
Abstract
Background
The plane leaf miner, Phyllonorycter platani is a widely distributed insect species on plane trees and has a well-documented colonisation history in Europe over the last century. However, phylogeographic data of the species are lacking.
Results
We analysed 284 individuals from 38 populations across Europe, Asia, and North America. A 1242 bp fragment of the mitochondrial COI gene and an 893 bp fragment of the 28S rDNA has been Sanger sequenced. Twenty-four haplotypes were detected on the COI gene, and two alleles were identified on the 28S rDNA. We revealed two distinct clades for both markers reflecting the geographic origins, Asia and Europe. The genetic distance between the two main clades is 2.08% on the COI gene and 0.10% on the nuclear DNA.
An overlapping zone of the two clades was found across Eastern Europe and the Anatolian Peninsula. We detected heterozygote individuals of the 28S rDNA gene in Moldavia, Ukraine and in the southern part of Turkey. These suggest that the two clades can hybridise. Furthermore, the presence of European type homozygote individuals has been confirmed in the southern part of Turkey as well.
Conclusions
We have shown that both post-glacial recolonization and recent expansion events influenced the present genetic structure of P. platani. The genetic patterns revealed at least two refugia during the last ice age: one in the Balkan Peninsula and the other in the Caucasus region. Recent expansion was detected in some European and Central Asian populations. The two main clades (Europe/Asia) show definite genetic differences; however, several hybrid individuals were found in the overlapping zone as well (stretching over Eastern Europe and the Anatolian Peninsula). Discrepancies in mitochondrial and nuclear data indicate introgressions in the southern part of the Anatolian Peninsula.
Similar content being viewed by others

Background
Biological invasions have been considered a major economic and conservation problem recently [1, 2]. The plane leaf miner, Phyllonorycter platani (STAUDINGER, 1870), is one of the most important invasive Gracillariidae species [3, 4]. Its colonisation history is well documented across Europe and its range expansion started in the second half of the nineteenth century [3, 5]. The colonisation process includes several jumps from the native origin (SE-Europe) to the Northern and North-Western Europe [5]. The dispersal occurred in anemochoral and antropochoral ways with passive transportation of mined leaves and/or saplings [5].
According to the Global Taxonomic Database of Gracillariidae [6] Phyllonorycter platani (STAUDINGER, 1870) has a large distribution area across Europe, the Anatolian Peninsula, Near-East and Central Asia. Šefrová [3, 5] suggests that it is native to Southern Europe and to Central Asia where the host plant (Platanus orientalis) is native. Lopez-Vaamonde et al. [4] considered the area of origin of P. platani as unknown.
Heinrich [7] described Phyllonorycter felinella HEINRICH, 1920 in California (US), but Deschka [8] synonymized this taxon with Lithocolletis platani STAUDINGER, 1870. The actual situation of the taxon was, until recently, uncertain. [8,9,10,11], [7, 12,13,14].
Leaf mining is a special kind of phytophagy where monophagous species are dominant. Host shifts are rare in the genus Phyllonorycter [15] and the plane leaf miner is considered oligophagous. Confirmed host plants are various plane trees (taxon names are according to the Catalogue of Life [16]): Platanus hybrida BROT., P. occidentalis L., P. orientalis L. and P. racemosa NUTTAL [11]. However, mines have been observed mainly on P. orientalis (Austria, Bulgaria, Canary Islands, France, Greece, Italy, Macedonia, Romania, Spain, Tajikistan, Turkey, Turkmenistan, and UK) and P. hybrida (Austria, Croatia, Denmark, France, Italy, Poland, Portugal, Slovakia and UK) (De Prins personal communication).
Platanus orientalis is native to the eastern part of the Mediterranean, across the Anatolian Peninsula and the Caucasus to Central Asia, especially in the coastal areas and river valleys [17,18,19,20]. The precise determination of native area boundaries is not possible anymore because the species was planted in ancient times [18, 20,21,22,23]. Plane trees are the most common ornamental and alley trees in temperate, Mediterranean and subtropical cities [24]. Therefore, aesthetic damage or early defoliation caused by P. platani is of high significance [7, 25, 26].
Most genetic analyses of Phyllonorycter species represent taxonomic works and use the mtDNA barcode fragment [27,28,29]. Others studied leaf miners and their host plant relationship using 28S rDNA marker [15, 30]. However, only a few studies used molecular data to reveal the colonisation history of Phyllonorycter [31, 32] or other Gracillariid leaf miner species [33].
Only a limited number of sequences were available in the various databases (e.g. NCBI, BOLD) for P. platani before this study. They include phylogenetic works of Phyllonorycter species [15, 30], outgroup data for Cameraria microsatellite markers [34], and parasitoid gut content [35]. In one of our previous works, 31 individuals were sequenced for the 520 bp long fragment of the 3′ end of the COI gene, which represented only 4 haplotypes for Europe [36].
Our aims were (i) to assess the current phylogeographic pattern of this species. (ii) To determine the origin of P. platani and its possible refugial areas. (iii) To reveal factors influencing the recent genetic pattern, especially the isolation by distance and/or geographic isolation of certain populations.
Materials and methods
Sampling and molecular methods
We collected larvae and pupae from 38 populations of P. platani, two population of P. issikii, and one of P. maestingella (Table 1, Fig. 1). The identification of the species was based on damage symptoms (type and locality of the mine) and the host plants. All samples were stored in 96% ethanol at 4 °C until DNA extraction. Voucher specimens and extracted DNA samples are stored at the institute’s collection.

