
Abstract
Background
The evolutionary origin and historical demography of extant Arcto-Tertiary forest species in East Asia is still poorly understood. Here, we reconstructed the evolutionary and population demographic history of the two extant Euptelea species in China (E. pleiosperma) and Japan (E. polyandra). Chloroplast/nuclear DNA sequences and microsatellite loci were obtained from 36 Euptelea populations to explore molecular structure and diversity in relation to past and present distributions based on ecological niche modelling (ENM). Time-calibrated phylogenetic/phylogeographic inferences and niche-identity tests were used to infer the historical process of lineage formation.
Results
Euptelea pleiosperma diverged from E. polyandra around the Late Miocene and experienced significant ecological differentiation. A near-simultaneous diversification of six phylogroups occurred during the mid-to-late Pliocene, in response to the abrupt uplift of the eastern Tibetan Plateau and an increasingly cooler and drier climate. Populations of E. pleiosperma seem to have been mostly stationary through the last glacial cycles, while those of E. polyandra reflect more recent climate-induced cycles of range contraction and expansion.
Conclusions
Our results illustrate how Late Neogene climatic/tectonic changes promoted speciation and lineage diversification in East Asia’s Tertiary relict flora. They also demonstrate for the first time a greater variation in such species’ responses to glacial cycles in Japan when compared to congeners in China.
Similar content being viewed by others

Background
Because of the lack of extensive glaciations there during the Quaternary [1], the warm temperate climate zones of East Asia are considered to be one of the most important areas for the long-term persistence of Arcto-Tertiary forest flora [2]. In fact, although there are c. 600 genera of the Arcto-Tertiary relict flora endemic to East Asia (e.g. Cercidiphyllum, Davidia, Euptelea, Ginkgo biloba, Tetracentron), their evolution has been less well elucidated [3, 4]. Our limited understanding of the evolution of these Arcto-Tertiary plants is partly attributable to the scarce vascular plant fossils in the Tertiary and Quaternary beds of East Asia [5]. In addition, Arcto-Tertiary relict species have persisted with little morphological change [6], which might mask more recent diversification events. Finally, the modern patterns of endemism and disjunction of these relicts (e.g. between China and Japan) provide only limited insight into how past changes in climate, tectonics, and/or sea-level influenced their range dynamics and population/species divergence [7–10].
Nonetheless, recent molecular phylogeographic studies suggest that most Arcto-Tertiary relicts endemic to China have been little affected by Quaternary glacial-interglacial alternation [4, 11, 12]. In those instances, species likely tracked their climatic niche boundaries locally through altitudinal migration in a topographically complex landscape [13–15]. As a possible exception, the Chinese-Japanese disjunct Cercidiphyllum japonicum responded to Quaternary climate change through latitudinal range shifts, in both regions, which likely reflects the species’ high(er) pollen/seed dispersal capacity, sprouting ability, and/or climatic (drought/cold) tolerance [3]. Hence, these different responses may be largely explained by taxon-specific differences in migratory capacity and/or eco-climatic niche preferences [13, 16, 17]. However, additional studies are needed to gain a better understanding of when Arcto-Tertiary relict plants in East Asia diversified and whether and how they have responded to Late Neogene/Quaternary environmental change in the extent and connectivity of their habitats.
Euptelea Sieb. et Zucc., is the only genus in Eupteleaceae, comprising two extant species, E. pleiosperma Hook. f. et Thoms. and E. polyandra Sieb. et Zucc. [18, 19] (Additional file 1: Figure S1a). Both are diploid (2n = 28) [20, 21], small- to medium-sized, broad-leaved deciduous trees with bisexual and wind-pollinated flowers that turn into winged fruitlets (‘samaras’), dispersed by gravity, wind and/or water [22]. E. pleiosperma has its main centre of distribution in regions bordering the eastern margin of the Qinghai-Tibetan Plateau (QTP)/Hengduan Mts. and southwest China (Sichuan Basin/Yungui Plateau), with scattered occurrences in central/east China and northeast India/Assam; its populations are distributed in isolated stands across a wide range of altitudes (c. 700–3600 m above sea level) in mountain riparian warm-temperate deciduous (hereafter ‘WTD’) forest [23]. By contrast, E. polyandra is restricted to the riparian WTD forest of south-central Japan (c. 100–1600 m above sea level) [24–27] (Additional file 1: Figure S1a).
Euptelea has extensive fossil records throughout the Northern Hemisphere, extending to at least the Palaeocene [28–32]. In East Asia, Euptelea-like fossils have been reported from the Mid-Miocene of southeast China [33] and from the Pliocene to the Mid-Pleistocene of central Japan [2, 34, 35] (Additional file 1: Figure S1b). Although E. polyandra had initially been classed as a member of Trochodendraceae [36], Smith [37] incorporated this species into the unigeneric family Eupteleaceae, consisting of only E. pleiosperma at that time. Previous phylogenetic analyses [38] supported E. pleiosperma and E. polyandra as separate species, but only two samples from each species were included. Moreover, Euptelea is part of the earliest-diverging lineage of Ranunculales (along with Circaeasteraceae, Papaveraceae, Lardizabalaceae, Menispermaceae, Berberidaceae, Ranunculaceae), which, in turn, appear at the base of eudicots [38–40]. However, no information is available on the divergence times between and within the two species, to say nothing about their genetic architecture, demographic history, and ecological (climatic) niche divergence. Notably, this latter aspect has rarely been examined in phylogeographic studies of East Asia’s temperate flora in general, and its Tertiary relict components in particular [3].
Here, we use an integrative approach by combining phylogeographic and fossil-calibrated phylogenetic analyses with (palaeo)climatic data and niche identity tests to clarify the time and mode of lineage divergence and historical demography of Euptelea. Our specific aims were to (i) reveal the genetic relationships of the two extant species (e.g. are they recognizable as separate genetic entities?), and approximate divergence times of interspecific and intraspecific lineages; (ii) determine climatic niche divergence of both species and the two major lineages identified in E. pleiosperma; and (iii) illustrate the genetic diversity and population demographic patterns of both species.
Methods
Population sampling
Leaf material was obtained from 26 populations (n = 350) of E. pleiosperma and 10 populations (n = 90) of E. polyandra (Additional file 2: Table S1, Additional file 1: Figure S1a), covering most of the geographical range of the genus in East Asia except northeast India/Assam [37]. Fresh leaves were collected from each individual and dried with silica gel.
DNA isolation, sequencing and microsatellite genotyping
Total genomic DNA was extracted from the silica-dried leaf tissue using DNA Plantzol (Invitrogen) following the manufacturer’s protocol. For the phylogeographic DNA surveys, two intergenic spacer (IGS) regions (psbA–trnH, rpoB–trnC) and the rpL16 intron of chloroplast (cp) DNA, revealing high levels of variation in a preliminary screen, and the entire internal transcribed spacer (ITS) region of nuclear ribosomal (nr) DNA were selected for sequencing. Subsets of the samples were sequenced at cpDNA regions (n = 204 and 82 for E. pleiosperma and E. polyandra, respectively; Additional file 2: Table S1) and ITS region (n = 113 and 48 for E. pleiosperma and E. polyandra, respectively; Additional file 2: Table S1). A subset of individuals was also sequenced at three additional cpDNA regions (petN–trnC, matK, rbcL) for phylogenetic analysis (35 individuals, representing 35 chlorotypes; see the Results section). The primers and methodology for PCR amplification of these cpDNA and ITS regions were described elsewhere [41–44]. The PCR products were directly sequenced in both directions with an ABI 3730XL DNA Analyzer (Applied Biosystems, Foster City, CA, USA). The raw sequence data were adjusted, assembled and aligned in geneious v.4.8.5 [45]. All cpDNA and ITS haplotype sequences have been deposited in GenBank (see Additional file 3: Table S2 for accession numbers).
