
Abstract
The origins of feral cats in Australia may be understood with the help of molecular studies, but it is important that hypotheses be tested with appropriate sampling and methodology. We point out several shortcomings in the analysis by Koch et al. (BMC Evol Biol 15:262, 2015; A voyage to Terra Australis: human-mediated dispersal of cats. Dryad Digital Repository, 2015), present a reanalysis of part of the study and discuss the challenges of elucidating the early history of feral cats.
Similar content being viewed by others


Main text
In a recent issue of this journal, Koch et al. [1] presented the first molecular study addressing a compelling evolutionary question – the origins of feral cats in Australia. Feral populations of domestic cats (Felis catus) are a serious problem in Australian conservation [2–4]. The intriguing possibility that cats were introduced to Australia from Asia prior to European settlement [1, 5], if true, would potentially decouple the decline and extinction of many native mammals from the rise of cats. Evidence from historical records contradicts this hypothesis, but does not offer definitive proof [6, 7]. While genetic data could potentially hold the key to this problem, such a study must be based on sound and appropriate techniques. We have identified two principal deficiencies in the paper by Koch et al. [1]. We describe both below, with support from a reanalysis of part of the study, and discuss the difficulty of firmly ruling out a pre-European introduction of feral cats using molecular data.
Identifying colonisation pathways
While we agree that a substantial portion of the early Australian feral cat population was probably supplied by Europeans, the model selection approach used Koch et al. [1] was not appropriate to the task of elucidating the origins of the population. The first hypothesis that they aimed to test was whether cats arrived from Asia prior to European settlement. This is a question that the software used, Migrate-N [8], appears unsuited for. Migrate-N fits an island model at migration-drift equilibrium [8, 9], making it suitable for studying patterns of ongoing, stable migration. The appropriate use of Migrate-N in phylogeography is to estimate migration rates and effective population sizes simultaneously, for example in the case of asymmetric migration rates between cave and surface populations in the Mexican blind cavefish complex [10]. It is not designed to distinguish between recent gene flow and historical colonisation [8, 9], or to identify primary and secondary invasion routes, as Koch et al. [1] have interpreted their results.
Even if the research question concerned migration rates alone, key assumptions of Migrate-N are likely to be broken in Australian feral cats. First, migration-drift equilibrium appears unlikely in this system: regardless of whether a population of Asian origin already existed in Australia, the bulk of the European contribution is likely to have occurred very recently (i.e. in the 228 years since British colonisation). Although the population may have been relatively stable, the rates of gene flow into the population are unlikely to have remained constant during that interval (e.g. recent trends in desexing; [11, 12]). Second, the dataset is missing populations of stray and domestic cats in Australia, as well as populations from Asian locations other than Malaysia and Sulawesi. A “ghost” population should be considered in the analysis to satisfy the assumption that all populations have been sampled [13]. Finally, each population is assumed to be panmictic, which more careful analysis of the microsatellite data would have shown to be false (see below). The lack of estimated migration matrices or probability intervals made it difficult to assess whether a realistic final model was obtained for feral cats [1].
Migrate-N is unable to determine whether ancestral polymorphism or gene flow is responsible for shared polymorphism among Europe, Asia and Australia, but more suitable coalescent-based methods for modelling isolation with migration and estimating divergence times require multiple loci, and some of the more popular methods (e.g. IMa2 or LAMARC; [14, 15]) also require the population tree to be specified a priori. More flexible Approximate Bayesian Computation approaches (e.g. PopABC, [16]) are likely to be needed to distinguish between the complex sets of possible demographic histories that could have produced Australia’s current feral cat population [17].
The interpretation of the mitochondrial phylogeny was more appropriate, despite the lack of intraspecific differentiation in the cat [18]. Although it is surprising that only a Bayesian tree was shown, without corroboration from other approaches such as maximum likelihood (ML), there is evidence in the phylogeny presented by Koch et al. [1] of lineage sorting in some populations; only Subclade A was found on the Cocos (Keeling) Islands, Dirk Hartog Island, Tasman Island and Flinders Island, as well as in several Western Australian populations (although the sample size was rather low in the Tasman Island and Flinders Island populations). The links to historical human movements highlighted by Koch et al. [1] support their suggestion that the island populations reflect the genetic composition of early cat introductions, and perhaps the same applies to the Western Australian populations.
