climate cooling and dropping of sea levels, during the Würm glaciations, might have led to the restriction of biotic exchange across the Gibraltar Strait and consequently to the retreatment of specimens of the warty crab to specific refugia within the East Atlantic and Mediterranean Sea. Subsequent postglacial recolonization of suitable habitats, following the rise of sea level and amelioration of climate conditions, might have led not only to the resumed connectivity across the Gibraltar Strait but also to the recent colonization of the eastern Mediterranean. The resulting gene flow restoration among historically isolated populations across the Gibraltar Strait (which might have not had enough time to accumulate local significant genetic differences as well as adaptations to the disparate environmental conditions of the East Atlantic and the Mediterranean Sea) is still likely maintained by the impact of contemporary factors including oceanographic features, larval behaviour, and large effective population size.
Conclusions
The results of the present study provide new insights into the phylogeography and evolutionary history of a common but poorly studied Atlanto-Mediterranean decapod species. Specifically, they contribute to the understanding of the impact of historical processes on shaping contemporary population genetic structure and diversity in intertidal marine species. Increase in the sample size and investigation of highly variable nuclear markers are highly required and recommended in future investigation in order to confirm the discerned pattern of phylogeographic structure and provide a more complete and nuanced view of the evolutionary history of this important species.
Methods
Sampling scheme and genomic DNA isolation
A total of 155 samples of Eriphia verrucosa were obtained from 35 locations across the whole distribution range stretching from the East Atlantic to the Black Sea (Table 1 and Fig. 1). Apart from incorporating newly collected material (143 specimens from 31 locations), all available Cox1 sequences in GenBank (12 sequences previously obtained from 4 locations) have been also included in the study in order to maximize delineation of phylogeographic patterns and reliably infer the evolutionary history of the examined decapod species. Details on the sampled locations, and the number of individuals analyzed per population, are reported in Fig. 1 and Table 1. In the field, a single pereiopod was removed from the sampled crabs and stored in absolute ethanol until genetic analysis (these appendages are re-gained in the successive moults). Subsequently, the animals were released in their original environments. Back to the laboratory, total genomic DNA was extracted from pereiopod muscle tissue by means of the Puregene kit (Gentra Systems: Minneapolis, MN55447, USA).
Amplification and sequencing of mitochondrial Cox1 gene
Amplification of the mitochondrial Cox1 gene was carried out with the specifically constructed decapod primers COL6a and COH6 and/or COL6a and COH1b [71]. Details on the used mixture for PCR reactions as well as thermocycling conditions were cited in Deli et al. [15]. Visualization of PCR products (loaded on 1.5% agarose gel with GelRed staining) was carried out under UV light. Sequencing of the resulting Cox1 amplicons was carried out with the forward primer COL6a at LGC Genomics or Macrogen Europe. Inspection and edition of the obtained sequences were assured with Chromas Lite 2.1.1 [72]. After their alignment with Clustal W, implemented in BIOEDIT [73], Cox1 sequences were adjusted to a gene fragment of a 453 basepairs (bp) length for further statistical analyses. Sequences corresponding to the characterized Cox1 haplotypes were submitted to GenBank (accession numbers: MK493758-MK493787).
Data and statistical analyses
Data analysis procedure
We tried to increase the sample size of the analyzed populations by combining collection sites featuring low number of specimens. In our pooling strategy, we decided to take into account two main criteria: (1) Assignment according to geographic origin of the specimens so that only geographically closed locations within the same region (East Atlantic or Mediterranean Sea) or sub-regions (western Mediterranean or eastern Mediterranean) can be combined. (2) Assignment considering the postulated barriers to gene flow (aimed to be investigated in this study for phylogeographic structure, see below). In the latter case, the pooling procedure was not applied on the geographically close locations lying across these biogeographic boundaries even if this pattern of combination may still lead to limited sample size as that obtained in the two defined populations of Northern Tunisia and Alboran Sea (Tables 1 and 2). Accordingly, samples from the 35 surveyed locations were combined into 15 populations (Table 2). It has to be mentioned that the adoption of this procedure in population genetic investigation may be problematic. It could seriously question the reliability of the outcome of the statistical analyses since any small change in some of the pooling decisions may change the significance of some statistical tests. However, in the absence of larger sample sizes that would surely avoid this rather inconvenient procedure, respecting the two above mentioned conditions for specimens pooling in our study would at least allow minimizing the drawback of the method. Moreover, the resulting initial genetic signals would be helpful for the build up to an in-depth further investigation and orientate the sampling focus if any specific issues need to be addressed.