Following Zhang et al. [46] and Wu et al. [47], eight nSSR markers (EP04, EP06, EP10, EP59, EP87, EP91, EP278, EP294) and amplification protocols, specially developed for Euptelea, were used for genotyping all samples (n = 440). The PCR products were also loaded on an ABI 3730XL DNA Analyzer, and the data were scored and compiled using genemarker v.2.2.0 (SoftGenetics, State College, PA, USA).
Phylogeographic analyses of cpDNA, ITS, and nSSRs
For both cpDNA and ITS, all indels and inversions were treated as single mutation events, and coded as substitutions with equal weight [48]. Haplotype (h) and nucleotide (π) diversities [49] were calculated for each population (h S , π S) and species (h T , π T) using dnasp v.5.1 [50]. tcs v.1.21 [51] was used to estimate an unrooted network of Euptelea haplotypes that illustrates all linkages with a > 95 % probability of being most parsimonious. Analysis of molecular variance (AMOVA) in arlequin v.3.1 [52] was performed using Φ-statistics to quantify the composition of total genetic (cpDNA, ITS) variance, with significance of fixation indices tested using 10,000 permutations [53]. Population differentiation for ordered (N ST) and unordered (G ST) haplotypes (cpDNA) were obtained with permut [54, 55] to test whether N ST is significantly larger than G ST (1000 permutations), indicating the presence of phylogeographic structure.
For the nSSR dataset, one E. pleiosperma and two E. polyandra populations with small sample sizes (n < 8) were removed from all population-level analyses of genetic diversity and differentiation, limiting those analyses to 33 populations (marked with an asterisk in Additional file 2: Table S1). All eight loci were checked for frequencies of null alleles using the Expectation Maximization (EM) algorithm [56] implemented in freena [57]. We measured unbiased genetic diversity [58, 59] across all loci for all populations with n ≥ 8, and calculated their allelic (A R) and private allelic (PA R) richness (standardized for 8 individuals using rarefaction) in hp-rare v.1.1 [59]. fstat v.2.9.3 [60] was used to estimate the total number of detected alleles (N A), gene diversity (H S), and genetic differentiation (F ST) [61] between populations (n ≥ 8) per-locus and overall.
Bayesian clustering of individuals was performed in structure v.2.3.4 [62, 63] for the entire nSSR dataset (i.e. 36 populations) and each species. We used the admixture model without prior information on population membership, and assumed independent allele frequencies among populations. The number of clusters (K) was set to vary depending on the data set. For each value of K, we conducted 10 independent simulations with both a burn-in and run length of 100,000 Markov chain Monte Carlo (MCMC) replications. The number of gene pools (i.e. the optimal K) was inferred by estimating lnP(D) [64] and ΔK [65]. AMOVAs were carried out in arlequin with R-statistics as described above.
Phylogenetic divergence time estimation and demographic inference
To relate the diversification of major cpDNA lineages of Euptelea to Neogene events, we adopted a two-step approach for estimating divergence times in beast v.1.7.5 [66] by taking advantage of calibration points used in previous studies of Ranunculales [38, 67–69]. Taxon sampling has been shown to affect dates among basal Eudicots; increased taxon sampling, particularly in underrepresented clades, will likely improve the accuracy of divergence time estimation [70]. In this study, 33 Ranunculales species, including 13 species of Ranunculaceae, 13 of Berberidaceae and 5 of Menispermaceae (core Ranunculales), and the two species of Eupteleaceae (basal Ranunculales), plus representatives of Sabiaceae (Meliosma, Sabia) and Proteales (Nelumbo, Platanus) of basal eudicots used as outgroups (Additional file 3: Table S2) [38], were selected for estimating the crown group age of Euptelea. These selected species thus represent all major lineages of Ranunculales based on more broadly sampled earlier phylogenetic studies [38]. First, we retrieved sequences of two cpDNA regions (rbcL, matK) and 26S nrDNA from GenBank for the 33 Ranunculales species. Sequences were assembled together and then Bayesian searches for tree topologies and node ages of this cpDNA + 26S nrDNA dataset were implemented in beast with a GTR + G + I substitution model selected by jmodeltest v.2.1.4 [71], and an uncorrelated lognormal relaxed clock [72]. A Yule process was chosen as tree prior. Four calibration points were used to determine minimum age constraints on specific node priors (see nodes 1–4 in Fig. 4a). The first calibration point is based on the oldest confirmed fruit fossil of Sabiaceae from the Cretaceous in Czech Republic [73], which is unambiguously assigned to Insitiocarpus moravicus. Following Anderson et al. [67], we assigned this fruit fossil to the stem of Sabiaceae (98 Ma; see node 1 in Fig. 4a). The flower fossil of Platanocarpus brookensis from the Mid-Cretaceous in North America [74] was taken as the second calibration point and was used to calibrate the crown age of Nelumbo and Platanus (98 Ma; see node 2 in Fig. 4a) [69]. Following Anderson et al. [67], we used the endocarp fossil of Prototinomiscium (Menispermaceae) from the Cretaceous of central Europe [73, 75] to calibrate the crown age of the Menispermeaceae-Berberidaceae-Ranunculaceae clade (91 Ma; see node 3 in Fig. 4a). Finally, following Wang et al. [68], we used a leaf fossil of Mahonia from the Mid-Eocene in North America [5] to calibrate the crown age of the Ranzania-Mahonia-Berberis clade (45 Ma; see node 4 in Fig. 4a). All calibration points were modeled using a lognormal prior with a zero offset 98, 98, 91 and 45 Ma, respectively; by default, lognormal mean of 0 and lognormal SD of 0.5 were used to constrain each node (see nodes 1–4 in Fig. 4a). To evaluate the robustness of the resulting age estimates, we removed each calibration sequentially while holding the rest three calibrations [76].
In a second step, we applied a similar beast analysis to all 35 cpDNA (psbA–trnH, rpoB–trnC, rpL16 intron, petN–trnC, matK, rbcL) haplotype sequences of Euptelea identified in the phylogeographic survey (see the Results section; Fig. 1) to determine intrageneric node ages, with Platanus occidentalis used as outgroup (Fig. 4b). We employed the same settings as in the first step, but for a GTR + G substitution model and a constant-size coalescent tree prior. To calibrate the root node (see node B in Fig. 4b), we used the median crown age of Euptelea (5.46 Ma) estimated from the Ranunculales phylogeny (see node A in Fig. 4a) with a normal distribution (mean, 5.46 Ma; SD, 2.5 Ma; 95 % confidence interval [CI], 1.35–9.58 Ma). For both beast analyses, MCMC runs were conducted, each of 2 × 107 generations, with trees sampled every 2000 generations, after the initial 10 % were discarded as burn-in. The MCMC samples were examined in tracer v.1.5 [77] to confirm sampling adequacy and convergence of the chains to a stationary distribution.

