
Abstract
Background
There is a general lack of information on the dispersal and genetic structuring for populations of small-sized deep-water taxa, including free-living nematodes which inhabit and dominate the seafloor sediments. This is also true for unique and scattered deep-sea habitats such as cold seeps. Given the limited dispersal capacity of marine nematodes, genetic differentiation between such geographically isolated habitat patches is expected to be high. Against this background, we examined genetic variation in both mitochondrial (COI) and nuclear (18S and 28S ribosomal) DNA markers of 333 individuals of the genus Sabatieria, abundantly present in reduced cold-seep sediments. Samples originated from four Eastern Mediterranean cold seeps, separated by hundreds of kilometers, and one seep in the Southeast Atlantic.
Results
Individuals from the Mediterranean and Atlantic were divided into two separate but closely-related species clades. Within the Eastern Mediterranean, all specimens belonged to a single species, but with a strong population genetic structure (ΦST = 0.149). The haplotype network of COI contained 19 haplotypes with the most abundant haplotype (52% of the specimens) shared between all four seeps. The number of private haplotypes was high (15), but the number of mutations between haplotypes was low (1–8). These results indicate intermediary gene flow among the Mediterranean Sabatieria populations with no evidence of long-term barriers to gene flow.
Conclusions
The presence of shared haplotypes and multiple admixture events indicate that Sabatieria populations from disjunct cold seeps are not completely isolated, with gene flow most likely facilitated through water current transportation of individuals and/or eggs. Genetic structure and molecular diversity indices are comparable to those of epiphytic shallow-water marine nematodes, while no evidence of sympatric cryptic species was found for the cold-seep Sabatieria.
Similar content being viewed by others


Background
The deep sea is one of the largest ecosystems on earth, covering two-thirds of the planet and containing a substantial proportion of the earth’s biodiversity. This ecosystem has long been considered a vast and continuous environment with stable biotic, physicochemical and hydrographical conditions [1] which enabled widespread distribution of several deep-sea invertebrate species [2]. However, the deep-sea bottom is also characterized by the presence of fragmented and strongly differentiated ecosystems such as seamounts, trenches, hydrothermal vents and cold seeps (e.g., [3]). The deep sea supports a highly diverse and endemic benthic invertebrate fauna (e.g., [4, 5]). Additionally, molecular evidence suggests the presence of cryptic species complexes (genetically distinct but morphologically identical species; see [4, 6, 7]) of which some are found to be widespread [4, 7, 8] while others appear to be geographically restricted [4].
While our knowledge on the identity, diversity, and distribution of extant species continues to grow, insights on the evolutionary processes that are at the heart of this diversity in the deep sea are lagging behind. Genetic divergence in the marine environment may result from contemporary and historical processes related to the different oceanic basins [9], changes in seafloor topography [10], current regimes [11, 12], depth differences [13, 14], geographic distances [15] and oxygen levels [16]. For instance, genetic divergence in several benthic species is greater between populations at different depths in the same region than between populations of different regions at the same depth (e.g., [14, 17,18,19]). Also, bathyal organisms generally exhibit much greater population structure than those from the deeper abyss [6, 14, 20].
Next to hydrothermal vents, cold seeps are among the most widely spread deep-sea reducing environments and are characterized by high levels of methane and sulphide, the latter being toxic to most metazoans. Associated fauna therefore need to be adapted to the complexity of the presence of biogenic structures, geochemical conditions and microbial processes. Active seeps have been reported all over the world, even in the Arctic and Antarctic polar regions [21, 22], and from shallow to hadal depths [22, 23]. Since their discovery in the mid-1980s, one of the most puzzling issues has been the influence of past and present connectivity on the nature of cold-seep species assemblages and their geographical distribution [24]. Within the meiofaunal component of cold-seep biota, free-living nematodes are the most prevailing taxon [25].
Nematodes are among the most numerous multicellular taxa on earth, successfully adapted to nearly every ecosystem, but many species are biologically still poorly understood. Different species feed on materials as varied as algae, fungi, small animals, faecal matter and dead organisms [26]. Most nematode species have separate male and female individuals with males using spicules to transfer sperm into the vulva of females. Most free-living nematodes are egg-laying organisms. Yet, a few species, such as Halomonhystera disjuncta, are known to be ovoviviparous [27, 28]. The eggs then hatch into larvae, which appear essentially identical to the adults, except for an underdeveloped reproductive system.
Compared to more common deep-sea environments, most cold seeps support highly productive ecosystems characterized by poor nematode species richness, high biomass and elevated dominance of a few genera able to withstand the high concentrations of toxic hydrogen sulphide (e.g., [27, 29,30,31,32,33]). Until now, the genetic structure of nematode species inhabiting different isolated reduced deep-sea systems remained largely unknown (but see [34]). In terms of connectivity and dispersal, nematodes lack a pelagic stage and are therefore considered poor dispersers due to their small size and low swimming ability [35]. However, free-living nematodes and their eggs can become passively suspended and transported in the water column via external forces (e.g., erosion by hydrodynamic forces; [35]), even by very weak bottom currents [36]. Moreover, some nematode species can actively emerge from the sediment and swim in the water column (e.g., [37]). Population genetic studies confirmed restricted nematode dispersal in shallow-water environments at large geographical scales (> 100 km) and revealed cryptic diversity in a wide range of marine nematodes [38,39,40]. Although some of these cryptic species showed restricted geographical distributions, others were widespread and several of them were distributed sympatrically [40, 41]. Widespread and even cosmopolitan species distributions have been reported in several nematode species [41,42,43,44].
Here, we investigate the genetic structure and connectivity in species of heterogeneously distributed cold seeps in the deep sea. We sampled the dominant Sabatieria species, S. mortenseni, in one cold seep in the Atlantic and four cold seeps in the Eastern Mediterranean Sea to investigate the genetic diversity patterns within and between reduced cold-seep environments.
The genus Sabatieria is one of the most common nematode genera in marine benthic habitats, especially in muddy and muddy sand sediments [45], and is one of the dominant deep-dwelling genera in muddy reduced sediments of shallow-water sites all over the world [46,47,48], including the shelf break and upper slope [25, 49, 50]. The genus can attain high proportions at oxygen minima but gradually disappears almost completely below 2000 m depth [49, 51]. Many of its species are considered eurytopic and tolerant to unstable, highly polluted environments [52, 53]. Currently, 95 Sabatieria species are considered valid [54] and are found in different localities around the world (e.g., [55,56,57]). Sabatieria species appear to be divided in five distinct subgroups according to morphological differences related to the type and distribution of precloacal supplements, characteristics of the gubernaculum and apophysis, the number of turns of the amphideal fovea, and the cephalic setae [45]. The specimens investigated in this study belong to the pulchra group, which consists of six species, including S. mortenseni. Species of this group are distinguished by the disposition of the precloacal supplements, which are usually conspicuous and relatively few in numbers (5–9), and by the characteristic gubernaculum median pieces and short paired cervical setae [45]. S. mortenseni has been found in reduced sediments of shallow-water sites in Brazil, USA, Antarctica [58,59,60,61] and the Strait of Magellan (Chile) at depths ranging between 8 and 550 m [62]. It is also the dominant species at the REGAB pockmark in the Gulf of Guinea at a depth of more than 3000 m [33]. Based on the particular isolated character of cold seeps and the strong genetic structure in shallow water nematodes, we expected to observe a strong genetic structure between populations of cold seeps located several hundreds of kilometers apart. In addition, based on the presence of substantial cryptic diversity in marine nematodes we also expected to find undiscovered cryptic diversity in these cold seeps.
Methods
Sample locations and characteristics
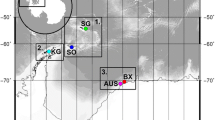
Sabatieria specimens were collected at five cold seeps, one located in the Atlantic Ocean and four in the Eastern Mediterranean area (Fig. 1). The REGAB cold seep in the Atlantic is located at 3150 m depth on the equatorial West African passive continental margin (Gulf of Guinea; [33]). This 800–1000 m wide and 15–20 m deep pockmark (crater-like depression) [33, 63] is characterized by methane seepage, gas hydrates and the presence of carbonate crusts [64, 65].