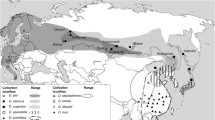
The native range of Platanus orientalis L.; current and previous distribution of Phyllonorycter platani (STAUDINGER 1870) and the sample locations in Europe. The native range of Platanus orientalis L. according to Feng et al. [3, 5]; current distribution of Phyllonorycter platani (STAUDINGER 1870) according to Global Taxonomic Database of Gracillariidae (www.gracillariidae.net); previous distribution as stated by Šefrová [3, 5]
DNA was extracted from entire bodies using: a) GenElute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich), b) E.Z.N.A.® Tissue DNA Kit and c) AquaGenomic Kit following the manufacturer’s protocol. Eluted DNA was stored at − 20 °C.
A 1242 bp-long region of the COI gene was amplified for 284 individuals by using Pat, (5’-TCCAA TGCACTAATCTGCCATATTA-3′) and Lep2F (5′- ATTCAACAATCATAAAGATATTGG-3′) primers [37, 38], as well as two additional primers Dick (5’-CCAACAGGAATTAAAATTTTTAGATGA-3′) [38] and Pc6F (5’-GCCCCAGATATAGCATTTCC-3′) [39]. PCR conditions included an initial denaturation step at 94 °C for 2 min, followed by 34 cycles at 94 °C for 30 s, 47 °C for 1 min and 72 °C for 1 min 30 s with a final extension step that lasted 10 min at 72 °C.
We amplified an 893 bp fragment of the 28S rDNA for 103 individuals from five selected populations (Table 1) using D1F (5’-ACCCGCTGAATTTAAGCATAT-3′) and D3R (5’-TAGTTCACCATCTTTCGGGTC-3′) primers [40]. We used the polymerase chain reaction as described by Lopez-Vaamonde et al. [40].
Sequences were generated (bidirectionally) at the Eurofin’s Laboratory (Ebersberg, Germany). Sequences are available via GenBank with accession numbers KY952988–KY953017.
Data analysis
For nuclear DNA (28S) analyses, 103 individuals were used and 284 individuals were used for mitochondrial DNA (COI) analyses (Table 1). Every specimen used for 28S rDNA analyses was amplified for COI too. Sequences were visualized using Sequence Scanner and then aligned using ClustalX [41]. After haplotypes were identified, those represented by only a single individual were verified by additional sequencing of an independent amplicon. P. issikii and P. maestingella were used as outgroups. Genetic distances were calculated with MEGA 5.02 [42].
Phylogenetic analyses
Maximum likelihood (ML) analysis was performed under GTR + I model with MEGA 5.02. The level of support for individual nodes was evaluated by bootstrapping with 5000 replicates. We used jModeltest 2.1.2 [43, 44] to select the best model of nucleotide substitution with Akaike Information Criterion (AIC) [45].
Population structure
Patterns of molecular diversity based on the mtDNA sequences between and within populations were assessed by estimating: nucleotide diversity (π) [46], transition/transversion ratio, haplotype diversity (h) [47, 48] using the software Arlequin version 3.5.1.2 [49].
Demographical expansion
Population dynamics analyses were performed on different geographical scales (all dataset, between continents, within continents, with special emphasis of European populations). For the estimation of Tajima’s D statistics [50] and Fu’s Fs [51] Arlequin 3.5.1.2 was used with 10,000 permutations [49]. On the small sample size (< 30 individuals) we additionally used DnaSp 5.10 [52] to estimate R2 [53].
Phylogeographical analysis
Spatial analysis of molecular variance (SAMOVA) was performed using SAMOVA v1.0 [54]. The program was run 1023 iterations. K values were tested, starting from two until the value for which FCT reached a plateau [55].
In addition, alternative geographical groups were tested with Analysis of Molecular Variance (AMOVA) [56,57,58] with Arlequin 3.5.1.2 [49]. The statistical significance of variance components in AMOVA was tested with 1000 permutations.
Isolation by distance was evaluated using Mantel test [59] with MANTEL NON-PARAMETRIC CALCULATOR ver. 2.0 [60]. Natural algorithms of geographical linear distances (km) between localities were correlated with the respective Tamura-Nei genetic distances [61] and were calculated with MEGA v.5.02 [42] with 1000 random iterations to obtain statistical inferences at α = 1%.
Statistical parsimony network (SP) [62] was created using a TCS 1.2.1 [63]. The nesting design was constructed on the SP network [64, 65].
Results
COI mtDNA
Twenty-four haplotypes were detected on the 1242 bp long fragment of the COI gene (Table 1, Fig. 2, Additional file 1). The number of variable sites was 43 (3.5%). Approximately the half of these were located on the barcode part of the gene.

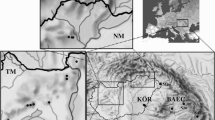
Distribution and phylogenetic relationship of Phyllonorycter platani mitochondrial haplotypes. a: Distribution of COI haplotypes in Europe; b: Statistical parsimony networks for all haplotypes (empty circles indicate missing or theoretical haplotypes); c: ML consensus tree of all COI haplotypes. Numbers above branches indicate ML probabilities (> 0.60)
There were 22 mutational steps between the Asian and European clades. Pairwise genetic distances between Asian (HT13–16) and European (HT1–12, 17–24) or between Asian (HT13–16) and North American (HT1, 11–12, 18) haplotypes are lower (1.80–2.30%) than the interspecific distance among related Phyllonorycter species (8.52–10.68%).