Analysis of genetic variability and assessment of regional genetic diversification
The nucleotide compositions as well as the number of variable and parsimony-informative nucleotide sites were estimated with MEGA version 7.0.18 [74]. Measurements of DNA polymorphism, including numbers of haplotypes (Nh) and polymorphic sites (Nps), haplotype (h) and nucleotide (π) diversities [75, 76], as well as mean number of nucleotide differences (K), were calculated for each population, each region (and sub-region), as well as for the total dataset by means of DnaSP version 5.10 [77].
Assessment and comparison of genetic diversification levels among the East Atlantic, western Mediterranean and eastern Mediterranean were carried out by the examination of seven genetic diversity indices (h, π, K, Hapr (haplotypic richness), Np (number of private haplotypes), Np/N (proportion of private haplotypes), and Np/Hapr (genetic endemism)). Owing to the difference among the sample sizes of the examined datasets, we applied a rarefaction procedure in order to discard the positive bias of sample size on inferred diversity. For each sample larger than the smallest one with the size N = 42, recorded for the East Atlantic region, a subsample of N individuals was drawn randomly and diversity indices were calculated. In order to infer reliable statistics, the procedure was repeated 30 times for each sample, and then the mean subsample diversity for each sample was calculated. In order to check for the consistency of the obtained results, and see whether the number of replicates could influence the outcome of comparison, analyses were re-carried out with 60 replicates. The unpaired t-test, as implemented in the online software T-Tests-Free Statistics and Forecasting Software (Calculators) version 1.2.1 [78], was used to assess inter-regional difference for each genetic diversity parameter.
Assessment of genealogical relationships among Cox1 haplotypes
Genealogical relationships among Cox1 haplotypes were examined and assessed through construction of the haplotype network with the statistical parsimony procedure included in the software TCS version 1.21 [79]. Correlation between genealogy and geographic distribution of Cox1 haplotypes was assessed by comparing the two measurements of population differentiation, GST (taking into account haplotype frequencies) and NST (considering relationship among haplotypes) [80, 81], in PERMUT & CPSRR version 2.0 [81].
Analyses of genetic differentiation and phylogeographic structure
Patterns of genetic differentiation within the total mitochondrial dataset (by means of one-level AMOVA [82]), and among pairs of populations, were assessed in ARLEQUIN version 3.1 [83]. Both nucleotide divergence (based on the Tajima-Nei model [84]) and haplotypic frequencies were used to infer these estimations. A total of 10,000 permutations were adopted to estimate significant genetic distances (P < 0.05). Accurate inference of the significance level was assured by the B-Y FDR correction [85] (based on the determined critical value of P < 0.00954).
The two-level AMOVA (as implemented in ARLEQUIN) was used to test hierarchical structuring of genetic variation within E. verrucosa, seeking for evidence of significant phylogeographic structure across postulated barriers to gene flow, such as the Gibraltar Strait, the Almería-Oran Oceanographic Front, the Siculo-Tunisian Stait, and the Peloponnese hydrographic break.
Evolutionary history and historical biogeography reconstruction
In order to reliably elucidate the evolutionary history of E. verrucosa, we specifically estimated the mutation rate of the Cox1 gene referring to the already surveyed phylogeny of the genus Eriphia by Lai et al. [86], whose findings suggest congruence between geography and phylogeny in Eriphia. We, therefore, implemented a calibration point derived from the biogeographic event of the Pleistocene glacial sea-level low stands, assuming that the vicariant speciation of Indian and Indo-West Pacific species of the genus Eriphia might have been triggered by the impact of dry exposure of the Sunda Shelf at the onset of the Pleistocene glaciation cycles which occurred approximately 2.588 million years ago (± 0.005) [87, 88]. The estimation of molecular clock rate was carried out in BEAST version 1.7.5 [89]. Before proceeding with the analysis, selection of the best-fit substitution model for the data was carried out with MODELTEST version 3.7 [90]. Cox1 sequences, corresponding to the eight examined species of the genus Eriphia by Lai et al. [86] (GenBank accession numbers: HM638034-HM638038; KC771023-KC771024; KC962408) as well as to the haplotypes retrieved in our study, were used in the analysis. The two parameter Birth-Death model (considered as an appropriate null model for species diversification [91]), along with an uncorrelated lognormal relaxed clock, were used as priors for the analysis. A total run of 30 million generations were designed for the Markov chain Monte Carlo (MCMC) simulations. The outcome of the Bayesian analysis was exhibited and checked in TRACER version 1.5 [92].