Map of the location of the investigated cold seeps and distances between them. Amsterdam MV, Amon MV, Cheops and PM in the Eastern Mediterranean area; REGAB in the Atlantic Ocean. Source maps: http://www.geography-site.co.uk; http://www.emersonkent.com/map_archive/eastern_mediterranean.htm
The four other cold seeps are located in the Eastern Mediterranean area (Fig. 1) at distances that vary from 131 to 405 km. The most northern cold seep, Amsterdam mud volcano (MV), is located on the southern flanks of the Anaximenes seamount (Anaximander Mountains, Southwestern Turkey; [66]) in the northern Levantine basin. This 2050 m deep MV expels a methane plume of more than 200 m high and harbors methane concentrations of up to 2000 nmol/kg [67]. The other investigated seeps are located at the opposite side of the Eastern Mediterranean sea, at the Nile Deep Sea Fan (NDSF) in the southern Levantine basin. Three major zones of fluid seepage were investigated: the eastern province, the central province (also called the pockmark area) and the Menes caldera. The very active Amon MV, located in the eastern province at ~1118 m depth, is a large volcano of 2.7 km in diameter and 90 m in height [68]. Investigations showed high methane and hydrocarbon emissions (1310 nmol/L) into the water column [69]. In the central province, ship-borne multibeam acoustic images revealed numerous small pockmarks and/or mounds [70, 71]. For this study, the fauna of two major pockmark sites (2A and 2B) and an area close to 2A but located at the lower slope (between ∼ 1650 and 2200 m water depth) were sampled and pooled as PM in the rest of the analyses. Finally, within the Menes caldera, the Cheops MV is an active MV located at 3000 m water depth at the base of the Egyptian continental slope. From the volcanoes flows a mixture of brine, fluids and mud. The brines are expelled through numerous sub-circular vents (a few meters in diameter) which are covered with white sulphidic material produced by sulphate-reducing bacteria [72].
Sample collection and molecular processing
The cold seeps investigated in this study were the subject of international projects, and were thoroughly investigated during multiple campaigns. Therefore, samples for both morphological and molecular research were available for these particular reduced environments. Samples were collected during MEDECO Leg 2 of RV Pourqoui Pas? (November 2007) and MSM13/3 – MSM13/4 campaign of the RV Maria S. Merian (November–December 2009) for the Mediterranean seeps, and during the Guineco cruise (2008) onboard RV Meteor for the Atlantic REGAB site (Table 1). At each cold seep, multiple cores of reduced sediments were gathered by means of Remotely Operated Vehicles (ROV), sliced per cm and preserved in ethanol or frozen at −20 °C. After extraction of nematodes from the sediment by density gradient centrifugation with Ludox (a colloidal silica polymer; specific gravity 1.18; [73]), Sabatieria individuals were identified under a stereomicroscope using diagnostic morphological characteristics. Specimens were randomly picked with a fine needle and put in MilliQ water to remove adhering particles. Prior to molecular analysis, multiple individuals per location (PM(2A): 45; PM(2B): 42; PM: 62; Cheops: 112; Amon MV: 115; Amsterdam MV: 108; REGAB: 34) were photographed using a Leica DMR microscope equipped with a Leica DFC 420 camera to serve as a morphological reference. Individuals examined here were assigned to Sabatieria mortenseni based on the number of turns of the amphideal fovea (this study: 3, [60]: 2.5; [61]: 2.5–3; [74]: 2.25–2.5); and for the males, based on the spicule shape, the presence of a sclerotized central part of the apophysis, and the shape of the precloacal supplements. The number of precloacal supplements showed substantial variation in our specimens: males from the Eastern Mediterranean had 6–8 precloacal supplements, and males from REGAB 5–6. The number of precloacal supplements in S. mortenseni is 6 according to [45] and 6–7 according to [74]. Multiple characteristics (e.g., amphid diameter, head diameter, anal body diameter) were measured in 18–48 males per cold seep (see Additional file 1: Table S1). Morphometric data were obtained to calculate ratios (e.g., De Man ratio ‘a’, amphid diameter as percentage of the corresponding body diameter) following the instructions found in [45, 74] to assure that specimens from both the Atlantic and Mediterranean area belonged to the same morphological species (see Additional file 1: Table S1). Representative specimens were deposited in the Ghent University Museum, Zoology Collection (voucher codes UGMD104325–104331). Individual nematodes were then immediately transferred in 20 μl Worm Lysis Buffer (WLB: 50 mM KCl, 10 mM Tris pH 8.3, 2.5 mM MgCl2, 0.45% NP40, 0.45% Tween20). In this way, 618 Sabatieria specimens were collected from the Mediterranean seeps, and 41 from samples of the REGAB seep. DNA was extracted by adding 1.5 μl proteinase K (10 mg/ml) to each WLB-stored specimen, followed by incubation for 1 h at 65 °C and 10 min at 95 °C, and finally centrifugation for 1 min at 14000 rpm prior to usage.
For the polymerase chain reaction (PCR), 1 μl of DNA template was used. In this study, 420 base pairs (bp) of the mitochondrial cytochrome c oxidase subunit 1 (COI) gene were amplified using the forward primer JB3 (5′-TTTTTTGGGCATCCTGAGGTTTAT-3′) [75] and the reverse primer JB5GED (5′-AGCACCTAAACTTAAAACATARTGRAARTG-3′) [39]. Standard PCR amplifications were conducted in 25 μl volumes for 35 cycles, each consisting of 30 s denaturation at 94 °C, 30 s annealing at 50 °C, and 45 s extension at 72 °C, with an initial denaturation step of 5 min at 94 °C and a final extension step of 10 min at 72 °C. Four μl of each PCR product was loaded on a 1% agarose gel to check the strength and size of the amplified product. After electrophoresis, 420 PCR products, each corresponding to a single nematode specimen, were sent to MacroGen (The Netherlands) for bidirectional sequencing with the primers JB3-JB5GED.
After identifying the different COI haplotypes, DNA from each haplotype was used to amplify two nuclear DNA gene regions: the 18S region of the small ribosomal subunit and the D2D3 region of the conserved 28S large ribosomal subunit. 752 bp of the D2D3 region was amplified with the forward primer D2A (5′-ACAAGTACCGTGAGGGAAAGTTG-3′) and the reverse primer D3B (5′-TCCTCGGAAGGAACCAGCTACTA-3′) [76], while ±950 bp of the 18S region was acquired with the forward primer G18S4 (5′-GCTTGTCTCAAAGATTAAGCC-3′) and the reverse primer 4R (5′-GTATCTGATCGCCKTCGAWC-3′) [77]. PCR conditions were similar to those for COI, except that 40 cycles were applied, and the annealing temperature was augmented to 54 °C. Next to this, an extension time of one minute was applied for the amplification of the 18S gene region. The PCR products of 36 nematodes for 18S and 34 nematodes for D2D3 were sent for sequencing.
Data analyses
Phylogenetic analysis and variation in COI and nuclear markers
COI sequences were checked and trimmed for further phylogenetic analysis in DNASTAR Lasergene Seqman Pro [78]. Primer ends were removed, and sequences which were too short or showed too many uncertain bp were removed from the dataset. The remaining sequences were aligned using CLUSTALW v2 with default parameters as incorporated in MEGA 6.0 [79] and translated using the invertebrate mitochondrial genetic code. A blastx search was performed against the non-redundant protein collection (nr) in GenBank to check the nematode origin of the sequences. Unique haplotypes were identified using a distance matrix based on uncorrected p-distances of untranslated sequences in MEGA. The number of conserved, variable and parsimony informative positions was determined. The best-fit substitution model was selected in jModelTest [80, 81] using the Bayesian Information Criterion (BIC). After that, phylogenetic relationships between haplotypes were constructed in MEGA using Neighbor-Joining (NJ, p-distance model) and Maximum Likelihood (ML, Hasegawa-Kishino-Yano model with gamma distribution as identified by jModelTest; [82]) tree-constructing algorithms, both with 1000 bootstrap replicates. The nematode species Praeacanthonchus punctatus, Ptycholaimellus carinatus and Odontophora setosa, all from the same subclassis (Chromadoria) as Sabatieria, were added to the dataset as outgroup (Derycke, unpublished data). The same strategy was followed for the two nuclear markers with the exception that a Tamura-Nei + G substitution model [83] was used in ML tree construction and that outgroups included both unpublished (Odontophora setosa and Praeacanthonchus punctatus) as well as published sequences extracted from GenBank (Ptycholaimellus sp.: accession JN968285-18S; Praeacanthonchus punctatus: accession AF210416-D2D3 and Odontophora sp.: accession DQ077756-D2D3). For D2D3, there were no Ptycholaimellus sequences available. The unique haplotypes found in this study have been submitted to GenBank and are available under accession numbers LT703318-LT703405. Representative DNA samples were deposited in the MoMentuM database of CeMoFe (Centre for Molecular Phylogeny and Evolution), UGent.