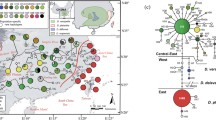
a Distribution of chlorotypes in 26 populations of E. pleiosperma (China) and 10 populations of E. polyandra (Japan) (see Additional file 2: Table S1 for population codes). Chlorotypes shared among populations are denoted by colour, while population-specific ones (‘E’) are white. The red and black dashed lines represent, respectively, the Sino-Himalayan/Sino-Japanese Forest boundary and the species’ boundary across the East China Sea region. The black dotted lines delimitate the four phylogroups of E. pleiosperma (i.e. population groups that share closely related chlorotypes). The black dotted line in Japan marks the boundary between ‘southern’ and ‘central’ phylogroups in E. polyandra. Abbreviations: TGMR, Three Gorges Mountain Region. b Dark-shaded areas in the inset indicate current mainland and island configurations, and light-shaded areas indicate exposed coastal areas and sea basins of East Asia during Late Pleistocene sea-level alterations owing to glaciations (modified after Park) [125]. c tcs-derived network of genealogical relationships between the 35 chlorotypes of the two species. Each circle denotes a single chlorotype with size proportional to frequency. Small open circles represent missing chlorotypes. The four clades (phylogroups) of E. pleiosperma identified by tcs were denoted as C1, C2, C3, C4, while those two clades (phylogroups) of E. polyandra were represented by J1 and J2. The baseline map was created by us using ArcGIS v.10.2.2
For each species, and major (cpDNA) phylogroups (see the Results section), we constructed Bayesian skyline plots (BSPs) in beast to date changes in effective population size (N e) over time [78]. We assumed an uncorrelated lognormal relaxed clock with substitution rates obtained from the intrageneric beast analyses described earlier. The MCMCs were run for 1 × 107 generations with trees sampled every 1000 generations, following a burn-in of the initial 10 % cycles. Three replicate runs were performed to confirm convergence.
Ecological niche modelling and niche identity tests
ENMs were generated using the maximum entropy method implemented in maxent v.3.2.1 [79] to predict the potentially suitable climate envelopes for each Euptelea species in the present, and in two past periods: the Last Glacial Maximum (LGM), c. 21 kya; and the Last Interglacial (LIG), c. 130 kya. Information on the geographic distribution of Euptelea was based on a total of 470 presence records, including 99 for E. pleiosperma [i.e. 26 sample sites of this study plus 73 records from the Chinese Virtual Herbarium (http://www.cvh.org.cn)] and 371 for E. polyandra [i.e. 10 sample sites of this study and 361 records from the S-Net data portal (http://science-net.kahaku.go.jp/)], with the removal of duplicate records within each pixel (2.5 arc-min; c. 5 km). We compiled 19 environmental variables for each period from the WORLDCLIM database with a resolution of 2.5 arc minutes [80] for each environment layer (Additional file 2: Table S1). Based on 99 records for E. pleiosperma and 371 records for E. polyandra, a current distribution model was developed for each species using five bioclimatic data layers (annual mean temperature, annual precipitation, temperature annual range, mean temperature of wettest quarter, and precipitation seasonality) at 2.5 arc-minutes resolution for the present (1950–2000). This limited bioclimatic dataset avoided containing highly correlated variables (data not shown), and therefore prevented potential overfitting [81]. By adopting default settings with 10 replicates, we used 75 % of species records to train the model and 25 % to test the model. The area under the ROC curve (AUC) was calculated for each run to assess the model accuracy. Values between 0.7 and 0.9 indicate good discrimination [82]. We then projected the established model onto the reconstructed climatic conditions for the LGM and LIG periods, as simulated by the community climate system model v.3.0 [83], provided by the Palaeoclimate Modelling Intercomparison Project (http://pmip2.lsce.ipsl.fr/). Following Sakaguchi et al. [84], the present-day and LGM palaeoclimate layers were prepared in 2.5 arc-minutes resolution. To account for changes in palaeo-coastlines in the East China Sea (ECS) region during the LGM (c. -130 m lower than at present) [85], we used the seafloor topography data (ETOPO1) from the National Geophysical Data Center of National Oceanic and Atmospheric Administration (NOAA, Washington, DC, USA).
To examine potential environmental factors associated with the divergence between E. pleiosperma and E. polyandra (and between two major cpDNA lineages of the former species; see Results section), the means and standard errors of those 19 bioclimatic variables were then calculated for each species (lineage) and compared to detect significant differences using two-tailed t-tests in R v.3.1.1 [86]. We also performed niche identity tests [87] in enmtools v.1.4.3 [88] with 500 pseudoreplicates based on (i) the five bioclimatic variables used for ENM (see above); and (ii) all the 19 BIOCLIM variables together, for testing the null hypothesis that the two species (lineages) are occupying identical climatic environments (‘niches’). To this aim, we compared their actual niches to a distribution of niche similarities obtained from pairs of pseudoniches based on randomly sampled occurrence points [89]. Niche overlap was quantified by both Schoener’s D [90] and the standardized Hellinger distance (I).
Results
CpDNA phylogeography and diversity
The total alignment of three cpDNA regions sequenced across the 286 individuals of Euptelea was 2078 bp in length, including 25 single-nucleotide variable sites, five indels (5–18 bp), and one inversion (23 bp; Additional file 4: Table S3). Together, these 31 polymorphisms identified 35 haplotypes (‘chlorotypes’, H1–35), of which 25 were specific to E. pleiosperma (H1–25) and 10 to E. polyandra (H26–35; Fig. 1a). Of the 35 chlorotypes identified in Euptelea, 24 were population-specific and nearly half of the populations of each species were fixed for a single chlorotype (Fig. 1a, Additional file 2: Table S1). Accordingly, non-hierarchical AMOVA (Table 1) revealed that most of the cpDNA sequence variation within each species resided among populations (E. pleiosperma/E. polyandra: Φ ST = 0.87/0.75; both P < 0.01), and this variation exhibited significant phylogeographic structure (E. pleiosperma/E. polyandra: N ST = 0.847/0.879 > G ST = 0.835/0.836; both P < 0.05). In the tcs network (Fig. 1b), these species-specific chlorotypes formed two clades separated by thirteen mutational steps (between H25 and H30). This pronounced subdivision was also indicated by hierarchical AMOVA with 72.52 % among-species variation (Table 1).
Among the 25 chlorotypes of E. pleiosperma, eight (H1–8) were specific to a group of six populations (JZ, BM, SJ, DY, TQ, EM) from the region bordering the eastern QTP/Hengduan Mts. (hereafter ‘C1’; see dotted lines in Fig. 1a). By contrast, the remaining chlorotypes were largely specific to three ‘central-eastern’ regions: (i) the northern and, in particular, southern flanks of the Sichuan Basin, including the Yungui Plateau (‘C4’: H10–11, H14–17); (ii) the mid-lower reaches of the Yangtze (‘C2’: H18–22); and (iii) areas north of this river (‘C3’: H23–25). The parsimony network (Fig. 1b) grouped these chlorotypes into two lineages (western vs central/eastern) and four distinct phylogroups (i.e. C1–C4) according to geography; the only exceptions were H9 and H13, which resided in a single population (TS) from the Sichuan Basin (hereafter ‘SB’) region (Fig. 1a and b; Additional file 2: Table S1), however, were assigned to two distinct phylogroups (C1 and C2, respectively). In E. polyandra, the two distinct phylogroups (J1 vs J2) identified segregated populations into the southern and central parts of Japan (i.e. Kyushu, Shikoku, south Honshu/Chubu vs north Honshu; see Fig. 1a, b).
ITS phylogeography and diversity
The total alignment of the ITS region sequenced across the 161 individuals of Euptelea was 691 bp in length, including 18 single-nucleotide variable sites. These polymorphisms yielded 10 ribotypes (R1–10; Additional file 5: Table S4), with five each specific to E. pleiosperma (R1 − 5) and E. polyandra (R6–10). The parsimony network (Fig. 2) grouped E. pleiosperma vs E. polyandra ribotypes into distinct clades (‘lineages’), separated by nine mutational steps, resulting in 80.08 % among-species variation (Φ CT = 0.80, P < 0.01; Table 1). In E. pleiosperma, the most basal ribotype (R1) was also the most frequent and widespread one, being fixed in 14 (out of 26) populations, while in E. polyandra almost all populations (8/10) were polymorphic. In contrast to cpDNA, most of the within-species ITS variation resided within populations (E. pleiosperma/E. polyandra: Φ ST = 0.36/0.31, both P < 0.01; Table 1).