However, results based on organelle markers, which represent a single linked locus, should be interpreted cautiously, and several important parameter choices, such as substitution model and tree prior, appear to be missing from the methods section [1]. Furthermore, the occurrence of both major mitochondrial subclades in both Asian and European populations in approximately equal frequencies implies little power to distinguish relationships with the Australian feral cat populations. Indeed, both subclades also occur in the wild relative of F. catus, F. silvestris lybica (Near Eastern Wildcat), and subclade A even occurs in the Central Asian Wildcat, F. s. ornata [18]. There is little resolution in the tree, but the few supported (HPD > 0.7) clades with > =3 samples include either a single geographic region or representatives of all three regions of interest (mainland/Tasmania, Europe and Asia). As a result, there is little power to infer population relationships. The “secondary introductions” favoured by Koch et al. [1] cannot be inferred from either the Migrate-N analysis or the mitochondrial phylogeny. An alternative explanation for the observed topology, suggested by a thoughtful reviewer, is that the Asian cats sampled from Singapore and Malaysia have European ancestry, resulting from the immigration of European cats documented in earlier work [18, 19]. Nuclear data are needed to distinguish between European and Asian random bred cats, however, as no phylogeographic association has been observed among mitochondrial subclades [18]. Without more Asian samples in the nuclear DNA analysis, it is impossible to verify whether these cats represent Asian heritage.
Genetic structure of Australian feral cat populations
Koch et al. [1] used the program STRUCTURE [20, 21] to study the genetic structure of 265 cats samples across mainland Australia and the surrounding islands (262 cats), as well as Malaysia (three cats). The lack of Asian samples in the nuclear data set prevented its use in testing the hypothesis of an Asian origin, which another recent study was able to do [5]. Instead, the key findings reported were a lack of structure within mainland Australia and high differentiation between mainland Australia and the offshore islands. The Malaysian samples clustered with the Australian mainland group and the Cocos (Keeling) Island population clustered with Tasman and Flinders Islands. The key flaws of the analysis were insufficient MCMC run length and number of runs, which did not follow recommendations of the program manual or those of a study of the reproducibility of STRUCTURE results [22]. Koch et al. [1] used 50,000 burn-in iterations, followed by the collection of data from 100,000 iterations, and supporting information suggested that convergence had not occurred in all runs.
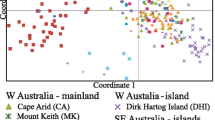
With longer runs and more replication, we were unable to reproduce either the number of populations (K) in the overall data set or the lack of structure within the Australian mainland using the data downloaded from Dryad [23]. Using the same model as Koch et al. [1], but with a burn-in of 500,000 iterations and a subsequent run length of 1,000,000, we found that the model specifying K = 5 outperformed that assuming K = 4 (Fig. 1). Among the 20 runs at each value of K (1 to 10), CLUMPAK [24] detected a single grouping pattern (“mode”) at values of 1–3, 5 and 7. In contrast, the value favoured by Koch et al. [1], K = 4, led to the offshore Tasmanian islands (Tasman and Flinders Islands) clustering with Dirk Hartog Island, rather than the Cocos (Keeling) Islands in three out of 20 runs (Fig. 1). Our reanalysis shows that these islands formed a stable grouping with Tasmania at K = 5, and at K = 7 there was evidence of a distinction between the Tasmania population and the two islands (in agreement with the PCA presented by Koch et al. [1]). The reanalysis also provided evidence of some clustering within the Australian mainland samples, as detected by Spencer et al. [5].

Log-likelihood (a) and ΔK (b) plots for reanalysis of all microsatellite data in Koch et al. [1], with 20 replicate runs of 500 000 burn-in iterations and 1 000 000 further iterations, summarised by Structure Harvester [31]. Below are ancestry coefficients (c) for major and minor modes (K = 4 to K = 7) identified and plotted by CLUMPAK [24, 32]
Koch et al. [1] employed the ΔK [25] statistic to identify the value of K best supported by the data, but did not follow the authors’ recommended procedure for detecting substructure. Evanno et al. [25] noted that their method does not perform well in the presence of hierarchical structure and suggested repeated rounds of analysis within the clusters detected at the optimal K, in order to detect further substructure. We favour the approach of Pritchard et al. [20], which also recommends reanalysing clusters that may harbour substructure. If Koch et al. [1] had examined substructure, they would have most likely detected groupings within the feral cats of mainland Australia (shown in red on their Fig. 2, [1]). Our reanalysis of the largest cluster in the global analysis (at K = 5) detected three groups (Fig. 2), corresponding to the “pan-Australian” and “coastal Western Australian” clusters identified by Spencer et al. [5], as well as a third cluster occurring in rubbish dump populations in southwestern Western Australia. Approximately half the cats of such provenance were estimated to have ~90 % ancestry belonging to the third cluster, suggesting that the third group is not an artefact. Malaysia was omitted from this analysis, as the sample size was extremely small (three animals).