Population genetic structure in COI
Analysis of Molecular Variance (AMOVA) was performed in ARLEQUIN version 3.5.1.2 [84] and used to analyze the population genetic structure of Sabatieria COI sequence variation across the different seeps. Population pairwise ΦST-statistics were calculated (10,000 permutations, 0.05 significance level) to determine which seeps were significantly differentiated from each other. Because the HKY model given by jModelTest is not available in ARLEQUIN, the more inclusive Tamura-Nei (TrN) model [83] with gamma distribution was used to calculate ΦST and other relevant statistics. Significance levels of the obtained p-values were corrected according to the sequential Bonferroni method [85]. Next to this, standard measures of genetic variation within populations, such as nucleotide diversity (π) [86] and haplotype diversity (h) [86, 87], and their standard deviation were calculated per cold seep. Tajima’s D [88] and Fu’s Fs [89] neutrality tests were performed to infer whether sequence evolution was neutral and whether geographical groups showed signs of deviations from neutrality. Significantly negative values for Tajima’s D and Fu’s Fs reflect an excess of rare polymorphisms in a population, which indicates either positive selection or an increase in population size [90]. These tests were performed both on the haplotype distribution of the separate seeps as well as on the whole dataset as one, with the number of simulated samples set at 1000.
To test whether genetic divergence between populations increased with geographic distance (i.e. isolation-by-distance IBD; [91]), geographic and genetic distances were compared using a Mantel test (1000 permutations; [92]). Both untransformed and log-transformed distances were used to test the IBD model. The geographic distance between populations was measured as the shortest continuous water-surface distance (Fig. 1). To explore the intraspecific relationships between observed haplotypes, a TCS network was constructed with the PopART software (http://popart.otago.ac.nz).
Results
Phylogenetic analysis of COI
Sequence alignment yielded a COI fragment of 420 bp long. This fragment contained 135 conserved and 264 variable positions, 193 of which were parsimony informative and 69 of which were singletons. Both the NJ and ML tree topology of COI haplotypes showed two well-supported clades (Fig. 2). The REG clade contained the two sequences from the REGAB cold seep (Table 1), while the EM clade included all 331 sequences from the Eastern Mediterranean cold seeps, except haplotype V. This latter sequence was highly divergent from the other sequences (p-distance EM: 0.400–0.408; REG: 0.444). P-distances between the two clades ranged between 0.234–0.269, while p-distances within clades were much lower (EM: 0.003–0.043; REG: 0.015). No subdivision according to geographic location could be discerned within the EM clade.

Phylogenetic ML tree of the COI haplotype sequences with indication of two clades. EM = Eastern Mediterranean clade; REG = REGAB clade. Values along branches are bootstrap values from the NJ algorithm based on p-distance and the ML procedure based on HKY + G, separated by a comma. Bootstrap values lower than 70 are not visualized in the figure. Each organism received a unique identification code (PCR code_Cold seep(s)_haplotype). In case haplotypes occurred at only one geographical location, their origin is indicated by a symbol and color (see legend). Haplotypes shared among several locations are not marked
Phylogenetic analysis of the nuclear markers
The 18S fragment was 942 bp long, contained 678 conserved and 262 variable positions, 98 of which were parsimony informative and 163 of which were singletons. Two insertions of a single bp were noted in two outgroup species (Praeacanthoncus punctatus and Ptycholaimellus sp.). The D2D3 fragment was 752 bp long with 206 conservative and 531 variable positions, of which 155 parsimony informative and 373 singletons. The NJ and ML analysis of both markers showed congruent tree topologies which were similar to the topology of the COI tree, with two well-supported clades (Fig. 3). P-distances between the two clades were 0.012–0.014 for 18S, and 0.120–0.172 for D2D3. As for COI, p-distances within clades were much lower (18S: EM: 0.000–0.002; REG: 0.000; D2D3: EM: 0.000–0.019, REG: 0.000). There was no clear subdivision according to geographic location in the nuclear markers. Only the sequences from the Amsterdam MV formed a separate group with high bootstrap support in the tree of the D2D3 fragment (Fig. 3b, group 28S B). Similar to COI, haplotype V was clearly separated from all other Sabatieria sequences with high p-distance values (18S: EM: 0.082–0.090, REG: 0.080; D2D3: EM: 0.627–0.643, REG: 0.631–0.636). A Blast search of its 18S sequence identified it as Terschellingia longicaudata (95–98% similarity), a species that is abundantly present at Amon MV. This haplotype was therefore excluded from further population genetic analyses.

Phylogenetic ML trees for (a) 18S and (b) the D2D3 marker of 28S. EM: Eastern Mediterranean clade; REG: REGAB clade. Values along branches are bootstrap values of both NJ (p-distance) and ML (TrN + G) procedures, separated by a comma. Bootstrap values lower than 70 are not visualized in the figure. Each organism received a unique identification code (PCR code_Cold seep_haplotype). Symbols and colors indicate the origin of the sequences
Similarly, since 5% COI sequence divergence is commonly observed between different nematode species [37], and because of the congruence between mitochondrial and nuclear markers, the REG clade was considered as an independently evolving lineage, and was omitted from the dataset to investigate population genetic structure within the Eastern Mediterranean clade.
Population genetic structure of COI
A total of 331 individuals were included in the population genetic analyses (only EM clade sequences), divided evenly over all four cold seeps from the Eastern Mediterranean area (Table 2). A fragment of 393 bp long was obtained, containing 33 variable positions, 15 parsimony informative and 18 singletons. In total, 21 haplotypes were found. The number of haplotypes per seep was comparable, with seven haplotypes found at Cheops and PM, eight at Amsterdam MV, and nine at Amon MV. At each seep, two or three haplotypes were found regularly, while the remaining haplotypes were rare (Table 2). Haplotype A was the most widely distributed and abundant haplotype, occurring in all cold seeps and in densities of at least twice that of the second most abundant haplotype. The only exception was Amsterdam MV, where haplotype A (18 times) was slightly less common than Q (23 times) and B (19 times). Next to haplotype A, five other haplotypes (B-F) were also found in multiple seeps. All other haplotypes were unique to one seep, or even to a single specimen (Table 2, Fig. 4).

TCS network constructed with the haplotypes found in the EM clade. Each circle represents a haplotype, while the different colors represent the cold seeps. The division and size of the circles shows the frequency of this haplotype in each cold seep. Hatch marks along connecting branches indicate the number of substitutions between two haplotypes. Small black circles represent missing haplotypes
The TCS network further indicated the dominance of haplotype A, which was found in all four seeps (Fig. 4). Two groups of haplotypes differing in only one mutation with the closest related haplotype could be identified, and both groups had haplotypes in all four cold seeps (group 1 and group 2 in Fig. 4). Group 1 contained 14 haplotypes, of which 5 were only found in Amsterdam MV, 4 only in Cheops and 1 only in Amon. Group 2 contained 5 haplotypes, with Cheops, PM and Amon each having one private haplotype. In addition to these two groups of haplotypes, haplotypes E and N showed 6 and 8 mutational differences with the closest (missing) haplotype.