a Distribution of ITS ribotypes in the 36 populations of Euptelea (see Additional file 2: Table S1 for population codes). Red and black dashed lines are identified in Fig. 1. b tcs-derived network of genealogical relationships between the 10 ribotypes of the two species. Each circle denotes a single ribotype, with size proportional to frequency. Small open circles represent missing ribotypes
Species subdivision and population genetic structure at nSSR loci
Across the 440 individuals of Euptelea genotyped at eight nSSR loci, we obtained altogether 150 alleles, ranging from 7 to 29 alleles per locus (for details see Additional file 6: Table S5). For the entire dataset, the ΔK statistic detected a rate change in lnP(D) corresponding to K = 3 (Additional file 7: Figure S2). Accordingly, individuals of E. pleiosperma segregated into cluster I (Fig. 3a and b) that was present at high frequency in western populations (except JZ) but also fixed in three populations (DF, HS, YN) from the southern SB region, while the rest of the central-eastern individuals were largely assigned to cluster II. By contrast, individuals of E. polyandra almost exclusively formed a distinct cluster (Fig. 3a and b). A separate structure analysis of E. pleiosperma resulted in similar patterns, while for E. polyandra individuals were variously assigned to three clusters without geographical structure (data not shown).

a Colour-coded grouping of the 36 Euptelea populations according to the three structure clusters (I–III) (see Additional file 2: Table S1 for population codes). Red and black dashed lines are identified in Fig. 1. b Histogram of the structure analysis for the model with K = 3 (according to both lnP(D) and ΔK; see Additional file 7: Figure S2). Each vertical bar represents one individual. The assignment ratio of each individual into one of the three clusters is shown along the y-axis. Each cluster is represented by a distinct colour
Hierarchical AMOVA (Table 1) supported significant nSSR variation between the two species (R CT = 0.23, P < 0.01). Within each species, most of this variation resided within populations, with somewhat stronger population subdivision in E. pleiosperma (R ST = 0.17, P < 0.01) than in E. polyandra (R ST = 0.10, P < 0.01).
Molecular dating and demographic analyses
The beast-derived cpDNA + 26S nrDNA chronogram of Ranunculales (Fig. 4a) recovered Euptelea as sister to core Ranunculales (PP = 0.81) and estimated the crown age of Euptelea as c. 5.46 Ma [95 % highest posterior density (HPD): 1.23–10.87 Ma; see node A in Fig. 4a] (Table 2). Using this estimation as root prior for the cpDNA chronogram of Euptelea (Fig. 4b), the time to the most recent common ancestor (TMRCA) of all 35 chlorotypes was estimated as c. 6.04 Ma (95 % HPD: 2.89–9.36 Ma; see node B in Fig. 4b), suggesting a Late Miocene/Early Pliocene split between the two species. The TMRCA for all chlorotypes of E. pleiosperma was estimated as c. 3.64 Ma (95 % HPD: 1.38–6.46 Ma; see node C in Fig. 4b), which in turn dates the species’ divergence into two lineages (see above) to around the Mid-Pliocene. The TMRCA for all chlorotypes of E. polyandra (viz. its divergence into J1 vs J2 phylogroups) was estimated to be only slightly younger, c. 3.20 Ma (95 % HPD: 1.34–5.56 Ma; see node D in Fig. 4b). By contrast, the coalescence times of all six phylogroups of Euptelea most likely fall into the Quaternary [C1: 1.66 (0.47–3.31) Ma; C2: 1.66 (0.42–3.50) Ma; C3: 1.15 (0.17–2.68) Ma; C4: 1.73 (0.48–3.47) Ma; J1: 1.50 (0.36–3.07) Ma; J2: 2.31 (0.88–4.13); see nodes C1–4 and J1–2 in Fig. 4b and Table 2].

a beast-derived chronogram of Ranunculales based on cpDNA (rbcL, matK) + 26S nrDNA sequences with calibration points denoted by nodes 1–4 (see text and Table 2 for details); and b the crown age of Euptelea and its phylogroups based on cpDNA (psbA-trnH, rpoB-trnC, rpL16, petN-trnC, matK, rbcL) sequences. Blue bars on each node indicate 95 % highest posterior density (HPD) confidence intervals for time estimates (in million yr ago, Ma). Posterior probabilities (PP > 0.5) are shown above nodes. Mean divergence times and 95 % HPDs are summarized in Table 2. Chlorotypes are represented by letter codes (H1–35). Codes of subclades (phylogroups) are identified in Fig. 1
For E. pleiosperma, the Bayesian skyline plots analysis (Additional file 8: Figure S3a) showed that N e kept steady growth up to the Late Pleistocene (c. 0.25 Ma), followed by stable population sizes until the present, while for each of the four phylogroups, N e kept steady through time (data not shown). By contrast, for E. polyandra and its two phylogroups, this analysis consistently indicated stable population sizes up until the Late Pleistocene (c. 0.5 Ma), followed by a slight growth until the present (Additional file 8: Figure S3b–d).
Ecological niche modelling and niche identity tests
The AUC values (± SD) for the distribution modelling, averaged over 50 replicates, were very high (0.958 ± 0.016 vs 0.971 ± 0.002 for E. pleiosperma and E. polyandra, respectively), indicating a good predictive model performance. The predicted distribution of Euptelea under current conditions (1950–2000) (Fig. 5a) was similar to its actual distribution, but there were also some predicted areas where either species does not occur at present, such as northern east China and south Korea (E. pleiosperma) or Hokkaido/north Honshu of Japan and central/east China (E. polyandra). During the LIG (c. 130 kya BP; Fig. 5c), the genus’ predicted distribution was more or less similar to that at present, but likely more favourable habitats especially for E. pleiosperma were in southwest China (Hengduan Mts.) and the surroundings of the Sichuan Basin. By contrast, at the LGM (c. 21 kya BP; Fig. 5b), the predicted suitable habitats of E. pleiosperma probably persisted around the latter basin, but also slightly expanded eastward, along the northern flanks of the middle-Yangtze River. At the same time, the predicted suitable habitats of E. polyandra became restricted to a narrow coastal strip along the Pacific Ocean side of southeast Japan. Notably, predicted suitable habitats likely existed for E. pleiosperma in the latter region as well, and conversely, for E. polyandra in central China (especially south of the middle-Yangtze River) and along the coasts of the exposed East China Sea continental shelf; however, for each species, hospitable habitats were apparently absent on the landbridge extending from east China across south Korea to south Japan (Fig. 5b).

Potential distribution probability (in logistic value) of occurrence for Euptelea in East Asia. a At the present (0 kya); b at the Last Glacial Maximum (LGM: c. 21 kya) and c during the Last Interglacial (LIG: c. 130 kya). Presence records of E. pleiosperma in China (n = 99) and E. polyandra in Japan (n = 371) are plotted as purple and dark blue points in the maps, respectively. See text for details
Out of the 19 bioclimatic variables, all but three (bio2: mean diurnal temperature range; bio8: mean temperature of wettest quarter; bio10: mean temperature of warmest quarter), showed significant differences in mean values between E. pleiosperma and E. polyandra (Additional file 9: Figure S4). Randomization tests of niche identity (based on nineteen bioclimatic variables or five non-correlated variables) indicated that these sister species are not ecologically equivalent (P < 0.01) (Fig. 6), regardless of the measure of similarity used (Schoener’s D or Hellinger’s I). Within E. pleiosperma, seven (out of 19) bioclimatic variables showed significant (P < 0.05) differences between the western and central-eastern cpDNA lineages, i.e. in terms of isothermality (bio3), temperature/precipitation seasonality (bio4/bio15), and aspects of temperature (bio6, bio7, bio9, bio11; see Additional file 10: Figure S5). However, niche identity tests could not reject the null hypothesis that the two lineages occupy identical climatic niches, as D and I were non-significant (P > 0.05) in either instance (Fig. 7).