Log-likelihood (a) and ΔK (b) plots for reanalysis of microsatellite data from populations belonging to the major cluster for K = 5 in Fig. 1 (omitting Malaysia), with 20 replicate runs of 500 000 burn-in iterations and 1 000 000 further iterations, summarised by Structure Harvester [31]. Below are ancestry coefficients (c) for K = 2 and K = 3, averaged across runs and plotted by CLUMPAK [24, 32]
In summary, we disagree with two of the main conclusions of the microsatellite analysis of Koch et al. [1]: genetic structure within the Australian mainland populations is detectable with this data set and there is serious doubt as to whether the Tasman Island and Flinders Island populations cluster with the Cocos (Keeling) Islands.
Elucidating the history of feral cats
Understanding the contributions of different sources of introductions to feral cat populations in Australia is interesting from an evolutionary perspective, as it could hold the key to differences in morphology and behaviour among bioregions. However, we must not jump to conclusions based on flawed analyses of a small dataset. Even careful analysis of a large microsatellite dataset [5] provides a somewhat simplified picture of what is likely to be a complex history. In our opinion, resolution of the history of the feral cat in Australia will probably require a powerful multilocus marker set, as well as improved sampling of potential source populations and domestic breeds whose genetic variation may have been added to the feral gene pool [26].
The difficulty of trying to detect pre-European introductions is the large amount of gene flow that has probably occurred in the intervening years from domestic cats of European origin. It is possible that only a small fraction of Asian ancestry would persist, so any analysis would have to be extremely powerful to effectively rule out an Asian contribution. Larger marker sets would be needed to confidently exclude an Asian origin, but even they might fail to detect a small amount of Asian ancestry without a suitable reference population of Asian cats. Genome-wide marker data would also enable testing of whether Asian ancestry persisted in the Australian feral cat population partly as a result of natural selection [27].
We hope to have shed some light on the structure that can be detected with microsatellites, as well as the reasons that more appropriate methods and data are needed to understand what we can of the history of feral cats in Australia.
Funding
This work did not require external funding.
Authors’ contributions
RA, JG and JK planned the response, RA wrote the first draft of the manuscript, DS conducted the STRUCTURE reanalysis, and all authors edited the manuscript.
Acknowledgements
We thank Fran Zewe and Guy Ballard for initial discussions of the study.
Ethics approvals and consent to participate
Not applicable.
Consent to publish
Not applicable.
Response
The authors thank Andrew et al. for their Correspondence and their additional interpretations with regard to the human-mediated dispersal of cats [1]. However, their criticism is misaddressed at least in part, and the suggested extra analyses do not provide substantial new information.
Andrew et al. identified two aspects of concern, the first one refers to the fact that we used a model selection approach (based on Migrate-N) to answer the question whether cats from Asia arrived prior to European settlements. We fully agree with Andrew et al. that this approach is not designed to study temporal aspects of migration routes. However, neither in the Introduction, nor in the Discussion, have we claimed that we intended to differentiate among current and past gene flow. Instead we studied the structure of gene flow routes. Furthermore, we based our overall interpretation of the invasion history of cats on a variety of analyses and datasets (e.g. phylogenetic tree, migration model selection approach, microsatellite data and analyses, and historical data). Any interpretation related to samples with a low number of individuals (e.g. Asia) have been adequately discussed.
The second aspect of concern is related to the analysis of DNA microsatellite data using the program STRUCTURE [20]. We agree with Andrew et al. that longer runs and more replication and most importantly adding a reanalysis using only a fraction of the samples (one cluster at K = 5) does result in an additional substructure (three groups instead of one). The outcome of this analysis, however, does not alter the main conclusion of the manuscript, rather adds a minor aspect to the overall picture. In addition, if such an ‘hierarchical STRUCTURE analysis’ is applied then q thresholds as well as alternative methods (e.g. BAPS) are required (see e.g. [28]).
In summary, we acknowledge that in addition to our results on the voyage of cats to Terra Australis [1, 29, 30] there may be differentiation that we did not detect among Australian mainland feral cat populations and that feral cat populations from Tasman and Flinders island might show differences from those of Cocos (Keeling) islands that we did not detect.
Abbreviations
- K :
-
The number of populations assumed or estimated in analysis of genetic data by the program STRUCTURE
References
Koch K, Algar D, Searle J, Pfenninger M, Schwenk K. A voyage to Terra Australis: human-mediated dispersal of cats. BMC Evol Biol. 2015;15(1):262.
Algar D, Burrows N. Feral cat control research: Western Shield review-February 2003. Conserv Sci W Aust. 2004;5(2):131–63.
McGregor H, Legge S, Jones ME, Johnson CN. Feral cats are better killers in open habitats, revealed by animal-borne video. PLoS ONE. 2015;10(8):e0133915.
Doherty TS, Davis RA, van Etten EJB, Algar D, Collier N, Dickman CR, Edwards G, Masters P, Palmer R, Robinson S. A continental-scale analysis of feral cat diet in Australia. J Biogeogr. 2015;42(5):964–75.