Haplotype diversity (h) was higher in Amsterdam MV (0.773 ± 0.022) than in the other seeps (between 0.576 ± 0.045 and 0.616 ± 0.052), while nucleotide diversity (π) was an order of magnitude lower in Cheops than in the three other seeps (Table 2). AMOVA resulted in a high ΦST-value (0.149, p < 0.001), attributing 15% of the genetic variability in the dataset to differences between locations (Table 3). All pairwise ΦST-values were high (0.111–0.283) and significantly different from zero after Bonferroni correction (Table 4). The mantel test did not show any significant correlation between genetic and geographic distances, for both untransformed (p = 0.390, correlation coefficient = 0.240) and log-transformed (p = 0.459, correlation coefficient = 0.204) distances. The same was true for the three seeps in the southern part of the East Mediterranean (Cheops, PM and Amon) when analyzed separately (p = 0.4995, correlation coefficient = 0.363). Tajima’s D was only significantly negative for Cheops (D = −1.649, p = 0.025). None of Fu’s Fs were significantly different from zero. Also the analysis of the whole dataset as one group did not yield significant results (Tajima’s D = −0.819, p = 0.213; Fu’s Fs = −1.672, p = 0.371).
Discussion
Our results show that Sabatieria mortenseni is present in all samples in cold seeps from the East Mediterranean. The continuous character and the presence of currents as a large-scale transportation mechanism of organisms are likely to facilitate dispersal and gene flow in the marine environment and may result in wide species distributions (e.g., deep-sea foraminifera in Arctic and Antarctic sediments [93, 94]; see also [2]). Also for nematodes, widespread distributions have been reported. For example, the nematode Litoditis marina maintains trans-Atlantic populations, and specimens have been found floating in macroalgae rafts in the North Sea [41]. Bik and co-authors [42] found evidence for cosmopolitan marine Oncholaimid species with identical gene sequences (18S, 28S and COI) at trans-Atlantic sample sites. Bhadury and co-workers [43] revealed that the majority of the morphologically defined Terschellingia longicaudata specimens shared a single 18S rRNA sequence and apparently belonged to a single species distributed from the British Isles to Malaysia in a wide range of marine habitats and ecological ranges. The high similarity of the T. longicaudata from Amon MV in this study with the shallow-water specimens described in [43] (98% similarity; Accession No.: AM941225, AM941226 and AM234716) is indicative of a wide distribution for this species. Nematodes can become passively suspended and transported in the water column by currents [35, 36]. This might not only apply to the organism itself, but also to the eggs, although this latter possibility has not been investigated yet. Next to passive transport, some nematode species can actively swim in the water column, with widespread species distributions as a result [41,42,43].
For Sabatieria, community analysis of the cold-seep environments analyzed in this study revealed that the genus (with S. mortenseni) was abundantly present in all reduced sediments sampled (relative abundances 10.6–88.6%; personal observation). The species was also present in the surrounding oxidized sediments, albeit in much lower abundances (0.0–6.4%; personal observation), indicating its preference for hypoxic or anoxic conditions. These data suggest a widespread distribution in the area under study. However, the extent of genetic similarity between Sabatieria mortenseni from the non-reduced environment and the cold seeps is not clear, since specimens from the non-reduced environment were preserved in formalin.
Genetic structure of Sabatieria mortenseni shows intermediate gene flow between cold seeps
The strong genetic structure revealed by AMOVA and the high pairwise Φst values suggest that suspension and transport by currents are not enough to establish extensive gene flow between the Sabatieria populations of the different cold seeps. Instead, several haplotypes are shared between seeps, the number of mutations between haplotypes is low and private haplotypes are present in each cold seep, all indicating that intermediate gene flow is occurring without long-term barriers to dispersal between cold seeps (cf. pattern V in [95]). The presence of S. mortenseni in the surrounding environment may indicate that the distribution of this species in the deep sea is much more homogeneous than anticipated. Evidently, widely distributed species that are not restricted to certain habitat types may have a larger capacity to disperse than narrowly distributed, specialized species. Even though the abundances of S. mortenseni in the surrounding sediment are low, such small local units may form part of a source-sink system facilitating gene flow between distant populations.
The East Mediterranean is one of two sub-basins of the Mediterranean Sea and hosts a complex array of current systems, including deep-water formation in the Adriatic Sea (Eastern Mediterranean Deep Water EMDW; [96]), cyclonic and anticyclonic gyres, and small energetic eddies [97, 98]. Main deep-water currents flow from west to east, along the North African coastline in the southern Ionian and Levantine basin (passing the Nile Deep Sea Fan, i.e. location of Cheops, PM and Amon MV) towards the northern Levantine basin (i.e. location of Amsterdam MV) [99]. Therefore, physical connections between the different cold seep environments of this study are theoretically possible and can explain the presence of several shared haplotypes between the cold seeps.
Even in the presence of currents able to mediate long-distance dispersal of organisms and eggs in relatively homogeneous habitats, dispersal success tends to decrease with increasing distance, resulting in larger genetic dissimilarity between populations that are more distant from each other (i.e. stepping-stone model; [91]). Given the scattered distribution of deep-sea cold seeps and hydrothermal vents, such distance effects have frequently been invoked to explain genetic divergence in chemosynthetic invertebrate species with pelagic larvae [100,101,102,103]. For the passively dispersing nematodes which lack such a larval stage, the isolation-by-distance effect might be even more pronounced. However, no significant isolation-by-distance for the Sabatieria populations of the East Mediterranean cold seeps was observed (see Mantel test results). This was also evident from the COI phylogenetic tree where populations did not cluster according to their geographic location (Fig. 2). Instead, genetic distances between closely located seeps (e.g., Amon MV and PM) were sometimes larger than between seeps further apart (e.g., Amsterdam MV and PM; Table 4). Such a pattern where genetic structuring does not correlate with geographic distance (i.e. chaotic genetic patchiness) is quite common in the marine environment for organisms with larval development [104,105,106,107,108], but also for nematodes [109], and can be explained by the multidirectional movement of organisms due to turbulent and nonlinear water currents, as well as by other ecological factors (e.g., variable local natural selection on settling larvae; [105]).
Few mutations between haplotypes point to the absence of long-term geographic barriers between cold seeps
Besides contemporary physical drivers for genetic population differentiation (currents, distance), historical events often leave their mark on current species distributions. On an evolutionary timescale, the Mediterranean deep-sea environment and its fauna are relatively young compared to that of other oceans. Our results show a significant population genetic structuring between the cold seeps within the Eastern Mediterranean sea which may reflect past isolation and (re-) colonization events. The haplotype network for COI consisted of a few central widespread and abundant haplotypes (A and B) surrounded by multiple private haplotypes (Fig. 4, group 1 and 2). Such a pattern is characteristic for population expansion, but statistical confirmation of such an event was limited: Tajima’s D was only significantly negative for Cheops MV. Instead, the two haplotype groups were separated by 2 or 3 mutations suggesting recent admixture between previously isolated populations (see also [95, 110]). The higher number of haplotypes in group 1 and the high abundance of the central haplotype A in all cold seeps indicate that this cluster of haplotypes has a longer evolutionary history in the four cold seeps than haplotypes from group 2, which was much less abundant and diverse than group 1 (Fig. 4). The high number of mutations in haplotypes E and N, their very low abundance and their restriction to the Amon cold seep (and PM for E) in the south suggest that individuals harboring these haplotypes most likely originated in locations outside our sampling area, in the southern part of the Mediterranean. In this scenario, the Amon cold seep has been colonized by four independent events (one event containing haplotypes from group 1, one containing haplotypes from group 2, one containing haplotype N and one containing haplotype E), whereas the other cold seeps were colonized by two (Cheops and Amsterdam MV) or three (PM) separate events. Overall, haplotype and nucleotide diversity for Sabatieria populations were similar to values reported for shallow-water nematodes (e.g., [38, 111]) and generally low (especially nucleotide diversity).
Finally, the genetic structure present in S. mortenseni may result from a strong adaptation to the particular local conditions at the different cold seep habitats. Such a scenario of differentiation by adaptation seems plausible considering the environmental heterogeneity typically found in chemosynthetic habitats [112]. In this case, local selective pressures could prevent the successful exchange of individuals (e.g., [113]), leading to isolation of the gene pools of the different populations. However, based on the data at hand it is difficult to assess whether such a mechanism can be invoked in the case of Sabatieria in the East Mediterranean.