Temporal patterns of Cox1 haplotypes diversification were estimated in the software BEAST version 1.7.5 with a coalescent tree prior, suggested as an appropriate null model for intraspecific data [93]. A strict molecular clock along with the generalized time reversible (GTR) model of sequence evolution [94] (as determined by MODELTEST) were also chosen as priors to carry out the analysis. The diversification time of the examined Cox1 dataset was inferred after calibrating the retrieved haplotype phylogeny with the determined Cox1 gene mutation rate for Eriphia. The Bayesian analysis was accomplished after running the MCMC simulations for 30 million generations (sampled every 1000 generations), checking the generated outputs for convergence (Effective Sample Sizes, ESS, of all parameters > 200) in TRACER version 1.5, and summarizing the resultant trees in TreeAnnotator version 1.7.5 [89]. The obtained calibrated Cox1 genealogy was checked and shown in FigTree version 1.4.0 [95].
The biogeographic history of E. verrucosa was inferred from ancestral area reconstruction based on Cox1 haplotypes using the S-DIVA (Statistical Dispersal-Vicariance Analysis) method, as implemented in RASP version 3.2 [96]. S-DIVA is a parsimony method of historical biogeography [97] and is recommended if there is some a priori knowledge of vicariance events that could have played an important role in the biogeographic history of the examined taxon. Taking into account the geographic distribution of E. verrucosa Cox1 haplotypes, three main biogeographic regions were defined: (A) Atlantic Ocean, (B) Mediterranean Sea; and (C) Atlantic-Mediterranean. The Most Likely States (MLS) function, as implemented in RASP, was used to highlight the likelihood of possible ancestral range at each generated node of the phylogeny. The MCMC output and condensed tree exported from BEAST and TreeAnnotator, as well as the distribution of the Cox1 haplotypes through all defined biogeographical areas were loaded as ‘trees file’ and ‘distribution file’ respectively in order to perform S-DIVA analysis in RASP.
Inference of demographic history
Signatures of past demographic changes in the warty crab E. verrucosa were detected by the analysis of three neutrality tests (Tajima’s D [98], Fu’s Fs [99], and Ramos-Onsins and Rozas’s R2 [100]). A total of 1000 coalescent simulations were employed in ARLEQUIN to estimate D and Fs statistics, as well as in DnaSP to calculate the R2 index. Significantly negative outputs of D and Fs are usually indicators of demographic expansion. In case of examined data with small size, the R2 test has more power and is more reliable than D and Fs to detect population expansion. Evidence of sudden demographic expansion event was also checked via analysis of the Harpending’s raggedness index rg [101] in ARLEQUIN (with the significance of the test assessed with 10,000 replicates). All these measures were determined for each population, each region (and sub-region), as well as for the overall dataset.
Both demographic and spatial expansion [102] models, as implemented in ARLEQUIN, were assessed in the whole dataset as well as in the East Atlantic and Mediterranean Sea (including its Western and Eastern basins). The sum of squared deviations (SSD) between observed and expected distributions under the tested expansion model were used as a measure of fit, and the probability of obtaining a simulated SSD greater than or equal to the expected was computed by 1000 random permutations. If this probability was > 0.05, the expansion model was accepted. Time since expansion, measured in mutational time units (τ), was inferred for each study case.
Detailed information on the amplitude of historical demographic events that the warty crab populations could have undergone was also retrieved from the outcome of the Bayesian Skyline Plot (BSP) method [103]. BSP analyses were carried out, in BEAST version 1.7.5, for the East Atlantic and Mediterranean regions as well as for whole Cox1 dataset. A GTR substitution model and a strict molecular clock were set as priors for the analysis. Time since expansion was estimated with the specifically determined Cox1 gene mutation rate for Eriphia. Two MCMC simulations were run independently for 40 million iterations. After discarding the first 10% iterations (4 millions) as burn-in, LogCombiner version 1.7.5 [89] was used to combine the remaining outputs. BSP plots (corresponding to the three analyzed datasets) were generated in TRACER version 1.5 after checking for the data convergence.
Abbreviations
- °C:
-
Degrees celsius
- DNA:
-
Deoxyribonucleic acid
- mtDNA:
-
Mitochondrial DNA
- PCR:
-
Polymerase chain reaction
- UV:
-
Ultra violet
References
Nikula R, Väinölä R. Phylogeography of Cerastoderma glaucum (Bivalvia: Cardiidae) across Europe: a major break in the eastern Mediterranean. Mar Biol. 2003;143:339350.