Average sequence divergence between Asian and European clades (2.08%) was higher than the intrapopulation level (0.11%; 0.20%). Divergence data shows that the population from the southern part of Anatolia (HT16) is closer to the Caucasian and Central Asian group (0.46%) than to the European group (1.88%). The genetic divergence between European and North American haplotypes was rather low (0.1–0.4%) in comparison to the outgroups (8.4–11.0%). ML tree support two clades with 100% probability: 1) the Asian and 2) the European (including the North American haplotypes) (Fig. 2). The HT1 was detected in 51.4% of the individuals and it is the most common haplotype in Europe, the northern part of the Anatolian Peninsula, and North America (Fig. 2). HT2 was frequent (21.5% of the total 288 individuals) in Western, North-Western, and Central Europe; we also found it in some Southern European populations (Croatia). HT3 (2.5%) was detected only in Central Europe. HT13 (10.6%) was detected from the Caucasus (Georgia), Eastern Europe (Moldavia, Ukraine) and it was common in the Central Asian populations (Uzbekistan, Kyrgyzstan). We detected the unique haplotype HT16 (3.6%) in the southern part of Anatolia. The Mediterranean part of Europe was represented by several unique haplotypes (HT4–10, HT19, HT20) similar to the northern part of Anatolia (HT17, HT21–24). The HT11 was detected from the northern part of Anatolia and North America. The HT12 and HT18 revealed only from North America. The HT14–15 were unique haplotypes from Caucasus (Georgia).
We observed moderate values of the diversity indices in the species (h = 0.68, π = 0.55%) (Table 2). Haplotype diversities were moderate and nucleotide diversities were low in both the Asian (h = 0.49, π = 0.26%) and the European clade (North American samples included) (h = 0.58, π = 0.08%). Based on the high rate of the Caucasian (Georgia) diversity indices (h = 0.51, π = 0.05%), and the homogeneity of Central Asian populations (Uzbekistan, Kyrgyzstan) a recent expansion to Central Asia from the Caucasus is assumed. The homogeneity of the population from the southern part of Anatolia (Antalya) suggests a founder effect. We observed high diversity indices (h = 0.60, π = 0.06%) for the North American population. We revealed 0.57 haplotype diversity and 0.08% nucleotide diversity in the European specimens.
Genetic population structure correlated with geographic distances (Mantel test). The correlation coefficient (r) indicates either a moderate (full dataset r = 0.361, Ρ = 0.005), or a weak correlation: European clade (including North American samples, r = 0.01, Ρ = 0.010); and European population only (without the Eastern European samples, r = 0.206, Ρ = 0.005).
As the FCT values reached a plateau at K = 2 and single-population groups were formed when K > 2, we used two as the optimal number of population groups. The two groups found by the SAMOVA are geographically consistent and correspond to regions (Table 3). On the full dataset, the two main groups (the first group contains populations from the south part of Turkey, Ukraine, Georgia, Uzbekistan, and Kyrgyzstan while the second group contains all others) actually match to the two main clades. Most of the molecular variance is found among groups (Va = 94.32%, p < 0.001), but ca. 2.55% (Vb) of variance is still found among populations within groups (p < 0.001). We can detect only slight gene flow between the two main groups. In the second arrangement, when we used the European clade (without the East-European samples which constitute the hybrid zone), the first group contains Northern and North-Western European populations (Brussels, Dresden, Freising, London, Rotterdam), and the second one contains Southern and Central European populations with the North American samples (Va = 51.57%; Vb = 40.64%). In the third arrangement we used only the European samples (without Eastern European and North American samples) and received similar results (Va = 51.23%, Vb = 40.71%).
Most of the Tajima’s D and Fu’s Fs indices are not significant (Table 2); we detect significant negative values only from the European clade (D = − 2.470, Fs = − 17.852), and from the Southern and Central European subgroup (D = − 2.553, Fs = − 16.455), which suggests recent population expansion. On the other hand, the R2 indices, which better fit small sample sizes, are significant (p = 0.00) in the populations from North America (R2 = 0.156), Caucasus (R2 = 0.162), and Eastern Europe (R2 = 0.132).
28S rDNA
Two alleles were identified on the 893 bp long fragment of the 103 specimens sequenced from 11 selected populations (Fig. 3). We revealed 0.10% divergence between the two alleles, which means one variable site (T/G transversion). These two alleles represent the two main clades. Populations from N-NW Europe, S-C Europe, N Anatolia, and North America were homogenous and contain the European allele only. The population from the Central Asia (Kyrgyzstan, Uzbekistan) and the Caucasus (Georgia) contains the Asian allele only while samples from Eastern Europe (Moldova, Ukraine) and from the southern part of Anatolia were represented by homo and heterozygote individuals as well (Table 4, Fig. 3).

Distribution of Phyllonorycter platani genomic (28S) alleles in Europe and Asia
Discussion
Inter and intraspecific diversity
The interspecific divergence on the COI gene is high in the Lepidopteran families [66,67,68,69]. In the Phyllonorycter genus especially, an extremely high interspecific divergence was observed (19.60% between P. obandai and P. salictella) [28]. In the Phyllonorycter ulicicolella species group, Laštůvka et al. [29] noted a minimum interspecific pairwise genetic distance of 3.1%. The average divergence between the Phyllonorycter species is 6.04% on the barcode fragment [31].