Cryptic speciation
Our results show the presence of cryptic species within Sabatieria mortenseni from seeps located in different ocean basins: genetic evidence pointed towards two independently evolving lineages, one in the Eastern Mediterranean and one in the Southeast Atlantic region. Yet, no differences in diagnostic morphological features were observed and morphometric characters overlapped with those of S. mortenseni reported in [45, 74] (Additional file 1: Table S1). In general, cryptic species are common in the marine environment (e.g., [114]). Numerous cryptic species exist among the vesicomyid clams [115, 116] and lepetodrilid limpets [117] that dominate vents and seeps worldwide. Cryptic species have also frequently been detected in free-living marine nematodes from intertidal environments with sympatric distributions [38, 39, 118]. In contrast, populations of the marine nematode Paracanthonchus gynodiporata living on macroalgae were genetically homogeneous across more than 1000 km Brazilian coastline but showed substantial phenotypic plasticity [44]. Cryptic species have also been reported in deep-sea nematodes: Halomonhystera disjuncta from the Håkon Mosby mud volcano is genetically distinct from the intertidal Halomonhystera disjuncta [34] and two cryptic species have recently been discovered in Sabatieria from the continental shelf in Antarctica [119]. The current study shows that cryptic diversity is also present in nematodes dominating cold seeps in the deep-sea floor, but apparently only at a larger geographic scale. Although morphometric characters overlapped with S. mortenseni from [45, 74], specimens from the REGAB cold seep have a larger body size than S. mortenseni reported from the literature (Additional file 1: Table S1). Yet, the body size of the REGAB specimens overlaps with that of specimens from Amon and Amsterdam. We measured a large number of males (18–28 per cold seep in the Mediterranean Sea) and find a much larger morphological plasticity in S. mortenseni from the cold seeps than observed in S. mortenseni from the subtidal area near Rio de Janeiro [74]. Unfortunately, no sequence data are available from specimens from Rio de Janeiro to verify the extent of genetic differentiation between S. mortenseni from both areas.
Surprisingly, there was no evidence for cryptic speciation within the East Mediterranean despite the fact that geographical distances were much larger than those in previous studies in marine nematodes [109]. Because of the young age of the Mediterranean Sea (5.3–2 Myr), speciation of the Sabatieria populations may not have been completed yet. As a reference, cryptic species found in the shallow-water nematode Litoditis marina are at least 6.5 Myr old [120]. Nevertheless, the high variability of local cold-seep conditions (e.g., heterogeneity in fluid composition of sulphide and methane; see Methods section) and low exchange between cold seeps could induce perfect conditions for ecological speciation. We therefore expected cryptic speciation in disjunct cold-seep areas. However, Sabatieria species are known to withstand fluctuations in environmental chemistry as they regularly are the dominant genus in various reduced ecosystems (e.g., [27, 30,31,32,33]). The lack of cryptic species in these Mediterranean cold seeps can thus be due to the fact that this species, after spreading in the Mediterranean area, still shows sufficient gene flow to homogenize gene pools.
Conclusion
The populations of the marine nematode Sabatieria mortenseni inhabiting cold seeps from the bathyal East Mediterranean Sea and Atlantic Ocean are genetically structured. The presence of shared haplotypes and the low number of mutations between haplotypes indicate that barriers to dispersal between these cold seep populations are relatively recent. The low abundances of S. mortenseni in the surrounding sediment together with water currents in the area may form part of a source-sink system facilitating gene flow between distant populations. Our results further show the presence of cryptic species within S. mortenseni from seeps located in different ocean basins: genetic evidence pointed towards two independently evolving lineages, one in the Eastern Mediterranean and one in the Southeast Atlantic region but no differences in diagnostic morphological features were observed. Our results expand our knowledge on genetic diversity in marine nematodes and confirm that a single nematode species can be widely distributed despite the lack of pelagic larval stages.
References
Gage JD, Tyler PA. Deep-Sea biology: a natural history of organisms at the Deep-Sea floor. Cambridge, UK: Cambridge University Press; 1991. p. 504.
Brandt A, Blazewicz-Paszkowycz M, Bamber RN, Mühlenhardt-Siegel U, Malyutina MV, Kaiser S, et al. Are there widespread pericarid species in the deep sea (Crustacea: malacostraca)? Polish Polar Research. 2012;33(2):139–62. doi:10.2478/v10183-012-0012-5.
Levin LA, Sibuet M. Understanding continental margin biodiversity: a new imperative. Annu Rev Mar Sci. 2012;4:79–112. doi:10.1146/annurev-marine-120709-142714.
Baird HP, Miller KJ, Stark JS. Evidence of hidden biodiversity, ongoing speciation and diverse patterns of genetic structure in giant Antarctic amphipods. Mol Ecol. 2011;20:3439–54. doi:10.1111/j.1365-294X.2011.05173.x.
Bik HM, Sung W, De Ley P, Baldwin JG, Sharma J, Rocha-Olivares A, et al. Metagenetic community analysis of microbial eukaryotes illuminates biogeographic patterns in deep-sea and shallow water sediments. Mol Ecol. 2012;21:1048–59. doi:10.1111/j.1365-294X.2011.05297.x.
France SC, Kocher TD. Geographic and bathymetric patterns of mitochondrial 16S rRNA sequence divergence among deep-sea amphipods, Eurythenes gryllus. Mar Biol. 1996;126(4):633–43.
Goffredi SK, Hurtado LA, Hallam S, Vrijenhoek RC. Evolutionary relationships of deep-sea vent and cold seep clams (Mollusca: Vesicomyidae) of the “Pacifica/lepta” species complex. Mar Biol. 2003;142:311–20. doi:10.1007/s00227-002-0941-3.
Peek AS, Gustafson RG, Lutz RA, Vrijenhoek RC. Evolutionary relationships of deep-sea hydrothermal vent and cold-water clams (Bivalvia: Vesicomyidae): results from the mitochondrial cytochrome oxidase subunit I. Mar Biol. 1997;130:151–61.
Weinberg JR, Dahlgren TG, Trowbridge N, Halanych KM. Genetic differences within and between species of deep-sea crabs (Chaceon) from the North Atlantic Ocean. Biol Bull. 2003;204:318–26.
Etter RJ, Boyle EE, Glazier A, Jennings RM, Dutra E, Chase MR. Phylogeography of a pan-Atlantic abyssal protobranch bivalve: implications for evolution in the deep Atlantic. Mol Ecol. 2011;20:829–43. doi:10.1111/j.1365-294X.2010.04978.x.
Le Goff-Vitry MC, Pybus OG, Rogers AD. Genetic structure of the deep-sea coral Lophelia pertusa in the northeast Atlantic revealed by microsatellites and internal transcribed spacer sequences. Mol Ecol. 2004;13:537–49.
Olu-Le Roy KOL, von Cosel R, Hourdez S, Carney SL, Jollivet D. Amphi-Atlantic cold-seep Bathymodiolus species complexes across the equatorial belt. Deep-Sea Research Part I Oceanographic Research Papers. 2007;54:1890–911.
Creasey S, Rogers AD, Tyler P, Young C, Gage J. The population biology and genetics of the deep-sea spider crab, Encephaloides armstrongi wood-Mason 1891 (Decapoda: Majidae). Philosophical Transactions of the Royal Society of London Series B-Biological Sciences. 1997;352:365–79.
Etter RJ, Rex MA, Chase MC, Quattro JM. Population differentiation decreases with depth in deep-sea bivalves. Evolution. 2005;59(7):1479–91.
France SC. Genetic population structure and gene flow among deep-sea amphipods, Abyssorchomene spp., from six California continental borderland basins. Mar Biol. 1994;118:67–77.
Rogers AD. The role of the oceanic oxygen minima in generating biodiversity in the deep sea. Deep-Sea Research Part II-Topical Studies in Oceanography. 2000;47:119–48.
Etter RJ, Rex MA, Chase MC, Quattro JM. A genetic dimension to deep-sea biodiversity. Deep-Sea Research I. 1999;46:1095–9.
Howell KL, Rogers AD, Tyler PA, Billett DSM. Reproductive isolation among morphotypes of the Atlantic seastar species Zoroaster fulgens (Asteroidea: Echinodermata). Mar Biol. 2004;144:977–84.
Quattro JM, Chase MR, Rex MA, Greig TW, Etter RJ. Extreme mitochondrial DNA divergence within populations of the deep-sea gastropod Frigidoalvania brychia. Mar Biol. 2001;139:1107–13. doi:10.1007/s002270100662.