Reuschel S, Cuesta JA, Schubart CD. Marine biogeographic boundaries and human introduction along the European coast revealed by phylogeography of the prawn Palaemon elegans. Mol Phyl Evol. 2010;55:765–75.
Penant G, Aurelle D, Feral J-P, Chenuil A. Planktonic larvae do not ensure gene flow in the edible sea urchin Paracentrotus lividus. Mar Ecol Prog Ser. 2013;480:155–70.
Deli T, Chatti N, Said K, Schubart CD. Concordant patterns of mtDNA and nuclear phylogeographic structure reveal Pleistocene vicariant event in the green crab Carcinus aestuarii across the Siculo-Tunisian Strait. Mediterr Mar Sci. 2016;17(2):533–51.
Avise JC. Phylogeography: the history and formation of species. Cambridge: Harvard University Press; 2000.
Hewitt GM. The genetic legacy of the quaternary ice ages. Nature. 2000;405:907–13.
Hewitt GM. Genetic consequences of climatic oscillations in the quaternary. Phil Trans R Soc B. 2004;359:183–95.
Deli T, Kalkan E, Karhan SÜ, Uzunova S, Keikhosravi A, Bilgin R, et al. Parapatric genetic divergence among deep evolutionary lineages in the Mediterranean green crab, Carcinus aestuarii (Brachyura, Portunoidea, Carcinidae), accounts for a sharp phylogeographic break in the eastern Mediterranean. BMC Evol Biol. 2018;18:53.
Provan J, Bennett KD. Phylogeographic insights into cryptic glacial refugia. Trends Ecol Evol. 2008;23:564–71.
Hewitt GM. Some genetic consequences of ice ages and their role in divergence and speciation. Biol J Linnean Soc. 1996;58:247–76.
Hewitt GM. Postglacial re-colonization of European biota. Biol J Linnean Soc. 1999;68:87–112.
Provan J, Wattier RA, Maggs CA. Phylogeographic analysis of the red seaweed Palmaria palmate reveals a Pleistocene marine glacial refugium in the English Channel. Mol Ecol. 2005;14:793–803.
Hu Z-M, Kantachumpoo A, Liu R-Y, Sun Z-M, Yao J-T, Komatsu T, et al. A late Pleistocene marine glacial refugium in the south-west of Hainan Island, China: Phylogeographical insights from the brown alga Sargassum polycystum. J Biogeogr. 2018;45:355–66.
Albaina N, Olsen JL, Couceiro L, Ruiz JM, Barreiro R. Recent history of the European Nassarius nitidus (Gastropoda): phylogeographic evidence of glacial refugia and colonization pathways. Mar Biol. 2012;159:1871–84.
Deli T, Pfaller M, Schubart CD. Phylogeography of the littoral prawn species Palaemon elegans (Crustacea: Caridea: Palaemonidae) across the Mediterranean Sea unveils disparate patterns of population genetic structure and demographic history in the two sympatric genetic types II and III. Mar Biodivers. 2018;48:1979–2001.
Weiss R, Torrecilla Z, González-Ortegón E, González-Tizón AM, Martínez-Lage A, Schubart CD. Genetic differentiation between Mediterranean and Atlantic populations of the common prawn Palaemon serratus (Crustacea: Palaemonidae) reveals uncommon phylogeographic break. J Mar Biol Assoc U.K. 2018;98:1425-1434. https://doi.org/10.1017/S0025315417000492.
Evangelisti F, Bellucci A, Sabelli B, Albano PG. The periwinkle Echinolittorina punctata (Mollusca: Gastropoda) tracked the warming of the Mediterranean Sea following the Last Glacial Maximum. Mar Biol. 2017;164:34. https://doi.org/10.1007/s00227-017-3071-7.
Bahri-Sfar L, Lemaire C, Ben Hassine OK, Bonhomme F. Fragmentation of sea bass populations in the western and eastern Mediterranean as revealed by microsatellite polymorphism. Proc R Soc Lond B. 2000;267:929–35.
Patarnello T, Volckaert FAM, Castilho R. Pillars of Hercules: is the Atlantic-Mediterranean transition a phylogeographic break? Mol Ecol. 2007;16:4426–44.