Niche identity test plots for five non-correlated (a, b) and nineteen (c, d) BIOCLIM variables between the two species of Euptelea as quantified by the standardized Hellinger distance (I) and Schoener’s D [91]. The vertical line in each plot represents the observed values of niche similarity (both D and I) while the histograms represent those of null distributions

Results of the niche identity tests for five non-correlated (a, b) and nineteen (c, d) BIOCLIM variables between western and central-eastern populations of E. pleiosperma. The vertical line in each map represents the observed values of niche similarity (both D and I) while the histograms represent those of null distributions
Discussion
Late Neogene speciation and diversification of Euptelea
Our phylogenetic and phylogeographic analyses based on cytoplasmic and nuclear (ITS, nSSR) data (Figs. 1, 2, 3 and 4) clearly support the monophyly of the genus Euptelea and provide the first evidence for the genetic distinctiveness of E. pleiosperma (China) and E. polyandra (Japan). In addition, although these ecological differences (Additional file 9: Figure S4; Fig. 6) could reflect evolutionary divergence of both species, they might simply reflect the fact that populations of both species are largely allopatric and thus are exposed to different environmental backgrounds. Hence, these species are diagnosable by geographical, morphological, molecular and ecological criteria.
Based on our cpDNA chronogram (Fig. 4b), this speciation event occurred c. 6.04 (95 % HPD, 2.89–9.36) Ma. Notably, this point estimate broadly coincides with an early stage of East China Sea seafloor exposure between the Eurasian mainland and the Japanese/Ryukyu Islands (7.0–5.0 Ma) [91, 92]. Hence, this landbridge could have allowed a common ancestor to expand its range from China to Japan, followed by range fragmentation either due to an increasingly cooler and drier climate around that time in Asia (c. 7–5.3 Ma) [4, 93, 94] and/or a subsequent rise in sea level. Of course, this vicariant scenario has to be treated with caution because of broad confidence limits around the above point estimate (2.89–9.36 Ma; Fig. 4b). Nonetheless, our data clearly indicate a long period of independent evolution of the two species, which ensured almost complete lineage sorting at chloroplast and nuclear loci. In support of this, our ENM for the LGM (and possibly earlier, colder periods) predicted climatically suitable areas in central/east China for E. polyandra and in southeast Japan for E. pleiosperma, but no such areas were predicted for either species on the ECS landbridge (Fig. 5b). Therefore, the East China Sea landbridge likely acted as a formidable barrier to the dispersal of Euptelea species during the LGM and earlier cold periods, despite its repeated exposure during the Quaternary (2.0–1.3 Ma, and 0.2–0.015 Ma) [91, 92, 95].
There is increasing evidence to suggest that the East China Sea landbridge acted as a ‘filter’ for East Asia’s temperate flora in selectively preventing or facilitating the dispersal of certain plant species [10, 96]. For instance, genetic breaks across the East China Sea have been observed in several understorey herbs or shrubs of WTD forest (e.g. Kirengeshoma palmata; Platycrater arguta; Ligularia hodgsonii) [89, 96–98]. However, in sharp contrast, no genetic breaks associated with the East China Sea were found in two widespread WTD forest tree species, namely Cercidiphyllum japonicum [3] and Kalopanax septemlobus [85]. As with Euptelea spp., C. japonicum is also an Arcto-Tertiary relict of riparian plant communities with similar reproductive characteristics (e.g. wind-dispersed pollen and seeds, vegetative propagation) [22, 99, 100]. However, C. japonicum, which is presently found up to north Japan, is relatively tolerant to cold and arid climates, which might have facilitated gene exchange across the glacially exposed ECS landbridge up until its latest submergence (<16 000 year bp; Fig. 5c) [3]. By contrast, Euptelea is less tolerant to cold, more drought-sensitive, and also much weaker in terms of sprouting ability [26, 100, 101]. We therefore conclude that the East China Sea landbridge provided a far less suitable (albeit unknown) environment for Euptelea relative to Cercidiphyllum due to taxon-specific differences in climate-related niche requirements and/or other biological (e.g. recruitment) properties [96].
Our molecular dating analyses (see Fig. 4b; Table 2) indicate that the onset of diversification in E. pleiosperma (node C, c. 3.64, 95 % HPD, 1.38–6.46 Ma) and E. polyandra (node D, c. 3.20, 95 % HPD, 1.34–5.56 Ma) occurred at the mid-to-late Pliocene. These broad HPD ranges preclude us from explaining any of these splits in terms of particular geological or palaeoclimatic events. Despite these caveats, we propose that our dates are still broadly consistent with the mid-Pliocene abrupt uplift of the eastern Tibetan Plateau and adjacent southwest China (c. 3.4 Ma) [102], and the intensification of Northern Hemisphere Glaciation (3.2–2.5 Ma) [103]. These geological and/or climatic changes had possibly acted as an isolating barrier between regional populations (i.e. western China vs central/eastern China; south Japan vs central Japan), and promoted the diversification of two lineages of E. pleiosperma (western vs central/eastern) and two phylogroups of E. polyandra (J1 vs J2). In fact, such Late Neogene tectonic/climate-induced vicariance has also been invoked to explain phylogeographic splits in other Tertiary-relic deciduous tree species from East Asia (e.g. Cercidiphyllum spp.; Tetracentron sinense; Davidia involucrata; Sargentodoxa cuneata) [3, 4, 12, 104]. However, our estimated coalescent times of the various cpDNA phylogroups within E. pleiosperma (nodes C1 and C2: 1.66 Ma; C3: 1.15 Ma; C4: 1.73 Ma) and E. polyandra (node J1: 1.50 Ma; J2: 2.31 Ma) are generally more recent (Fig. 4b; Table 2), implying that this haplotype diversity and structure was most likely shaped by historical processes during the (Late) Quaternary (see below).
Relative demographic stability of E. pleiosperma in China
In contrast with the profound influence of Quaternary climatic fluctuations on the range dynamics of both Cercidiphyllum japonicum [3] and Sargentodoxa cuneata [104] in China, our results suggest a rather limited effect of such climate change on the demographic history and LGM distribution of E. pleiosperma in this region. This inference is based on three lines of evidence. First, E. pleiosperma exhibits strong phylogeographic structure in cpDNA (N ST = 0.847 > G ST = 0.835, P < 0.05), which largely reflects the presence of four montane phylogroups, one in the west (C1), and three (C2–4) in the Central-East (Figs. 1 and 4b). In fact, the only evident instances of inter-region seed flow pertain to the occurrence of both a western (H9) and C2 chlorotype (H13) in a single population (TS) from the SB region (Fig. 1); this is probably best explained by two independent migration events from the western/mid-lower Yangtze River regions, followed by the extinction of H9 and H13 in their respective source areas. Second, the BSP analysis for E. pleiosperma (Additional file 8: Figure S3a) indicates population growth up to the Late Quaternary (c. 0.25 Ma), followed by population stability until the present. Finally, our ENM for the LGM (Fig. 5b) suggests only slightly larger and more contiguous distributions of E. pleiosperma, particularly in areas around the Sichuan Basin and, to a lesser extent, along the northern flanks of the Yangtze River, when compared to the LIG or the present (Fig. 5a and c).