A wide range of varying intraspecific divergence exists for the different Lepidoptera species: e.g. 0.00–0.50% within Macrosaccus species, 0.49% within Cameraria ohridella, 0.17–2.07% Choristoneura sp., 0.11–3.22% Hyles sp., 1.30–2.50% Sciadia tenebraria, 1.50–4.10% Epirrita autumnata, 1.93% within P. salictella [28, 33, 66, 68,69,70]. Average intraspecific divergences are 0.94% among Phyllonorycter species [31]. The genetic distances values (0.10–2.30%) between P. platani haplotypes are similarly high to another plane feeding Lepidopteran species of Acalyptris platani (1.80–2.30%) [71]. In the case of P. issikii, the average distance (5.13 ± 0.003%) between the two clades is approximately two times higher [32] than between our two major clades of P. platani (2.08 ± 0.369%). The maximum intraspecific divergence is 2.96% for P. issikii and 1.38% for the putative new species [32], while we revealed 0.39% for the Asian and 0.25% for the European clade of P. platani.
The interspecific genetic divergence on 28S rDNA varies 0.20–25.00%, while the average intraspecific divergence is lower than 0.50% in the genus Phyllonorycter [30]. We detect much lower divergence between Asian and European alleles (0.10%). This was represented by a single variable position (T/G transversion). In comparison, P. issikii revealed 7 diagnostic substitution differences on this fragment of the 28S rDNA [32]. The high intraspecific divergence on mtDNA with low nuclear divergence does not support the presence of a cryptic species in our case [68]. In addition, we revealed heterozygote individuals from three populations for 28S rDNA, which shows that the individuals from the two clades can hybridize.
The haplotype diversity (h) of the plane leaf miner (0.68) compared to other Lepidopteran species (Aglais urticae 0.96) is medium-high [72]. The nucleotide diversity (π) of the full dataset (0.55%) is also medium-high (Dioryctria species 0.03–0.35%, Hyles genus 0.03–2.71%, Papilio species 0.26–2.71%) [33, 66, 72]. On the other hand, the 0.08% nucleotide diversity of the European samples compared to another leaf miner Lepidoptera, Cameraria ohridella (π = 0.17% in the natural area; and π = 0.09% in the other European places) is lower [33].
Phylogeographic pattern
Past and recent gene flow events determine the geographical pattern of populations [73]. One of them can be the influence of ice ages followed by species recolonization [74]. The other can be recent expansion or invasion. The latter is well described for the P. 12platani populations [3, 5, 75]; however, possible glacial refugial areas of the species remain unclear [4]. In our study, we demonstrated that both post-glacial recolonization and recent expansion events influenced the present genetic structure of P. platani.
According to the coalescent theory, the most frequent haplotype is supposed to be the most ancient one [76]. However, some authors [77] infirm this for Lepidopteran species. In our case these the modal haplotypes are HT1 (51.4%), HT2 (21.5%), HT13 (10.6%), HT3 (2.5%) (Fig. 2). The analysis of the population dynamics (Table 2, Tajima’s D, Fu’s Fs) and the geographical distribution pattern (Fig. 2) of both HT2 and HT3 suggest that these are rare haplotypes at the edge of the original distribution area of the species and that these haplotypes have gone to fixation under the range expansion and occurred more frequently in the recently colonized area [73, 78]. The diversity indices (Table 2) of the population from the southern part of the Anatolian Peninsula also show a possible recent expansion effect. Based on our results, there were likely two glacial refugial areas during the last ice age: one in the Balkan Peninsula and the other in the Caucasus. Analysis of further populations from this region, especially from the Caucasus and the south coast of the Caspian Sea, could provide a better resolution of the geographic patterns and the intermediate haplotypes between the two clades.
All methods used for the statistical analyses (ML, divergence data, SAMOVA) support the existence of two main clades (European and Asian) and the further differentiation of the Asian clade. The genetic divergence between the European and the Asian clade is high (2.08%), but this is typical for the members of the family Gracillariidae [31, 69]. Haplotype diversity is moderate (h = 0.49) and nucleotide diversity is low (π = 0.26%) for the Asian clade. Values for the European clade (h = 0.58, π = 0.08%) show only moderate difference. Rapid expansion after bottlenecks causes similar diversity patterns [33, 79, 80]. We surmise that the last glacial period caused this bottleneck. Several studies deal with the effects of the ice ages on diversity and effective population size [74, 80,81,82]. Plane trees and their herbivore communities may have survived in only a few refuges in Southern Europe, the coastal part of the Anatolian Peninsula, the east coast of the Black Sea, and the south coast of the Caspian Sea [20, 83,84,85,86]. The Mediterranean refugial area was fragmented consisting of several small, dispersed areas with warm and relative humid microclimates such as rivers floodplains, 400–800 m elevations, seaside, deep valleys etc. [83, 84, 86]. Médail and Diadema [84] describe 52 Mediterranean plant refugias in Europe. This may be the major reason for the high variability we found in the population from the Caucasus. Furthermore, it may also be the reason why only some of the Mediterranean populations were represented with high variations and why we found homogenous populations from the intermediate locations.
The HT16 (which is represented in the southern part of Anatolia) is linked more closely to the Asian haplotypes (HT13–15) than to the European. This suggests that there may have been a connection among the refuges of plane leaf miner populations during the interglacial periods.
Mantel test results and SAMOVA also support the view that the species survived the ice ages in several refugia because the isolation by distance values are moderate (r = 0.3605), and the variability value is high (Va = 94.32%) between the main two major clade.
Recent expansion
The Asian clade is well differentiated, so we analysed the dynamics of the subgroups: the Caucasus, Central Asia and the southern part of Anatolia (Antalya). The population from Antalya has a homogenous haplotype pattern, which refers to a founder effect. Several little plant refugia are described from the Mediterranean Basin [84], but the refugia of the Anatolian Peninsula have not been located exactly in the surroundings of Antalya. Climate simulations predicted possible refugia mainly from the northern part of Anatolia for the warm summer-green trees such as the host plant [83]. The homogeneity of the populations from Central Asia (Uzbekistan, Kirghizistan) and the high diversity values (h = 0.5111, π = 0.0536%) of the Caucasian (Georgia) population suggest that P. platani may spread from the Caucasus to Central Asia recently.