Ragionieri L, Schubart CD. Population genetics, gene flow and biogeographic boundaries of Carcinus aestuarii (Crustacea: Brachyura: Carcinidae) along the European Mediterranean coast. Biol J Linn Soc. 2013;109:771–90.
Fratini S, Ragionieri L, Deli T, Harrer A, Marino IAM, Cannicci S, et al. Unravelling population genetic structure with mitochondrial DNA in a notional panmictic coastal crab species: sample size makes the difference. BMC Evol Biol. 2016;16:150.
Pannacciulli FG, Maltagliati F, de Guttry C, Achituv Y. Phylogeography on the rocks: the contribution of current and historical factors in shaping the genetic structure of Chthamalus montagui (Crustacea, Cirripedia). PLoS One. 2017;12(6):e0178287.
Pérez-Losada M, Nolte MJ, Crandall KA, Shaw PW. Testing hypotheses of population structuring in the Northeast Atlantic Ocean and Mediterranean Sea using the common cuttlefish Sepia officinalis. Mol Ecol. 2007;16:2667–79.
Zulliger DE, Tanner S, Ruch M, Ribi G. Genetic structure of the high dispersal Atlanto-Mediterranean Sea star Astropecten aranciacus revealed by mitochondrial DNA sequences and microsatellite loci. Mar Biol. 2009;156:597–610.
Borrero-Pérez GH, Gonzalez-Wangüemert M, Marcos C, Pérez-Ruzafa A. Phylogeography of the Atlanto-Mediterranean Sea cucumber Holothuria (Holothuria) mammata: the combined effects of historical processes and current oceanographical pattern. Mol Ecol. 2011;20:1964–75.
Sanna D, Cossu P, Dedola GL, Scarpa F, Maltagliati F, Castelli A, et al. Mitochondrial DNA reveals genetic structuring of Pinna nobilis across the Mediterranean Sea. PLoS One. 2013;8(6):e67372.
Lambeck K, Esat TM, Potter EK. Links between climate and sea levels for the past three million years. Nature. 2002;419:199–205.
Maggs CA, Castilho R, Foltz D, Henzler C, Jolly MT, Kelly J, et al. Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology. 2008;89:108–22.
Wilson AB, Veraguth IE. The impact of Pleistocene glaciations across the range of a widespread European coastal species. Mol Ecol. 2010;19:4535–53.
Woodall LC, Koldewey HJ, Shaw PW. Historical and contemporary population genetic connectivity of the European short-snouted seahorse Hippocampus hippocampus and implications for management. J Fish Biol. 2011;78:1738–56.
Schunter C, Carreras-Carbonell J, Macpherson E, Tintoré J, Vidal-Vijande E, Pascual A, et al. Matching genetics with oceanography: directional gene flow in a Mediterranean fish species. Mol Ecol. 2011;20:5167–81.
Galarza JA, Carreras-Carbonell J, Macpherson E, Pascual M, Roques S, Turner GF, et al. The influence of oceanographic fronts and early-life-history traits on connectivity among littoral fish species. Proc Natl Acad Sci U S A. 2009;106:1473–8.
Millot C, Taupier-Letage I. Circulations in the Mediterranean Sea. Handb Environ Chem. 2005;5:29–66.
Millot C. Circulation in the Mediterranean Sea: evidences, debates and unanswered questions. Sci. 2005;69:5–21.
Provan J. The effects of past, present and future climate change on rangewide genetic diversity in northern North Atlantic marine species. Front Biogeogr. 2013;5(1):60–6.
Silva AC, Silva IC, Hawkins SJ, Boaventura DM, Thompson RC. Cheliped morphological variation of the intertidal crab Eriphia verrucosa across shores of differing exposure to wave action. J Exp Mar Biol Ecol. 2010;391:84–91.
Silva AC, Shapouri M, Cereja R, Dissanayake A, Vinagre C. Variations in crab claw morphology and diet across contrasting inter-tidal habitats. Mar Ecol. 2017;38:e12374.
d’Udekem d’Acoz C. Inventaire et distribution des crustacés décapodes de l’Atlantique nord-oriental, de la Méditerranée et des eaux continentales adjacentes au nord de 25°N. Paris: Muséum national d’Histoire naturelle; 1999. p. 383. (Collection Patrimoines Naturels, 40)
Flores AAV, Paula J. Intertidal distribution and species composition of brachyuran crabs at two rocky shores in Central Portugal. Hydrobiologia. 2001;449:171–7.