Zardus JD, Etter RJ, Chase MR, Rex MA, Boyle EE. Bathymetric and geographic population structure in the pan-Atlantic deep-sea bivalve Deminucula atacellana (Schenck, 1939). Mol Ecol. 2006;15:639–51. doi:10.1111/j.1365-294X.2005.02832.x.
Domack E, Ishman S, Leventer A, Sylva S, Willmott V, Huber B. A chemotrophic ecosystem found beneath Antarctic ice shelf. Eos. 2005;86(29):269–76.
Levin LA. Ecology of cold seep sediments: interactions of fauna with flow, chemistry and microbes. Oceanogr Mar Biol Annu Rev. 2005;43:1–46.
Sibuet M, Olu-Le Roy K. Cold seep communities on continental margins: structure and quantitative distribution relative to geological and fluid venting patterns. In: Wefer G, Billett D, Hebbeln D, Jorgensen BB, Schlüter M, van Weering TCE, editors. Ocean Margin Systems. Berlin, Germany: Springer-Verlag Berlin Heidelberg; 2002. p. 235–251.
Corliss JB, Dymond J, Grodon LI, Edmond JM, von Herzen RP, Ballard RD, et al. Submarine thermal springs on the Galápagos rift. Science. 1979;203(4385):1073–83.
Vanreusel A, De Groote A, Gollner S, Bright M. Ecology and biogeography of free-living nematodes associated with chemosynthetic environments in the deep sea: a review. PLoS One. 2010;5(8):e12449. doi:10.1371/journal.pone.0012449.
Moens T, Verbeeck L, De Maeyer A, Swings J, Vincx M. Selective attraction of marine bacterivorous nematodes to their bacterial food. Mar Ecol Prog Ser. 1999;176:165–78. doi:10.3354/meps176165.
Van Gaever S, Olu-LeRoy K, Derycke S, Vanreusel A. Metazoan meiofaunal communities at cold seeps along the Norwegian margin: influence of habitat heterogeneity and evidence for connection with shallow-water habitats. Deep-Sea Research I. 2009;56:772–85.
Van Gaever S, Moodley L, de Beer D, Vanreusel A. Meiobenthos of the Arctic Håkon Mosby mud volcano, with a parental-caring nematode thriving in sulphide-rich sediments. Mar Ecol Prog Ser. 2006;321:143–55. doi:10.3354/meps321143.
Guilini K, Levin LA, Vanreusel A. Cold seep and oxygen minimum zone associated sources of margin heterogeneity affect benthic assemblages, diversity and nutrition at the Cascadian margin (NE Pacific ocean). Prog Oceanogr. 2012;96:77–92. doi:10.1016/j.pocean.2011.10.003.
Lampadariou N, Kalogeropoulou V, Sevastou K, Keklikoglou K, Sarrazin J. Influence of chemosynthetic ecosystems on nematode community structure and biomass in the deep eastern Mediterranean Sea. Biogeosciences. 2013;10:5381–98. doi:10.5194/bg-10-5381-2013.
Pape E, Nara Bezerra T, Vanneste H, Heeschen K, Moodley L, Leroux F, et al. Community structure and feeding preference of nematodes associated with methane seepage at the Darwin mud volcano (Gulf of Cádiz). Mar Ecol Prog Ser. 2011;438:71–83. doi:10.3354/meps09278.
Portnova D, Mokievsky V, Soltwedel T. Nematode species distribution patterns at the Håkon Mosby mud volcano (Norwegian Sea). Mar Ecol. 2011;32:24–41. doi:10.1111/j.1439-0485.2010.00403.x.
Van Gaever S, Galéron J, Sibuet M, Vanreusel A. Deep-sea habitat heterogeneity on meiofaunal communities in the Gulf of Guinea. Deep-Sea Research II. 2009;56:2259–69.
Van Campenhout J, Derycke S, Tchesunov A, Portnova D, Vanreusel A. The Halomonhystera disjuncta population is homogeneous across the Håkon Mosby mud volcano (Barents Sea) but is genetically differentiated from its shallow-water relatives. J Zool Syst Evol Res. 2014;52(3):203–16. doi:10.1111/jzs.12054.
Palmer MA. Dispersal of marine meiofauna: a review and conceptual model explaining passive transport and active emergence with implications for recruitment. Mar Ecol Prog Ser. 1988;48:81–91.
Boeckner MJ, Sharma J, Proctor HC. Revisiting the meiofauna paradox: dispersal and colonization of nematodes and other meiofaunal organisms in low- and high-energy environments. Hydrobiologia. 2009;624:91–106.
Jensen P. Phyto-chemical sensitivity and swimming behaviour of the free-living marine nematode Chromadorita tenuis. Mar Ecol Prog Ser. 1981;4:203–6.
Derycke S, Remerie T, Vierstraete A, Backeljau T, Vanfleteren J, Vincx M, et al. Mitochondrial DNA variation and cryptic speciation within the free-living marine nematode Pellioditis marina. Mar Ecol Prog Ser. 2005;300:91–103.
Derycke S, Backeljau T, Vlaeminck C, Vierstraete A, Vanfleteren J, et al. Spatiotemporal analysis of population genetic structure in Geomonhystera disjuncta (Nematoda, Monhysteridae) reveals high levels of molecular diversity. Mar Biol. 2007;151:1799–812.
Derycke S, De Ley P, De Ley IT, Holovachov O, Rigaux A, Moens T. Linking DNA sequences to morphology: cryptic diversity and population genetic structure in the marine nematode Thoracostoma trachygaster (Nematoda, Leptosomatidae). Zool Scr. 2010;39(3):276–89.
Derycke S, Remerie T, Backeljau T, Vierstraete A, Vanfleteren J, Vincx M, et al. Phylogeography of the Rhabditis (Pellioditis) marina species complex: evidence for long-distance dispersal, and for range expansions and restricted gene flow in the northeast Atlantic. Mol Ecol. 2008;17:3306–55. doi:10.1111/j.1365-294X.2008.03846.x.
Bik HM, Thomas WK, Lunt DH, Lambshead PJD. Low endemism, continued deep-shallow interchanges, and evidence for cosmopolitan distributions in free-living marine nematodes (order Enoplida). BMC Evol Biol. 2010;10:389. doi:10.1186/1471-2148-10-389.
Bhadury P, Austen MC, Bilton DT, Lambshead PJD, Rogers AD, Smerdon GR. Evaluation of combined morphological and molecular techniques for marine nematode (Terschellingia spp.) identification. Mar Biol. 2008;154:509–18. doi:10.1007/s00227-008-0945-8.
Apolônio Silva de Oliveira D, Decraemer W, Moens T, Amadeu Paiva dos Santos G, Derycke S. Low genetic but high morphological variation over more than 1000 km coastline refutes omnipresence of cryptic diversity in marine nematodes. BMC Evol Biol. 2017;17:71.
Platt HM. The freeliving marine nematode genus Sabatieria (Nematoda: Comesomatidae). Taxonomic revision and pictorial keys. Zool J Linnean Soc. 1985;83:27–78.
Ott JA, Novak R, Schiemer F, Hentschel U, Nebelsick M, Polz M. Tackling the sulfide gradient: a novel strategy involving marine nematodes and chemoautotrophic ectosymbionts. Mar Ecol. 1991;12:261–79.
Vincx M. Free-living marine nematodes from the southern bight of the North Sea. I. Notes on species of the genera Gonionchus cobb, 1920, Neochromadora Micoletzky, 1924 and Sabatieria Rouville, 1903. Hydrobiologia. 1986;140:255–86.
Warwick RM, Gee JM. Community structure of estuarine meiobenthos. Mar Ecol Prog Ser. 1984;18:97–111.
Soetaert K, Heip C. Nematode assemblages of deep-sea and shelf break sites in the North Atlantic and Mediterranean Sea. Mar Ecol Prog Ser. 1995;125:171–83.
Muthumbi AW, Vanreusel A, Vincx M. Taxon-related diversity patterns from the continental shelf to the slope: a case study on nematodes from the western Indian Ocean. Mar Ecol. 2011;32:453–67. doi:10.1111/j.1439-0485.2011.00449.x.