Together, these cpDNA data suggest that populations of E. pleiosperma largely persisted in their separate mountainous refugia over periods of Quaternary climate change up to the present [4, 12, 105]. Such population persistence might have been facilitated through periodic episodes of up-slope contraction during interglacials, and down-slope expansion during glacials, with populations tracking favourable humidity conditions as imposed by the East Asian monsoon in areas of high relief [106]. During glacials, genetic exchange between neighboring montane lineages might have occurred through lower-elevation populations, as has been suggested for other mid- and high-elevation plant and animal taxa elsewhere [107–110]. In fact, the majority of E. pleiosperma populations belonging to the three central-eastern phylogroups share the same nuclear (nSSR) gene pool (II), excepting three from the SB region (DF, HS, YN) that share gene pool I with most western populations (Fig. 3). This pattern of nuclear cohesion, which is also reflected in the ITS data (Fig. 2), is thus not entirely unexpected if we assume that inter-region gene exchange during glacials mainly occurred via pollen in low-elevation populations that were also probably the first to be extirpated during elevation shifts caused by postglacial climate warming [110, 111]. However, when compared with the situation in the Central-East, it also appears (at least from the nSSR data) that pollen less readily dispersed between the western and central-eastern phylogroups; this is most likely caused by topographical effects of the eastern QTP/Hengduan Mts. rather than the minor (and overall non-significant) differences in eco-climatic space found between these groups (Additional file 10: Figure S5; Fig. 7). Notably, this most evident genetic break within E. pleiosperma around the Sichuan Basin coincides with the boundary between the Sino-Himalayan and Sino-Japanese Forest subkingdoms [112], and is also apparent in several other plant taxa [10, 113, 114].
Quaternary range shifts and cryptic glacial survival of E. polyandra in Japan
Our cpDNA results show that E. polyandra is comprised of two phylogroups with unique sets of chlorotypes and distinct geographic distributions, one in south Japan (Kyushu, Shikoku, south Honshu/Chubu) and the other in central Japan (north Honshu) (Fig. 1a, b). Given that E. polyandra is a moisture-dependent and drought-sensitive species, the deterioration of Tertiary warm, moist climates during the mid-to-late Pliocene (see above) likely triggered ecological changes [115], and hence drove the diversification of E. polyandra. Accordingly, this phylogeographic break in central Honshu, which previously has also been observed in several other biota [96, 116–118], must have been maintained in spite of an apparently profound influence of Late Quaternary climate change on the species demographic history and distribution. The latter is inferred from two lines of evidence. The first is the BSP analysis, which indicates that E. polyandra (and its phylogroups) experienced long-term demographic stability before N e started to increase only relatively recently, c. 0.5 Ma (Additional file 8: Figure S3b–d). This population growth may have been triggered by climate change at the beginning of China’s ‘Penultimate Interglacial Period’ (c. 0.46–0.33 Ma) [119] and the intensification of the warm, wet summer monsoon in East Asia since the mid-late Pleistocene (c. 1.0–0.78 Ma) [120]. For moisture-dependent island trees, such as E. polyandra [99], this may have also led to an increase of suitable habitats throughout the warmer periods of the Late Quaternary. The second line of evidence comes from our ENM (Fig. 5), suggesting that, during the LGM, E. polyandra experienced a dramatic contraction of its (present-like) LIG range into a narrow coastal strip along the Pacific Ocean side of southeast Japan (Fig. 5). Notably, the single population surveyed from Kyushu (MI) harbours an phylogenetically basal chlorotype (H31) that also predominates in south Honshu, whereas its mutational derivative (H34) is private to Shikoku (Fig. 1a and b). It is feasible, therefore, that southeast Japan (most likely Kyushu) provided the source populations for postglacial (re-)colonization of these latter areas. Supportive evidence for this scenario comes from the fossil pollen record, demonstrating an increase of WTD forest in south Honshu/Chugoku during early postglacial times [121]. However, in contrast to the ENM results, there are two lines of genetic evidence for the glacial survival of E. polyandra in north Honshu: first, this region exclusively harbours private cpDNA haplotypes (Fig. 1a and b), and second, based on the nSSRs, there is no decreasing trend of genetic diversity (in terms A R, PA R) compared with areas further south (see Additional file 2: Table S1), as would be expected under a leading edge model of northward colonization [122]. Rather, these data suggest that extant populations in north Honshu are derived from a cryptic glacial refuge that occurred within local favourable microclimates [123], but which cannot be detected by ENM-based palaeodistribution reconstructions [85, 124]. Evidently, both nuclear markers (nSSR, ITS) failed to detect this refuge, and hence the south-central divide of E. polyandra in cpDNA (Figs. 1, 2 and 3). It appears unlikely that this discordance reflects incomplete nuclear lineage sorting, given the timeframe of the phylogroup divergence. Rather, a more tenable explanation is that the species’ range-wide nuclear cohesion results from periodic secondary contact and admixture via pollen during inter-/postglacials.
Conclusions
The speciation and lineage diversification events of Euptelea reflect the primary influence of successive climate/tectonic-induced vicariance during the Late Miocene and Pliocene periods. By contrast, the haplotype diversity and structure at the within-group level of each Euptelea species are most likely shaped by the climatic cycles of the (Late) Quaternary. During this time, E. pleiosperma populations seem to have been mostly stationary within their multiple mountain refugia, whereby topographical constraints to seed/pollen dispersal fostered the divergence among ‘western’ and ‘central-eastern’ cpDNA phylogroups. Contemporaneously, E. polyandra populations in south Japan apparently underwent repeated range contractions/expansions, while those in central Japan may have persisted in a cryptic glacial refuge in north Honshu. Overall, our results (1) illustrate how Late Neogene climatic changes promoted speciation and lineage diversification in East Asia’s Tertiary relict flora; and (2) demonstrate for the first time a greater variation in such species’ responses to glacial cycles in Japan when compared to congeners in China. Overall, this study should contribute to a better understanding of the potential historical (e.g. climatic, geomorphological) processes that may have generally impacted the East Asia’s Arcto-Tertiary flora.
Availability of supporting data
DNA sequences: see Genbank accessions for E. pleiosperma and E. polyandra in Additional file 3: Table S2 in supporting information). In addition, The sequence alignments and phylogenetic trees were deposited in TreeBASE (http://www.treebase.org/) under the submission number 18919 (URL: http://purl.org/phylo/treebase/phylows/study/TB2:S18919).
References
Frenzel B, Pecsi B, Velichko AA. Atlas of Palaeoclimates & Palaeoenvironments of the Northern Hemisphere. Budapest: INQUA/Hungarian Academy of Sciences; 1992.
Manchester SR, Chen ZD, Lu AM, Uemura K. Eastern Asian endemic seed plant genera and their paleogeographic history throughout the Northern Hemisphere. J Syst Evol. 2009;47:1–42.
Qi XS, Chen C, Comes HP, Sakaguchi S, Liu YH, Tanaka N, Sakio H, Qiu YX. Molecular data and ecological niche modelling reveal a highly dynamic evolutionary history of the East Asian Tertiary relict Cercidiphyllum (Cercidiphyllaceae). New Phytol. 2012;196:617–30.
Sun YX, Moore MJ, Yue LL, Feng T, Chu HJ, Chen ST, Ji YH, Wang HC, Li JQ. Chloroplast phylogeography of the East Asian Arcto-Tertiary relict Tetracentron sinense (Trochodendraceae). J Biogeogr. 2014;41:1721–32.
Manchester SR. Biogeographical relationships of North American Tertiary floras. Ann Mo Bot Gard. 1999;86:472–522.
Nagalingum NS, Marshall CR, Quental TB, Rai HS, Little DP, Mathews S. Recent synchronous radiation of a living fossil. Science. 2011;334:796–9.
Qian H, Ricklefs RE. Large-scale processes and the Asian bias in species diversity of temperate plants. Nature. 2000;407:180–2.