The star-like shape of the SP network (Fig. 2) refers to recent demographical expansion from low effective population size [33, 73]. NCPA results are in accordance with Šefrová’s [3, 5] results; Šefrová stated that the plane leaf miner spread with jumps through Europe.
The populations of P. platani in Europe and in the north of the Anatolian Peninsula may have gone through a rapid range expansion after bottleneck (neutrality testes D = − 2.459, Fs = − 14.403). Populations from the France-Germany borderline, the eastern Alps and the eastern border of Germany compose the edges of the “W-NW” population supported by SAMOVA (“W-NW” and “S-C”), which are common barriers within Europe [85]. The low diversity values (h = 0.00, π = 0.00%) of “W-NW” group are consequences of the founder effect [79]. In the case of the “S-C” group, high haplotype diversities (0.466) with low nucleotide diversities (0.071%) resulted from a rapid expansion from small effective population size [79, 87]. Presumably, the HT2 and the HT3 were rare mutations that evolved on the edges of the original area and, after population expansion, they became fixed in the new populations [73]. The outcome of neutrality tests (R2 = 0.156) – similar to the diversity indices – suggests that sudden demographic expansion shaped the current pattern of intraspecific diversity of the North American population.
However, while analysing our dataset we have to take into consideration that three factors (1: population structure, 2: genetic diversity and 3: sampling scheme) might have major influence on the quantification of population size changes (see Chikhi et al. for further details [88]). In our case both the sampling scheme and the various genetic structure of the different populations may have an effect.
The results of the COI and 28S rDNA sequences show that the two main clades can hybridize. We found two possible hybrid zones. One of them is located in the eastern part of Europe; populations from Moldavia, Ukraine contain both of Asian and European haplotypes. In addition, we detected hybrid individuals from the Moldavian population with the nuclear marker. The other hybrid zone is located in the southern part of Anatolia. The detected unique COI haplotype (HT16) is more closely linked to the Asian haplotypes than to the European haplotypes. The revealed allelic pattern with the 28S rDNA marker shows the presence of heterozygotes and European-type homozygote individuals. This discrepancy with mitochondrial and nuclear data shows that there were introgressions in the southern part of Anatolia. In most cases, mito-nuclear discrepancies are the results of possible secondary contact zones after isolation [89, 90]. The extension of the hybrid zones is unknown at the moment because of the low number of sampled populations from these regions.
Our results also confirmed that the synonymization of Lithocolletis felinella HEINRICH, 1920 to Phyllonorycter platani (STAUDINGER, 1870) is required, however a morphotaxonomic approval is desirable.
Conclusions
We have shown that both post-glacial recolonization and recent expansion events influenced the present genetic structure of P. platani. The genetic patterns revealed at least two refugia during the last ice age: one in the Balkan Peninsula and the other in the Caucasus region. Recent expansion was detected in some European and Central Asian populations. The two main clades (Europe/Asia) show definite genetic differences; however, several hybrid individuals were found in the overlapping zone as well (stretching over Eastern Europe and the Anatolian Peninsula). Discrepancies in mitochondrial and nuclear data indicate introgressions in the southern part of the Anatolian Peninsula.
Abbreviations
- ML:
-
maximum likelihood
- NCPA:
-
nested clade analysis
- SP:
-
statistical parsimony
References
Roques A, Rabitsch W, Rasplus JY, Lopez-Vaamonde C, Nentwig W, Kenis M. Alien terrestrial invertebrates of Europe. In: Drake JA, editor. Handbook of alien species in Europe. vol. 3. Berlin: Springer; 2009. p. 63–79.
Biedrzycka A, Zalewski A, Bartoszewicz M, Okarma H, Jedrzejewska E. The genetic structure of raccoon introduced in Central Europe reflects multiple invasion pathways. Biol Invasions. 2014;16(8):1611–25.
Šefrová H. Invasions of Lithocollatinae species in Europe – causes, kinds, limits and biological impact (Lepidoptera, Gracillariidae). Ekológia (Bratislava). 2003;22(2):132–42.
Lopez-Vaamonde C, Agassiz D, Augustin A, De Prins J, De Prins W, Gomboc S, Ivinskis P, Karsholt O, Koutroumpas A, Koutroumpa F, Laštůvka Z, Marabuto E, Olivella E, Przybylowicz L, Roques A, Ryrholm N, Šefrová H, Šima P, Sims I, Sinev S, Skulev B, Tomov R, Zilli A, Lees D. Lepidoptera. In: Roques A, Kennis M, Lees D, Lopez-Vaamonde C, Rabitsch W, Rasplus JY, Roy DB, editors. Alien terrestrial arthropods of Europe. vol. 4. Sofia–Moscow: Pensoft; 2010. p. 603–68.
Šefrová H. Phyllonorycter platani (Staudinger) - a review of its dispersal history in Europe (Lepidoptera, Gracillariidae). Acta Universitatis agriculturae et silviculturae Mendelianae Brunensis. 2001;49(8):71–5.
Global Taxonomic Database of Gracillariidae (Lepidoptera). World Wide Web electronic publication (http://www.gracillariidae.net) Date accessed 04 Aug 2018.