Vanaverbeke J, Soetaert K, Heip C, Vanreusel A. The metazoan meiobenthos along the continental slope of the Goban spur (NE Atlantic). J Sea Res. 1997;38:93–107.
Jensen P. Ecology of benthic and epiphytic nematodes and brackish waters. Hydrobiologia. 1984;108:201–17.
Jensen P, Aagaard I, Burke RA, Dando PR, Jorgensen N, Kuijpers A, et al. ‘bubbling reefs’ on the Kattegat: submarine landscapes of carbonate-cemented rocks support a diverse ecosystem at methane seeps. Mar Ecol Prog Ser. 1992;83:103–12.
World register of Marine Species (WoRMS). http://www.marinespecies.org/
Gagarin VG, Thanh NV. A new genus and three new species of free-living nematodes from mangroves of the Red River estuary, Vietnam. Tạp chí Sinh Học. 2008;30:3–11.
Leduc D. Seven new species and one new species record of Sabatieria (Nematoda: Comesomatidae) from the continental slope of New Zealand. Zootaxa. 2013;3693:1–35.
Ansari KGMT, Lyla PS, Khan A. New records of six Sabatieria species (Nematoda: Comesomatidae) from Indian waters. Indian Journal of Geo-Marine Science. 2015;44(4):599–608.
Allgén CA. West American Marine Nematodes (Papers from Dr. Th. Mortensen's Pacific Expedition 1914–16. LXXV).Vidensk. Medd. Fra. Dansk naturh. Foren.1947;110:65–219.
Allgén CA. Freeliving marine nematodes. Further zoological results of the Swedish Antarctic expedition, 1901–1903 under the direction of Dr. Otto Nordenskjold. V(2) P.A. Stockholm, Sweden: Norstedt & Söner; 1959. p.293.
Ditlevsen H. Papers from Dr. Th. Mortensens Pacific Expedition 1914–16. III Marine free-living Nematodes from the Auckland and Campbell Islands. Videnskabelige Meddelelser fra Dansk naturhistorisk Forening i Kjøbenhavn. 1921;73:1–39.
Pastor de Ward CT. Nematodes marinos de la Ria Deseado (Leptolaimina, Leptolaimidae, Haliplectidae) Santa Cruz, Argentina. Physus. Secc A. 1984;42(103):87–92.
Chen GT, Herman RL, Vincx M. Meiofauna communities from the straits of Magellan and the Beagle Channel. Sci Mar. 1999;63:123–32.
Ondréas H, Olu K, Fouquet Y, Charlou JL, Gay A, Dennielou B, et al. ROV study of a giant pockmark on the Gabon continental margin. Geo-mar letters. 2005;25:281–92. doi:10.1007/s00367-005-0213-6.
Charlou JL, Donval JP, Fouquet Y, Ondreas H, Knoery J, Cochonat P, et al. The ZAIROV leg 2 scientific Party. Physical and chemical characterization of gas hydrates and associated methane plumes in the Congo-Angola Basin. Chem Geol. 2004;205:405–25. doi:10.1016/j.chemgeo.2003.12.033.
Treignier C, Derenne S, Saliot A. Terrestrial and marine n-alcohol inputs and degradation processes relating to a sudden turbidity current in the Zaire canyon. Org Geochem. 2006;37:1170–84. doi:10.1016/j.orggeochem.2006.03.010.
Lykousis V, Alexandri S, Woodside J, de Lange G, Dählmann A, Perissoratis C, et al. Mud volcanoes and gas hydrates in the Anaximander mountains (eastern Mediterranean Sea). Mar Pet Geol. 2009;26:854–72. doi:10.1016/j.marpetgeo.2008.05.002.
Charlou JL, Donva JP, Zitter T, Roy N, Jean-Baptiste P, Foucher J-P, et al. Evidence of methane venting and geochemistry of brines on mud volcanoes of the eastern Mediterranean Sea. Deep-Sea Research I. 2003;50:941–58. doi:10.1016/S0967-0637(03)00093-1.
Dupré S, Woodside J, Foucher J-P, de Lange G, Mascle J, Boetius A, et al., the NAUTINIL Scientific Party. Seafloor geological studies above active gas chimneys off Egypt (Central Nile Deep Sea fan). Deep-Sea Research I. 2007;54:1146–72. doi:10.1016/j.dsr.2007.03.007.
Mastalerz V, de Lange GJ, Dählmann A. Differential aerobic and anaerobic oxidation of hydrocarbon gases discharged at mud volcanoes in the Nile deep-sea fan. Geochim Cosmochim Acta. 2009;73:3849–63. doi:10.1016/j.gca.2008.12.030.
Bayon G, Loncke L, Dupré S, Caprais J-C, Ducassou E, Duperron S, et al. Multi-disciplinary investigation of fluid seepage on an unstable margin: the case of the Central Nile deep sea fan. Mar Geol. 2009;261:92–104. doi:10.1016/j.margeo.2008.10.008.
Loncke L, Mascle J, Parties FS. Mud volcanoes, gas chimneys, pockmarks and mounds in the Nile deep-sea fan (eastern Mediterranean): geophysical evidences. Mar Pet Geol. 2004;21:669–89.
Huguen C, Foucher JP, Mascle J, Ondréas H, Thouement M, Gontharet S, et al., the NAUTINIL Scientific Party. Menes caldera, a highly active site of brine seepage in the eastern Mediterranean sea: “in situ” observations from the NAUTINIL expedition (2003). Mar Geol. 2009;261:138–52. doi:10.1016/j.margeo.2009.02.005.
Heip C, Vincx M, Vranken G. The ecology of marine nematodes. Oceanography and Marine Biology - an Annual Review. 1985;23:399–489.
Botelho AP, Esteves AM, da Fonseca-Genevois V. Known and new species of Sabatieria Rouville, 1903 (Araeolaimida: Comesomatidae) from the southwest Atlantic (Campos Basin, Brazil). Mar Biol Res. 2014;10(9):871–91. doi:10.1080/17451000.2013.866249.
Bowles J, Blair D, McManus DP. Genetic variants within the genus Echinococcus identified by mitochondrial DNA sequencing. Mol Biochem Parasitol. 1992;54:165–74.
De Ley P, Félix MA, Frisse LM, Nadler SA, Sternberg PW, Thomas WK. Molecular and morphological characterization of two reproductively isolated species with mirror-image anatomy (Nematoda: Cephalobidae). Nematology. 1999;2:591–612.
Blaxter ML. A molecular evolutionary framework for the phylum Nematoda. Nature. 1998;392:71–5.
Swindell SR, Plasterer TN. SEQMAN. In: Swindel SR, editor. Methods in Molecular Biology, vol. 70. Sequence Data Analysis Guidebook. New York, US: Humana Press; 1997. p. 75–89.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772.
Guindon S, Gascuel O. A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst Biol. 2003;52:696–704.
Hasegawa M, Kishino K, Yano T. Dating the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–74.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–26.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour. 2010;10:564–7.
Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–5.
Nei M. Molecular evolutionary genetics. New York, NY, USA: Columbia University Press; 1987. 512p.
Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105(2):437–60.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Fu Y-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Aris-Brosou S, Excoffier L. The impact of population expansion and mutation rate heterogeneity on DNA sequence polymorphism. Mol Biol Evol. 1996;13(3):494–504.
Slatkin M. Isolation by distance in equilibrium and non-equilibrium populations. Evolution. 1993;47:264–79.
Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27(2):209–20.
Brandt A, Gooday AJ, Brandão SN, Brix S, Brökeland W, Cedhagen T, et al. First insights into the biodiversity and biogeography of the Southern Ocean deep sea. Nature. 2007;447:307–11.
Pawlowski J, Fahrni J, Lecroq B, Longet D, Cornelius N, Excoffier L, et al. Bipolar gene flow in deep-sea benthic foraminifera. Mol Ecol. 2007;16(19):4089–96. doi:10.1111/j.1365-294X.2007.03465.x.
Avise JC, Arnold J, Ball RM, Bermingham E, Lamb T, Neigel JE, et al. Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Annu Rev Ecol Syst. 1987;18:489–522.
Tanhua T, Hainbucher D, Schroeder K, Cardin V, Alvarez M, Civitarese G. The Mediterranean Sea system: a review and an introduction to the special issue. Ocean Sci. 2013;9:789–803.