Spencer PBS, Yurchenko AA, David VA, Scott R, Koepfli K-P, Driscoll C, O’Brien SJ, Menotti-Raymond M. The population origins and expansion of feral cats in Australia. J Hered. 2015;107(2):104–14.
Abbott I. Origin and spread of the cat, Felis catus, on mainland Australia, with a discussion of the magnitude of its early impact on native fauna. Wildlife Res. 2002;29(1):51–74.
Abbott I. The spread of the cat, Felis catus, in Australia: re-examination of the current conceptual model with additional information. Conserv Sci W Aust. 2008;7:1–17.
Beerli P, Felsenstein J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci U S A. 2001;98(8):4563–8.
Beerli P, Palczewski M. Unified framework to evaluate panmixia and migration direction among multiple sampling socations. Genetics. 2010;185(1):313–26.
Bradic M, Beerli P, García-de León FJ, Esquivel-Bobadilla S, Borowsky RL. Gene flow and population structure in the Mexican blind cavefish complex (Astyanax mexicanus). BMC Evol Biol. 2012;12(1):1–17.
Lilith M, Calver M, Styles I, Garkaklis M. Protecting wildlife from predation by owned domestic cats: Application of a precautionary approach to the acceptability of proposed cat regulations. Austral Ecol. 2006;31(2):176–89.
Toribio J-ALM, Norris JM, White JD, Dhand NK, Hamilton SA, Malik R. Demographics and husbandry of pet cats living in Sydney, Australia: Results of cross-sectional survey of pet ownership. J Feline Med Surg. 2009;11(6):449–61.
Beerli P. Effect of unsampled populations on the estimation of population sizes and migration rates between sampled populations. Mol Ecol. 2004;13(4):827–36.
Kuhner MK. LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics. 2006;22(6):768–70.
Hey J. Isolation with migration models for more than two populations. Mol Biol Evol. 2010;27(4):905–20.
Lopes JS, Balding D, Beaumont MA. PopABC: a program to infer historical demographic parameters. Bioinformatics. 2009;25(20):2747–9.
Csilléry K, Blum MGB, Gaggiotti OE, François O. Approximate Bayesian Computation (ABC) in practice. Trends Ecol Evol. 2010;25(7):410–8.
Driscoll CA, Menotti-Raymond M, Roca AL, Hupe K, Johnson WE, Geffen E, Harley EH, Delibes M, Pontier D, Kitchener AC, et al. The Near Eastern origin of cat domestication. Science. 2007;317(5837):519–23.
Lipinski MJ, Froenicke L, Baysac KC, Billings NC, Leutenegger CM, Levy AM, Longeri M, Niini T, Ozpinar H, Slater MR, et al. The ascent of cat breeds: genetic evaluations of breeds and worldwide random-bred populations. Genomics. 2008;91(1):12–21.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–87.
Gilbert KJ, Andrew RL, Bock DG, Franklin MT, Kane NC, Moore J-S, Moyers BT, Renaut S, Rennison DJ, Veen T, et al. Recommendations for utilizing and reporting population genetic analyses: the reproducibility of genetic clustering using the program STRUCTURE. Mol Ecol. 2012;21(20):4925–30.
Koch K, Algar D, Searle J, Pfenninger M, Schwenk K. A voyage to Terra Australis: human-mediated dispersal of cats. Dryad Digit Repository. 2015. http://dx.doi.org/10.5061/dryad.6t066.
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I. Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour. 2015;15(5):1179–91.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Bianco E, Soto HW, Vargas L, Pérez-Enciso M. The chimerical genome of Isla del Coco feral pigs (Costa Rica), an isolated population since 1793 but with remarkable levels of diversity. Mol Ecol. 2015;24(10):2364–78.
Racimo F, Sankararaman S, Nielsen R, Huerta-Sanchez E. Evidence for archaic adaptive introgression in humans. Nat Rev Genet. 2015;16(6):359–71.
Vaha JP, Erkinaro J, Niemela E, Primmer CR. Life-history and habitat features influence the within-river genetic structure of Atlantic salmon. Mol Ecol. 2007;16(13):2638–54.
Koch K, Algar D, Schwenk K. Feral Cat Globetrotters: genetic traces of historical human-mediated dispersal. Ecol Evol. 2016. doi:10.1002/ece3.2261.
Koch K, Algar D, Schwenk K. Population structure and management of invasive cats on an Australian Island. J Wildl Manag. 2014;78(6):968–75.
Earl DA, von Holdt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2011;4(2):359–61.
Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23(14):1801–6.
Authors’ contributions
KK and KS equally contributed to the writing of this correspondence. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Andrew, R.L., Smith, D., Gorrell, J.C. et al. Voyage of discovery? A comment on Koch et al. “A voyage to Terra Australis: human-mediated dispersal of cats”. BMC Evol Biol 16, 270 (2016). https://doi.org/10.1186/s12862-016-0813-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-016-0813-y