Heinrich C. On some forest Lepidoptera with descriptions of new species, larvae, and pupae. Proc United States National Museum. 1920;57(2305):53–96. 51-33
Deschka G. Schmetterlinge als Einwanderer. Stapfia. Publikation der botanischen Arbeitsgemeinschaft am Ober-oesterreichischen Landesmuseum. Linz. 1995;37:77-128.
Vasil'eva EA, Shkarlet OD. Lithocolletis platani in the Crimea. Lesnoe Khozyaĭstvo. 1989;11:47.
Kuznetzov VJ. On the fauna and biology of Lepidoptera of the western Kopet-Dagh. Fauna i Ekologya Nasekomykh Turkmenii. 1960;27:11–93.
Lees D. Phyllonorycter platani (Staudinger, 1987) (Lepidoptera, Gracillariidae). In: Roques A, Kennis M, Lees D, Lopez-Vaamonde C, Rabitsch W, Rasplus JY, Roy DB, editors. Alien terrestrial arthropds of Europe. Vol. 4. Moscow: Sofia; 2010. p. 1002–3.
Burke HE. Some important insect enemies of shade trees in central and southern California. In: Proceedings of the National Shade Tree Conference, 9th 1933. New York: New York Botanical Garden; 1933. p. 49–59.
Koehler CS, Campbell RL. Trunk-implanted Systemics for control of foliage insects on shade trees in California. J Econ Enthomology. 1968;61(3):778–83.
Gates MW, Heraty JM, Schauff ME, Wagner DL, Whitfield JB, Wahl DB. Survey of the parasitic hymenoptera on Leafminers in California. J Hymenopt Res. 2002;11(2):213–70.
Lopez-Vaamonde C, Winkström N, Labandeira C, Godfray HCJ, Goodman SJ, Cook JM. Fossil-calibrated molecular phylogenies reveal that leaf-mining moths radiated millions of years after their host plants. J Evol Biol. 2006;19(4):1314–26.
Species 2000 & ITIS Catalogue of Life, 2017 Annual Checklist. Species 2000: Naturalis, Leiden, the Netherlands. Digital resource at http://www.catalogueoflife.org. Accessed 05 Mar 2018.
Tutin TG, Edmontson JR. Platanaceae. In: Tutin TG, Burges NA, Chater A, Edmondson JR, Heywood VH, Moore DM, Valentine DH, Walters SM, Webb DA, editors. Flora Europaea. vol. 1, 2nd edn. Cambridge: Cambridge University Press; 1993. p. 463.
Caruso G, Gangale C, Uzunov D, Pignotti L. Chorology of Platanus orientalis(Platanaceae) in Calabria (S Italy). Phytologia Balcanica. 2008;14(1):51–6.
Karamanoğlu K. Türkiye Bitkileri cilt.1. Ankara Üniversitesi Eczacılık Fakültesi Yayınları: Ankara; 1976.
Feng Y, Oh S-H, Manos PS. Phylogeny and historical biogeography of the genus Platanus as inferred from nuclear and chloroplast DNA. Syst Bot. 2005;30(4):786–99.
Tamanyan K, Fayvush G, Nanagyulyan S, Danielyan T. Red book of plants of the Republic of Armenia. Higher plants and fungi. Yerevan: Ministry of Nature protection of Republic of Armenia; 2010.
Santamour FS, Jackot McArdle JA. Checklist of cultivated Platanus (planetree). J Arboric. 1986;12(3):79–83.
World Conservation Monitoring Centre: Platanus orientalis. The IUCN Red List of Threatened Species. 1998(e.T33951A9824290).
Liu G, Bao M. Adventitious shoot regeneration from in vitro cultured leaves of London plane tree (Platanus acerifolia Willd.). Plant Cell Rep. 2003;21:640–4.
Győrfi J. A Lithocolletis platani Stgr. és parazitái. Erdészeti Kísérletek. 1941;42:224–35.
Halperin J. Arthropod fauna and main insect pests of plane trees in Israel. Phytoparasitica. 1990;18(4):309–19.
Kawahara AY, Ohshima I, Kawakita A, Regier JC, Mitter C, Cummings MP, Davis DR, Wagner DL, De Prins J, Lopez-Vaamonde C. Increased gene sampling strengthens support for higher-level groups within leaf-mining moths and relatives (Lepidoptera: Gracillariidae). BMC Evol Biol. 2011;11(182):1–14.
De Prins J, Mozūraitis R, Lopez-Vaamonde C, Rougerie R. Sex attractant, distribution and DNA barcodes for the Afrotropical leaf-mining moth Phyllonorycter melanosparta (Lepidoptera: Gracillariidae). Zootaxa. 2009;2281:53–67.
Laštůvka Z, Laštůvka A, Lopez-Vaamonde C. A revision of the Phyllonorycter ulicicolella species group with description of a new species (Lepidoptera: Gracillariidae). SHILAP Revista de lepidopterología. 2013;41(162):251–65.
Lopez-Vaamonde C, Godfray HCJ, Cook JM. Evolutionary dynamics of host-plant use in a genus of leaf-mining moths. Evolution. 2003;57(8):1804–21.
De Prins J, De Prins W, De Coninck E, Kawahara AY, Milton MA, Hebert PDN. Taxonomic history and invasion biology of two Phyllonorycter leaf miners (Lepidoptera: Gracillariidae) with links to taxonomic and molecular datasets. Zootaxa. 2013;3709(4):341–62.
Kirichenko N, Triberti P, Ohshima I, Haran J, Byun BK, Li H, Augustin S, Roques A, Lopez-Vaamonde C. From east to west across the Palearctic: Phylogeography of the invasive lime leaf miner Phyllonorycter issikii (Lepidoptera: Gracillariidae) and discovery of a putative new cryptic species in East Asia. PLoS One. 2017;12(2):e0171104.