The POEM group, Robinson AR, Malanotte-Rizzoli P, Hecht A, Michelato A, Roether W, et al. General circulation of the eastern Mediterranean. Earth Sci Rev. 1992;32:285–309.
Robinson AR, Leslie WG, Theocharis A, Lascaratos A. Mediterranean Sea Circulation. Ocean currents: a derivative of the Encyclopedia of Ocean Sciences 2001; RWOS 0376:1689–1705. doi:10.1006/rwos.2001.0376
The MerMex Group: Durrieu de Madron X, Guieu C, Sempéré R, Conan P, Cossa D, D’Ortenzio F, Estournel C, Gazeau F, Rabouille C, Stemmann L, Bonnet S, Diaz F, Koubbi P, Radakovitch O, Babin M, Baklouti M, Bancon-Montigny C, Belviso S, Bensoussan N, Bonsang B, et al. Marine ecosystems’ responses to climatic and anthropogenic forcings in the Mediterranean. Prog Oceanogr 2011;91:97–166. doi:10.1016/j.pocean.2011.02.003.
Audzijonyte A, Vrijenhoek RC. Why gaps really are gaps: statistical phylogeography of hydrothermal vent invertebrates. Evolution. 2010;64(8):2369–84. doi:10.1111/j.1558-5646.2010.00987.x.
Shank TM, Halanych KM. Toward a mechanistic understanding of larval dispersal; insights from genomic fingerprinting of the deep-sea hydrothermal vent tubeworm Riftia pachyptila. Marine Ecology – An Evolutionary Perspective. 2007;28(1):25–35. doi:10.1111/j.1439-0485.2006.00146.x.
Vrijenhoek RC. Gene flow and genetic diversity in naturally fragmented metapopulations of deep-sea hydrothermal vent animals. J Hered. 1997;88(4):285–93.
Won Y, Young CR, Lutz RA, Vrijenhoek RC. Dispersal barriers and isolation among deep-sea mussel populations (Mytilidae: Bathymodiolus) from eastern Pacific hydrothermal vents. Mol Ecol. 2003;12:169–84.
Broquet T, Viard F, Yearsley JM. Genetic drift and collective dispersal can result in chaotic genetic patchiness. Evolution. 2013;67(6):1660–75.
Hogan JD, Thiessen RJ, Heath DD. Variability in connectivity indicated by chaotic genetic patchiness within and among populations of a marine fish. Mar Ecol Prog Ser. 2010;417:263–75. doi:10.3354/meps08793.
Iacchei M, Ben-Horin T, Selkoe KA, Bird CE, García-Rodríguez FJ, Toonen RJ. Combined analyses of kinship and Fst suggest potential drivers of chaotic genetic patchiness in high gene-flow populations. Mol Ecol. 2013;22:3476–94. doi:10.1111/mec.12341.
Muths D, Jollivet D, Gentil F, Davoult D. Large-scale genetic patchiness among NE Atlantic populations of the brittle star Ophiothrix fragilis. Aquat Biol. 2009;5:117–32. doi:10.3354/ab00138.
Selkoe KA, Watson JR, White C, Horin TB, Iacchei M, Mitarai S, et al. Taking the chaos out of genetic patchiness: seascape genetics reveals ecological and oceanographic drivers of genetic patterns in three temperate reef species. Mol Ecol. 2010;19:3708–26. doi:10.1111/j.1365-294X.2010.04658.x.
Derycke S, Backeljau T, Moens T. Dispersal and gene flow in free-living marine nematodes. Front Zool. 2013;10:1.
Borrero-Pérez GH, González-Wangüemert M, Marcos C, Pérez-Ruzafa A. Phylogeography of the Atlanto-Mediterranean sea cucumber Holothuria (Holothuria) mammata: the combined effects of historical processes and current oceanographical pattern. Mol Ecol. 2011;20:1964–75.
Derycke S, Backeljau T, Vlaeminck C, Vierstraete A, Vanfleteren J, Vincx M, et al. Seasonal dynamics of population genetic structure in cryptic taxa of the Pellioditis marina Complex (Nematoda: Rhabditida). Genetica. 2006;128:307–21.
Faure B, Jollivet D, Tanguy A, Bonhomme F, Bierne N. Speciation in the Deep Sea: multi-locus analysis of divergence and Gene flow between two hybridizing species of hydrothermal vent mussels. PLoS One. 2009;4(8):e6485. doi:10.1371/journal.pone.0006485.
Hilbish TJ, Koehn RK. Exclusion of the role of secondary contact in an allele frequency cline in the mussel Mytilus edulis. Evolution. 1985;39(2):462–46. doi:10.2307/2408375.
Knowlton N. Sibling species in the sea. Annu Rev Ecol Syst. 1993;24:189–216. doi:10.1146/annurev.ecolsys.24.1.189.
Peek AS, Gaut BS, Feldman RA, Barry JP, Kochevar RE, Lutz RA, et al. Neutral and non-neutral mitochondrial genetic variation in deep-sea clams from the family Vesicomyidae. J Mol Evol. 2000;50(2):141–53.
Vrijenhoek RC, Schutz SJ, Gustafson RG, Lutz RA. Cryptic species of deep-sea clams (Mollusca: Bivalvia: Vesicomyidae) from hydrothermal vent and cold-water seep environments. Deep-Sea Research I. 1994;41(8):1171–89.
Johnson SB, Waren A, Vrijenhoek RC. DNA barcoding of Lepetodrilus limpets reveals cryptic species. J Shellfish Res. 2008;27(1):43–51. doi:10.2983/0730-8000(2008)27[43:DBOLLR]2.0.CO;2.
De Oliveira DAS, Decraemer W, Holovachov O, Burr J, De Ley IT, De Ley P, et al. An integrative approach to characterize cryptic species in the Thoracostoma trachygaster hope, 1967 complex (Nematoda: Leptosomatidae). Zoological Journal of Linnean Society. 2012;164:18–35. doi:10.1111/j.1096-3642.2011.00758.x.
Hauquier F, Leliaert F, Rigaux A, Derycke S, Vanreusel A. Distinct genetic differentiation and species diversification within two marine nematodes with different habitat preference in Antarctic sediments. Accepted in BMC Evolutionary Biology. 2017; doi:10.1186/s12862-017-0968-1.
Grosemans T, Morris K, Thomas W, Rigaux A, Moens T, Derycke S. Mitogenomics reveals high synteny and long evolutionary histories of sympatric cryptic nematode species. Ecology and Evolution. 2016;6(6):1854–70.
Acknowledgements
The authors wish to thank the captain and crew of the RV Pourqoui Pas?, RV Merian and RV Meteor, as well as the scientific crew onboard all expeditions for their help and support in sample acquisition. Special thanks go to the pilots of the ROV Quest and ROV Victor 6000 for their technical support during sampling. We are grateful to Annelien Rigaux for the molecular support in the CeMoFE lab, Ghent University. We thank two anonymous reviewers for their critical and constructive remarks.
Funding
This work was funded by the BOF-project No. 01 J14909 and by the project ‘Hotspot Ecosystem Research and Man’s Impact on European Seas’ (HERMIONE), funded by the European Commission under the Framework Program VII (Contract No. 226354).
Availability of data and materials
Sequences of the different genetic markers have been deposited to the European Nucleotide Archive (ENA: http://www.ebi.ac.uk/ena) and GenBank and can be found under accession numbers LT703318-LT703405. All other data are included in this published article.
Author information
Authors and Affiliations
Contributions
ADG, AV and SD conceived the ideas and concept for this study; ADG performed the laboratory analyses; ADG, FH, AV and SD analyzed the data; ADG, FH, AV and SD contributed to the writing. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The manuscript does not report on or involve the use of any vertebrate or human data or tissue, therefore this section is not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1: Table S1.
Morphometric analysis of S. mortenseni. (DOC 44 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
De Groote, A., Hauquier, F., Vanreusel, A. et al. Population genetic structure in Sabatieria (Nematoda) reveals intermediary gene flow and admixture between distant cold seeps from the Mediterranean Sea. BMC Evol Biol 17, 154 (2017). https://doi.org/10.1186/s12862-017-1003-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-017-1003-2