Rossi AC, Parisi V. Experimental studies of predation by the crab Eriphia verrucosa on both snail and hermit crab occupants of conspecific gastropod shells. Boll Zool. 1973;40:117–85.
Dumitrache C, Konsulova T. Eriphia verrucosa Forskal, 1755. Black Sea red data book. Nairobi: United Nations Environment Programme; 2009.
Erkan M, Balkıs H, Kurun A, Tunalı Y. Seasonal variations in the ovary and testis of Eriphia verrucosa (Forskal, 1775) from Karaburun, SW Black Sea. Pak J Zool. 2008;40:217–21.
Karadurmuş U, Aydin M. An investigation on some biological and reproduction characteristics of Eriphia verrucosa (Forskål, 1775) in the South Black Sea (Turkey). Turk J Zool. 2016;40:461–70.
Lumare F, Gozzo S. Sviluppo larvale del crostaceo Xantideae Eriphia verrucosa (Forskal, 1775) in condizioni di laboratorio. Boll Pesca Piscic Idrobiol. 1972;17:185–209.
Avise JC. Molecular markers, natural history and evolution. New York and London: Chapman and Hall; 1994.
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am. 1994;87:651–701.
Knowlton N, Weigt LA. New dates and new rates for divergence across the isthmus of Panama. Proc R Soc Lond. 1998;265:2257–63.
Schubart CD, Diesel R, Hedges SB. Rapid evolution to terrestrial life in Jamaican crabs. Nature. 1998;393:363–5.
Marino IAM, Pujolar JM, Zane L. Reconciling deep calibration and demographic history: Bayesian Inference of post glacial colonization patterns in Carcinus aestuarii (Nardo, 1847) and C. maenas (Linnaeus, 1758). PLoS ONE. 2011;6(12):e28567.
Crandall ED, Sbrocco EJ, DeBoer TS, Barber PH, Carpenter KE. Expansion dating: calibrating molecular clocks in marine species from expansions onto the Sunda shelf following the last glacial maximum. Mol Biol Evol. 2012;29(2):707–19.
Clark PU, Dyke AS, Shakun JD, Carlson AE, Clark J, Wohlfarth B, et al. The last glacial maximum. Science. 2009;325:710–4.
Béranger K, Mortier L, Gasparini GP, Gervasio L, Astraldi M, Crépon M. The dynamics of the Sicily Strait: a comprehensive study from observations and models. Deep-Sea Res PT II. 2004;51:411–40.
Serena F. Field identification guide to the sharks and rays of the Mediterranean and Black Sea. p. 97. In: FAO Species identification Guide for Fishery Purposes. Rome: FAO; 2005.
Eckman JE. Closing the larval loop: linking larval ecology to the population dynamics of marine benthic invertebrates. J Exp Mar Biol Ecol. 1996;200:207–37.
Wing SR, Largier JL, Botsford LW, Quinn JF. Settlement and transport of benthic invertebrates in an intermittent upwelling zone. Limnol Oceanogr. 1995;40:316–29.
Deli T, Ben Mohamed A, Ben Attia MH, Zitari-Chatti R, Said K, Chatti N. High genetic connectivity among morphologically differentiated populations of the black sea urchin Arbacia lixula (Echinoidea: Arbacioida) across the central African Mediterranean coast. Mar Biodiv. 2019;49:603-620.
Sá-Pinto A, Branco M, Sayanda D, Alexandrino P. Patterns of colonization, evolution and gene flow in species of the genus Patella in the Macaronesian Islands. Mol Ecol. 2008;17:519–32.
Pérez-Portela R, Villamor A, Almada V. Phylogeography of the sea star Marthasterias glacialis (Asteroidea, Echinodermata): deep genetic divergence between mitochondrial lineages in the north-western mediterranean. Mar Biol. 2010;157:2015–28.
Gonzalez-Wangüemert M, Froufe E, Pérez-Ruzafa A, Alexandrino P. Phylogeographical history of the white seabream Diplodus sargus (Sparidae): implications for insularity. Mar Biol Res. 2011;7:250–60.
Alves M, Gaillard F, Sparrow M, Knoll M, Giraud S. Circulation patterns and transport of the Azores front current system. Deep-Sea Res PT II. 2002;49:3983–4002.
Juliano MF, Alves MLR. The Atlantic subtropical front / current Systems of Azores and St. Helena. J Phys Oceanogr. 2007;37:2573–98.
Chevolot M, Hoarau G, Rijnsdorp AD, Stam WT, Olsen JL. Phylogeography and population structure of thornback rays (Raja clavata L., Rajidae). Mol Ecol. 2006;15:3693–705.