Valade R, Kenis M, Hernandez-Lopez A, Augustin S, Mari Mena N, Magnoux E, Rougerie R, Lakatos F, Roques A, Lopez-Vaamonde C. Mitochondrial and microsatellite DNA markers reveal a Balkan origin for the highly invasive horse-chestnut leaf miner Cameraria ohridella (Lepidoptera, Gracillariidae). Mol Ecol. 2009;18:3458–70.
Mari Mena N, Valade R, Magnoux E, Augustin S, Lopez-Vaamonde C. Microsatellites for the highly invasive Cameraria ohridella (Lepidoptera: Gracillariidae) and their amplification in related species. Eur J Entomol. 2008;105:945–8.
Rougerie R, Smith MA, Fernandez-Triana J, Lopez-Vaamonde C, Ratnasingham S, Hebert PDN. Molecular analysis of parasitoid linkages (MAPL): gut contents of adult parasitoid wasps reveal larval host. Mol Ecol. 2011;20(1):179–86.
Lakatos F, Augustin S, Lopez-Vaamonde C. Genetic patterns of Phyllonorycter platani - an invasive leaf miner moth with Mediterranean origin. In: International congress of entomology, 6-12 July 2008. Durban: XXIII. International Congress of Entomology; 2008.
Lunt DH, Zhang D-X, Szymura JM, Hewitt GM. The insect COI gene: evolutionary patterns and conserved primers for phylogenetic studies. Insect Mol Biol. 1996;5:153–65.
Dinca V, Zakharov EV, Hebert PDN, Vila R. Complete DNA barcode reference library for a country’s butterfly fauna reveals high performance for temperate Europe. Proc R Soc B. 2011;278:347–55.
Avtzis DN, Arthofer W, Stauffer C. Identifying haplotypes of Pityogenes chalcographus(Col., Scolytidae) by single strand conformation polymorphism (SSCP). Mitteilungen der Deutschen Gesellschaft für allgemeine und angewandte Entomologie. 2006;15:173–6.
Lopez-Vaamonde C, Rasplus JY, Weiblen GD, Cook JM. Molecular phylogenies of fig wasps: partial Cocladogenesis of pollinators and parasites. Mol Phylogenet Evol. 2001;21(1):55–71.
Thompson JD, Higgins DG, Gibbson TJ. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9.
Guindon S, Gascuel O. A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst Biol. 2003;52:696–704.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772.
Akaike H. A new look at the statistical model identification. IEEE Trans Autom Control. 1974;19:716–23.
Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987.
Ewens WJ. The sampling theory of selectively neutral alleles. Theor Popul Biol. 1972;3:87–112.
Zouros E. Mutation rates, population sizes and amounts of electrophoretic variation of enzyme loci in natural populations. Genetics. 1979;92:623–46.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to performpopulation genetics analyses under Linux and windows. Mol Ecol Resour. 2010;10:564–7.
Tajima F. The effect of change in population size on DNA polymorphism. Genetics. 1989;123:597–601.
Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992;9(3):552–69.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Ramos-Onsins SE, Rozas J. Statistical properties of new neutrality tests against population growth. Mol Biol Evol. 2002;19(12):2092–100.
Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002;11(12):2571–81.
Chiari Y, van der Meijden A, Mucedda M, JM L¸o, Hochkirch A, Veith M. Phylogeography of Sardinian cave salamanders (genus Hydromantes) is mainly determined by geomorphology. PLoS One. 2012;7(3):e32332.
Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–70.
Weir BS. Genetic data analysis II: methods for discrete population genetic data. Sunderland, MA, USA: Sinauer Assoc., Inc.; 1996.
Excoffier L, Smouse P, Quattro J. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–91.
Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27(2):209–20.
Liedloff AC. Mantel Nonparametric Test Calculator. Version 2.0. Australia: School of Natural Resource Sciences, Queensland University of Technology; 1999.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–26.
Templeton AR, Crandall KA, Sing CF. A Cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III Cladogram Estimation Genetics. 1992;132:619–33.
Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9(10):1657–60.
Templeton AR, Routman E, Phillips CA. Separating population structure from population history: a cladistic analysis of the geographic distribution of mitochondrial DNA haplotypes in the tiger salamander, Ambystoma tigrinum. Genetics. 1995;140:767–2.
Templeton AR. Nested clade analysis of phylogeographic data: testing hypothesis about gene flow and population history. Mol Ecol. 1998;7:381–97.
Roe AD, Sperling FAH. Patterns of evolution of mitochondrial cytochrome c oxidase I and II DNA and implications for DNA barcoding. Mol Phylogenet Evol. 2007;44:325–45.
Hajibabaei M, Janzen DJ, Burns JM, Hallwachs W, Hebert PDN. DNA barcodes distinguish species of tropical Lepidoptera. Proc Natl Acad Sci U S A. 2006;103:968–71.
Kvie KS, Hogner S, Aarvik L, Lifjeld JT, Johnsen A. Deep sympatric mtDNA divergence in the autumnal moth (Epirrita autumnata). Ecol Evol. 2013;3(1):126–44.
Davis DR, De Prins J. Systematics and biology of the new genus Macrosaccus with descriptions of two new species (Lepidoptera, Gracillariidae). ZooKeys. 2011;98:29–82.
Huemer P, Hausmann A. A new expanded revision of the European high mountain Sciadia tenebraria species group (Lepidoptera: Geometridae). Zootaxa. 2009;2117:1–30.
van Nieukerken EJ. Acalyptris Meyrick: revision of the platani and staticis groups in Europe and the Mediterranean (Lepidoptera: Nepticulidae). Zootaxa. 2007;1436:1–48.