Alberto F, Massa S, Manent P, Diaz-Almela E, Arnaud-Haond S, Duarte CM, et al. Genetic differentiation and secondary contact zone in the seagrass Cymodocea nodosa across the Mediterranean–Atlantic transition region. J Biogeogr. 2008;35:1279–94.
Wangensteen OS, Turon X, Pérez-Portela R, Palacin C. Natural or naturalized? Phylogeography suggests that the abundant sea urchin Arbacia lixula is a recent colonizer of the Mediterranean. PLoS One. 2012;7(9):e45067.
Pérez-Portela R, Rius M, Villamor A. Lineage splitting, secondary contacts and genetic admixture of a widely distributed marine invertebrate. J Biogeogr. 2017;44(2):446–60.
Kukla G. Saalian supercycle, Mindel/Riss interglacial and Milankovitch's dating. Quat Sci Rev. 2005;24:1573–83.
Collina-Girard J. Atlantis off the Gibraltar Strait? Myth and geology. Comptes Rendus l’Acad Sci Ser II A Earth Planet Sci. 2001;333(4):233–40.
Roman J, Palumbi SR. A global invader at home: population structure of the green crab Carcinus maenas in Europe. Mol Ecol. 2004;13:2891–8.
Comps B, Gömöry D, Letouzey J, Thiébaut B, Petit RJ. Diverging trends between heterozygosity and allelic richness during postglacial colonization in the European beech. Genetics. 2001;157:389–97.
Coyer JA, Diekmann OE, Serrão EA, Procaccini G, Milchakova N, Pearson GA, et al. Population genetics of dwarf eelgrass Zostera noltii throughout its biogeographic range. Mar Ecol Prog Ser. 2004;281:51–62.
Schubart CD. Mitochondrial DNA and decapod phylogenies: The importance of pseudogenes and primer optimization. In: Martin JW, Crandall KA, Felder DL, editors. Crustacean issues 18: decapod crustacean Phylogenetics. Boca Raton, Florida: Taylor and Francis/CRC Press; 2009. p. 47–65.
Technelysium Pty Ltd. Chromas lite ver. 2.1.1., 2012. Available from: http://www.technelysium.com.au/chromas_lite.html.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetic analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987.
Tajima F. Evolutionary relationships of DNA sequences in finite populations. Genetics. 1983;105:437–60.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Holliday IE. T-Tests (v1.0.3) in Free Statistics Software (v1.2.1), Office for Research Development and Education, 2017. Available from: https://www.wessa.net/rwasp_Tests%20to%20Compare%20Two%20Means.wasp.
Clement M, Posada D, Crandall K. TCS: A computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–60.
Pons O, Petit RJ. Estimation, variance and optimal sampling of gene diversity. Theor Appl Genet. 1995;90:462–70.
Pons O, Petit RJ. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics. 1996;144:1237–45.
Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–91.
Excoffier L, Laval G, Schneider S. Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinformatics Online. 2005;1:47–50.
Tajima F, Nei M. Estimation of evolutionary distance between nucleotide sequences. Mol Biol Evol. 1984;1:269–85.
Narum SR. Beyond Bonferroni: less conservative analyses for conservation genetics. Conserv Genet. 2006;7(5):783–7.
Lai JCY, Thoma BP, Clark PF, Felder DL, Ng PKL. Phylogeny of eriphioid crabs (Brachyura, Eriphioidea) inferred from molecular and morphological studies. Zool Scr. 2014;43(1):52–64.
Hui M, Kraemer WE, Seidel C, Nuryanto A, Joshi A, Kochzius M. Comparative genetic population structure of three endangered giant clams (Cardiidae: Tridacna species) throughout the indo-West Pacific: implications for divergence, connectivity and conservation. J Molluscan Stud. 2016;82:403–14.
Riccardi AC. IUGS ratified ICS recommendation on redefinition of Pleistocene and formal definition of base quaternary. International Union of Geological Sciences; 2009.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8.
Nee S, May RM, Harvey PH. The reconstructed evolutionary process. Philos Trans R Soc Lond B. 1994;344:305–11.
Rambaut A, Drummond AJ. Tracer v 1.4.8. Institute of Evolutionary Biology: University of Edinburg; 2007. Available from: http://beast.community/tracer
Kingman JFC. The coalescent. Stoch Process Their Appl. 1982;13:235–48.
Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures on mathematics in the life sciences. Amer Math Soc. 1986;17:57–86.
Rambaut A. FigTree v1.3.1, 2009. Computer program available at: http://tree.bio.ed.ac.uk/software/figtree/.
Yu Y, Harris AJ, Blair C, He X. RASP (reconstruct ancestral state in phylogenies): a tool for historical biogeography. Mol Phylogenet Evol. 2015;87:46–9.
Ronquist F. Dispersal-Vicariance analysis: a new approach to the quantification of historical biogeography. Syst Biol. 1997;46(1):195–203.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Ramos-Onsins SE, Rozas J. Statistical properties of new neutrality tests against population growth. Mol Biol Evol. 2002;19:2092–100.
Harpending HC. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol. 1994;66(4):591–600.
Excoffier L. Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol. 2004;13:853–64.
Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22:1185–92.
Acknowledgements
We would like to thank Daou Haddoud for his help with crab sampling from the Libyan coast, Salud Deudero for sending specimens from Mallorca Island, Micha Ilan for providing materials from Sdot Yam and Korbinian Eckel, Ruth Jesse, Lapo Ragionieri, Silke Reuschel, Henrik and Sven Schubart, for their help while collecting in the field and to S. Reuschel, Tobias Platzek, and Alexandra Harrer for generating the first sequences. Some of the field work was conducted during DAAD academic exchange projects of the last author with Sonya Uzunova from the Institute of Fish Resources in Varna (Bulgaria; PPP program D/08/02059) and with Marco Vannini and Sara Fratini from the University of Florence (Italy; PPP program Vigoni-DAAD D/04/47157). Special thanks are also extended to the Institute of Zoology and the chair Prof. Jürgen Heinze at the University of Regensburg (Germany) for hosting the first author during a research visit and for financially supporting this research. We are also very grateful to the editor and two anonymous reviewers for their very interesting comments and suggestions that greatly helped with improving the quality of the manuscript.
Funding
This study was financially supported by the Institute of Zoology and the chair Prof. Jürgen Heinze at the University of Regensburg (Germany). The funding body specifically helped with the experimental design of the genetic investigation and sample collection. Some of the field work was conducted during DAAD academic exchange projects of the University of Regensburg (Germany) with the Institute of Fish Resources in Varna (Bulgaria; PPP program D/08/02059) and with the University of Florence (Italy; PPP program Vigoni-DAAD D/04/47157). We acknowledge support of the publication fee by the University of Regensburg.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and in the Additional file 1: Table S1, Additional file 2: Table S2, Additional file 3: Table S3, Additional file 4: Table S4 and Additional file 5: Figure S1.
Author information
Authors and Affiliations
Contributions
TD and CDS conceived and funded the study. TD and CK participated in laboratory work. TD carried out the statistical analyses. TD and CDS wrote the manuscript. All authors read, edited and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
None of the sampled populations of Eriphia verrucosa is endangered or protected by any international or national legal framework. Also, no specific permissions were required for sampling activities.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Estimation of genetic diversity parameters in Atlantic and Mediterranean specimens of Eriphia verrucosa, based on two analyzed datasets (including and excluding retrieved sequences from GenBank). (DOCX 13 kb)
Additional file 2:
Table S2. Pattern of assignment of the twelve Cox1 sequences of Eriphia verrucosa (retrieved from GenBank) to the detected haplotypes in this study. (DOCX 11 kb)
Additional file 3:
Table S3. Geographic distribution of the 30 haplotypes of E. verrucosa recorded at the 15 defined populations within the East Atlantic Ocean and Mediterranean Sea. (DOCX 16 kb)
Additional file 4:
Table S4. Analysis of pairwise genetic differentiation in a total dataset of 143 specimens of Eriphia verrucosa (excluding the twelve sequences from GenBank). (DOCX 17 kb)
Additional file 5.
Figure S1. Analysis of the historical biogeography of E. verrucosa based on a total dataset of 143 sequences (excluding the GenBank sequences). (DOCX 304 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Deli, T., Kiel, C. & Schubart, C.D. Phylogeographic and evolutionary history analyses of the warty crab Eriphia verrucosa (Decapoda, Brachyura, Eriphiidae) unveil genetic imprints of a late Pleistocene vicariant event across the Gibraltar Strait, erased by postglacial expansion and admixture among refugial lineages. BMC Evol Biol 19, 105 (2019). https://doi.org/10.1186/s12862-019-1423-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-019-1423-2