Vandewoestijne S, Baguette M, Brakefield PM, Saccheri IJ. Phylogeography of Aglais urticae (Lepidoptera) based on DNA sequences of the mitochondrial COI gene and control region. Mol Phylogenet Evol. 2004;31(2):630–46.
Excoffier L, Foll M, Petit RJ. Genetic consequences of range expansions. Annu Rev Ecol Evol Syst. 2009;40:481–501.
Hewitt GM. Post-glacial re-colonization of European biota. Biol J Linn Soc. 1999;68:87–112.
Deschka G. Phyllonorycter (= Lithocolletis) platani (Staudinger 1870) in Oberösterreich. Jber Steyrer Ent Runde. 1983;17:21–31.
Fry AJ, Zink RM. Geographic analysis of nucleotide diversity and song sparrow (Aves: Emberizidae) population history. Mol Ecol. 1998;7:1303–13.
Montano V. Coalescent inferences in conservation genetics: should the exception become the rule? Biol Lett. 2016;12(6):20160211.
Excoffier L, Ray N. Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol. 2008;23:347–51.
Grant WAS, Bowen BW. Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered. 1998;89(5):415–26.
Durka W, Babik W, Ducroz JF, Heidecke D, Rosell F, Samjaa R, Saveljev AP, Stubbe A, Ulevicus A, Stubbe M. Mitochondrial phylogeography of the Eurasian beaver Castor fiber L. Mol Ecol. 2005;14:3843–56.
Hewitt GM. The genetic legacy of the quaternary ice ages. Nature. 2000;405:907–13.
Petit RJ, Aguinagalde I, de Beaulieu J-L, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Gerhard Müller-Starck G, Demesure-Musch B, Palmé A, Martín JP, Rendell S, Vendramin GG. Glacial Refugia: hotspots but not melting pots of genetic diversity. Science. 2003;300(6):1563–5.
Leroy SAG, Arpe K. Glacial refugia for summer-green trees in Europe and south-West Asia as proposed by ECHAM3 time-slice atmospheric model simulations. J Biogeogr. 2007;34:2115–28.
Médail F, Diadema K. Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J Biogeogr. 2009;36:1333–45.
Schmitt T. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front Zool. 2007;4(11):1–13.
Tzedakis PC, Lawson IT, Frogley MR, Hewitt GM, Preece RC. Buffered tree population changes in a quaternary Refugium: evolutionary implications. Science. 2002;297:2044–7.
de Jong MA, Wahlberg N, Eijk M, Brakefield PM, Zwaan BJ. Mitochondrial DNA signature for range-wide populations of Bicyclus anynana suggests a rapid expansion from recent Refugia. PLoS One. 2011;6(6):e21385.
Chikhi L, Sousa VC, Luisi P, Goossens B, Beaumont MA. The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes. Genetics. 2010;186(3):983–95.
Toews DP, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol. 2012;21(16):3907–30.
Chan KM, Levin SA. Leaky prezygotic isolation and porous genomes: rapid introgression of maternally inherited DNA. Evolution. 2005;59(4):720–9.
Acknowledgements
We thank Christian Stauffer, Carlos Lopez-Vaamonde, Natalia Kirichenko, Coralie Bertheau, Susanne Krumböck, Gyula Hoffmann, Norbert Frank, György Csóka, Katalin Tuba, Bálint Horváth, Bálint Mészáros, Balázs Velekei, Judit Sárándi-Kovács, Dániel Winkler for their helpful comments and Frank Berger for the language corrections.
We would like to thank Olga Antuhova, Dimitrios Avtzis, Sezgin Ayan, Rossitza Dimitrova, Wojciech Grodzki, Boris Hrasovec, Jan Kollar, Carlos Lopez-Vaamonde, Valentyna Meshkova, Nicolai Olenici, Ralf Petercord, Judit Sárándi-Kovács, Lidia Sukovata, Katalin Tuba, Marek Turcani, Balázs Velekei, foresters of Evia-island, and the students of University of Sopron, Faculty of Forestry for assistance in collecting some samples.
Funding
Funding was provided by TÁMOP–4.2.1.B-09/1/KONV (collection of samples, support to VT); TÁMOP–4.2.2.B–10/1–2010–0018 (implementation of lab works, support to VT); VKSZ_12-1-2013-0034 (publication, support to VT) programs and the COST-STSM-FP1002–10569 grant (implementation of lab works).
Availability of data and material
Collected insect individuals and extracted DNA samples are deposited at the Institute of Silviculture and Forest Protection, University of Sopron. Our basic data are stored at DRYAD: doi:https://doi.org/10.5061/dryad.6r1t61j. All sequence data that represent a new haplotype are publicly available at NCBI (KY952988–KY953017 accession numbers).
Declarations
Supplementary data will be available via Dryad.
Author information
Authors and Affiliations
Contributions
FL designed the study. FL and VT collected field sample. VT performed laboratory work, genetic and statistical analyses. VT and FL drafted the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
COI haplotype distribution of Phyllonorycter platani. Number of COI haplotypes (HT1–24) in the countries/locations investigated. (DOCX 32 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tóth, V., Lakatos, F. Phylogeographic pattern of the plane leaf miner, Phyllonorycter platani (STAUDINGER, 1870) (Lepidoptera: Gracillariidae) in Europe. BMC Evol Biol 18, 135 (2018). https://doi.org/10.1186/s12862-018-1240-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-018-1240-z