Qian H, Ricklefs RE. Diversity of temperate plants in East Asia – Reply. Nature. 2001;413:130.
Harrison SP, Yu G, Takahara H, Prentice IC. Palaeovegetation: diversity of temperate plants in East Asia. Nature. 2001;413:129–30.
Qiu YX, Fu CX, Comes HP. Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol Phylogenet Evol. 2011;59:225–44.
Gong W, Chen C, Dobes C, Fu CX, Koch MA. Phylogeography of a living fossil: Pleistocene glaciations forced Ginkgo biloba L. (Ginkgoaceae) into two refuge areas in China with limited subsequent postglacial expansion. Mol Phylogenet Evol. 2008;48:1094–105.
Chen JM, Zhao SY, Liao YY, Gichira AW, Gituru RW, Wang QF. Chloroplast DNA phylogeographic analysis reveals significant spatial genetic structure of the relictual tree Davidia involucrata (Davidiaceae). Conserv Genet. 2015;16:583–93.
Hewitt GM. The genetic legacy of the Quaternary ice ages. Nature. 2000;405:907–13.
Willis KJ, Niklas KJ. The role of Quaternary environmental change in plant macroevolution: the exception or the rule? Philos Trans R Soc Lond B Biol Sci. 2004;10:159–72.
Graham CH, VanDerWal J, Phillips SJ, Moritz C, Williams SE. Dynamic refugia and species persistence: tracking spatial shifts in habitat through time. Ecography. 2010;33:1062–9.
Davis MB, Shaw RG. Range shifts and adaptive responses to Quaternary climate change. Science. 2001;292:673–9.
Gavin DG, Fitzpatrick M, Gugger PF, Heath KD, Rodríguez-Sánchez F, Dobrowski SZ, Hampe A, Hu FS, Ashcroft MB, Bartlein PJ. Climate refugia: joint inference from fossil records, species distribution models and phylogeography. New Phytol. 2014;204:37–54.
Fu DZ, Endress PK. Eupteleaceae. In: Wu ZY, Raven PH, Hong D, eds. Flora of China, vol. 6. Beijing, China: Science Press; St. Louis, MO, USA: Missouri Botanical Garden Press; 2001. 123.
Ohaba H. Eupteleaceae. In: Iwatsuki K, Boufford DE, Ohaba H, editors. Flora of Japan, vol. IIb. Tokyo: Kodansha; 2001. p. 256.
Ratter JA, Milne C. Chromosome counts in primitive angiosperms: II. Edinburgh: Notes from the Royal Botanic Garden; 1976. p. 143–5.
Pan KY, Lu AM, Wen J. Chromosome number and development of gametophytes in Euptelea pleiospermum (Eupteleaceae). Acta Phytotax Sin. 1991;29:439–44.
Wei XZ, Jiang MX. Limited genetic impacts of habitat fragmentation in an “old rare” relict tree, Euptelea pleiospermum (Eupteleaceae). Plant Ecol. 2012;213:909–17.
Fu LK, Jin JM. China plant red data book: rare and endangered plants. Beijing: Science Press; 1992.
Sakai A, Ohsawa M. Topographical pattern of the forest vegetation on a river basin in a warm-temperate hilly region, central Japan. Ecol Res. 1994;9:269–80.
Sakai A, Ohsawa T, Ohsawa M. Adaptive significance of sprouting of Euptelea polyandra, a deciduous tree growing on steep slopes with shallow soil. J Plant Res. 1995;108:377–86.
Sakai A, Sakai S, Akiuama F. Do sprouting tree species on erosion-prone sites carry large reserves of resources? Ann Bot. 1997;79:625–30.
Hisabae M, Sone S, Inoue M. Breakdown and macroinvertebrate colonization of needle and leaf litter in conifer plantation streams in Shikoku, southwestern Japan. J Forest Res. 2005;16:108–15.
Scott RA, Barghoorn ES. The occurrence of Euptelea in the Cenozoic of western North America. J A Arboretum. 1955;36:259–65.
Krutzseh W. Zur Kenntnis der präquartären periporaten Pollenformen. Geologie. 1966;15:16–71.
Wolfe JA. Fossil forms of Amentiferae. Britionia. 1974;23:334–55.
Wolfe JA. Paleogene floras from the Gulf of Alaska Region. Geol Surv Prof Pap. 1977;997:1–107.
Averyanova AL. Fossil leaves of Euptelea (Eupteleaceae) from the late Eocene of the Eastern Kazakhstan. Botanicheskiĭ Zhurnal. 2009;94:577–80.
Zheng Y, Wang W. Sequence of Miocene Fotan Group in SE Fujian and its palyno-assemblages. Acta Phytotax Sin. 1994;33:200–16 (in Chinese with English abstract).
Onoe T. Palaeoenvironmental analysis based on the Pleistocene Shiobara flora in the Shiobara volcanic basin, central Japan. Report, Geological Survey of Japan, Tsukuba. 1989;269:1–207 (In Japanese with English abstract and systematic descriptions).
Ozaki K. Late Miocene and Pliocene floras in central Honshu, Japan. Yokohama: Bulletin of Kanagawa Prefectural Museum Natural Science Special Issue; 1991.
Nast CG, Bailey LW. Morphology of Euptelea and comparison with Trochodendron. J A Arboretum. 1946;27:186–92.
Smith AC. A taxonomic review of Euptelea. J A Arboretum. 1946;27:175–85.
Wang W, Lu AM, Ren Y, Endress ME, Chen ZD. Phylogeny and classification of Ranunculales: evidence from four molecular loci and morphological data. Perspect Plant Ecol. 2009;11:81–110.
Kim S, Soltis DE, Soltis PS, Zanis MJ, Suh Y. Phylogenetic relationships among early-diverging eudicots based on four genes: were the eudicots ancestrally woody? Mol Phylogenet Evol. 2004;31:16–30.
Soltis DE, Smith SA, Cellinese N, Wurdack KJ, Tank DC, Brockington SF, Refulio-Rodriguez NF, Walker JB, Moore MJ, Carlsward BS. Angiosperm phylogeny: 17 genes, 640 taxa. Am J Bot. 2011;98:704–30.
Li RQ, Chen ZD, Lu AM, Soltis DE, Soltis PS, Manos PS. Phylogenetic relationships in Fagales based on DNA sequences from three genomes. Int J Plant Sci. 2004;165:311–24.
Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 2007;94:275–88.
Geleta M, Bekele E, Dagne K, Bryngelsson T. Phylogenetics and taxonomic delimitation of the genus Guizotia (Asteraceae) based on sequences derived from various chloroplast DNA regions. Plant Syst Evol. 2010;289:77–89.
Barniske AM, Borsch T, Müller K, Krug M, Worberg A, Neinhuis C, Quandt D. Phylogenetics of early branching eudicots: comparing phylogenetic signal across plastid introns, spacers, and genes. J Syst Evol. 2012;50:85–108.
Drummond AJ, Ashton B, Cheung M, Heled J, Kearse M, Moir R, Stones-Havas S, Thierer T, Wilson A. Geneious v4.8. 2009. http://www.geneious.com.
Zhang J, Yao X, Wei X, Chen L, Jiang M. Development and characterization of 14 polymorphic microsatellite loci in the endangered tree Euptelea pleiospermum (Eupteleaceae). Mol Ecol Resour. 2008;8:314–6.
Wu BY, Kurokochi H, Hogetsu T. Development of 12 microsatellite markers in Euptelea polyandra by a random tailed genome-walking method using Phi29 DNA polymerase. Conserv Genet Resour. 2009;1:59–61.
Simmons MP, Ochoterena H. Gaps as characters in sequence-based phylogenetic analyses. Syst Biol. 2000;49:369–81.
Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987.
Rozas J, Sánchez-De I, Barrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–7.
Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–9.
Excoffier L, Laval G, Schneider S. Arlequin ver.3.1: an integrated software package for population genetics data analysis. Bern: Computational and Molecular Population Genetics Laboratory, Institute of Zoology, University of Bern; 2006.
Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–91.
Pons O, Petit RJ. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics. 1996;144:1237–45.
Burban C, Petit RJ, Carcreff E, Jactel H. Range wide variation of the maritime pine bast scale Matsucoccus feytaudi Duc. (Homoptera: Matsucoccidae) in relation to the genetic structure of its host. Mol Ecol. 1999;8:1593–602.
Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc B. 1977;39:1–38.
Chapuis MP, Estoup A. Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol. 2007;24:621–31.
Kalinowski ST. Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv Genet. 2004;5:539–43.
Kalinowski ST. HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes. 2005;5:187–9.
Goudet J. FSTAT, version 2.9.3.2. A program to estimate and test gene diversities and fixation indices. 2001. http://www2.unil.ch/popgen/softwares/fstat.htm.
Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–70.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–87.
Pritchard JK, Wen X, Falush D. Documentation for Structure software: Version 2.3. 2009. Available at http://pritchardlab.stanford.edu/structure_software/release_versions/v2.3.4/html/structure.html.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214.
Anderson CL, Bremer K, Friis EM. Dating phylogenetically basal eudicots using rbcL sequences and multiple fossil reference points. Am J Bot. 2005;92:1737–48.
Wang W, Chen ZD, Liu Y, Li RQ, Li JH. Phylogenetic and biogeographic diversification of Berberidaceae in the Northern Hemisphere. Syst Bot. 2007;32:731–42.
Bell CD, Soltis DE, Soltis PS. The age and diversification of the angiosperms re-revisited. Am J Bot. 2010;97:1296–303.
Milne RI. Effects of taxon sampling on molecular dating for within-genus divergence events, when deep fossils are used for calibration. J Syst Evol. 2009;47:383–401.
Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8.
Drummond AJ, Nicholls GK, Rodrigo AG, Solomon W. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics. 2002;161:1307–20.
Knobloch E, Mai DH. Monographie der Früchte und Samen in der Kreide von Mitteleuropa. Rozpravy ústredního ústavu geologickénho, Praha. 1986;47:1–219.
Crane PRKR, Pedersen EM, Friis, Drinnan AN. Early Cretaceous (early to middle Albian) platanoid inflorescences associated with Sapindopsis leaves from the Potomac Group of North America. Syst Bot. 1993;18:328–44.
Knobloch E, Mai DH. Neue Gattungen nach Früchten und Samen aus dem Cenoman bis Maastricht (Kreide) von Mitteleuropa. Feddes Repertorium. 1984;95:3–41.
Wahlberg N, Wheat CW, Pena C. Timing and patterns in the taxonomic diversification of Lepidoptera (butterflies and moths). Plos One. 2013;8:e80875.
Rambaut A, Drummond AJ. Tracer v1.5. 2009. http://tree.bio.ed.ac.uk/software/tracer.
Heled J, Drummond AJ. Bayesian inference of population size history from multiple loci. BMC Evol Biol. 2008;8:289.
Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modeling of species geographic distributions. Ecol Model. 2006;190:231–59.
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. Very high resolution interpolate climate surfaces for global land areas. Int J Climatol. 2005;25:1965–78.
Peterson AT, Nakazawa Y. Environmental data sets matter in ecological niche modelling: an example with Solenopsis invicta and Solenopsis richteri. Global Ecol Biogeogr. 2008;17:135–44.
Swets JA. Measuring the accuracy of diagnostic systems. Science. 1988;240:1285–93.
Collins WD, Bitz CM, Blackmon ML, Bonan GB, Bretherton CS, Carton JA, Chang P, Doney SC, Hack JJ, Henderson TB. The Community Climate System Model version 3 (CCSM3). J Clim. 2006;19:2122–43.
Sakaguchi S, Sakurai S, Yamasaki M, Isagi Y. How did the exposed seafloor function in postglacial northward range expansion of Kalopanax septemlobus? Evidence from ecological niche modelling. Ecol Res. 2010;25:1183–95.
Sakaguchi S, Qiu YX, Liu YH, Qi XS, Kim SH, Han JG, Takeuchi Y, Worth JRP, Yamasaki M, Sakurai S, Isagi YJ. Climate oscillation during the Quaternary associated with landscape heterogeneity promoted allopatric lineage divergence of a temperate tree Kalopanax septemlobus (Araliaceae) in East Asia. Mol Ecol. 2012;21:3823–38.
Pinheiro J, Bates D, DebRoy S, Sarkar D. R Development Core Team nlme: R package version 3.1–92, 2009. http://cran.r-project.org/web/packages/nlme/index.html.
Warren DL, Glor RE, Turelli M. Environmental niche equivalency versus conservatism: quantitative approaches to niche evolution. Evolution. 2008;62:2868–83.
Warren DL, Glor RE, Turelli M. ENMTools: a toolbox for comparative studies of environmental niche models. Ecography. 2010;33:607–11.
Wang JF, Gong X, Chiang YC, Kuroda C. Phylogenetic patterns and disjunct distribution in Ligularia hodgsonii Hook. (Asteraceae). J Biogeogr. 2013;40:1741–54.
Schoener TW. The Anolis lizards of Bimini: resource partitioning in a complex fauna. Ecology. 1968;49:704–26.
Kimura M. Quaternary paleogeography of the Ryukyu Arc. J Geoge Sci. 1996;105:259–85.
Kimura M. Land connections between Eurasian continent and Japanese Islands – related to human migration. Migration and Diffusion. 2003;4:14–33.
Cerling TE, Harris JM, MacFadden BJ, Leakey MG, Quade J, Eisenmann V, Ehleringer JR. Global vegetation change through the Miocene/Pliocene boundary. Nature. 1997;389:153–8.
Li JJ, Fang XM, Song CH, Pan BT, Ma YZ, Yan MD. Late Miocene-Quaternary rapid stepwise uplift of the NE Tibetan Plateau and its effects on climatic and environmental changes. Quaternary Res. 2014;81:400–23.
Dobson M, Kawamura Y. Origin of the Japanese land mammal fauna: allocation of extant species to historically-based categories. Quaternary Res. 1998;37:385–95.
Qi XS, Yuan N, Comes HP, Sakaguchi S, Qiu YX. A strong ‘filter’ effect of the East China Sea land bridge for East Asia’s temperate plant species: inferences from molecular phylogeography and ecological niche modelling of Platycrater arguta (Hydrangeaceae). BMC Evol Biol. 2014;14:41.
Qiu YX, Sun Y, Zong M, Zhang XP, Lee J, Murata J, Fu CX, Comes HP. Molecular phylogeography of East Asian Kirengeshoma (Hydrangeaceae) in relation to Quaternary climate change and landbridge configurations. New Phytol. 2009;183:480–95.
Qiu YX, Qi XS, Jin XF, Tao XY, Fu CX, Naiki A, Comes HP. Population genetic structure, phylogeography, and demographic history of Platycrater arguta (Hydrangeaceae) endemic to East China and South Japan, inferred from chloroplast DNA sequence variation. Taxon. 2009;58:1226–41.
Wei XZ, Jiang MX, Huang HD, Yang JY, Yu J. Relationships between environment and mountain riparian plant communities associated with two rare tertiary-relict tree species, Euptelea pleiospermum (Eupteleaceae) and Cercidiphyllum japonicum (Cercidiphyllaceae). Flora. 2010;205:841–52.
Wei XZ, Jiang MX. Contrasting relationships between species diversity and genetic diversity in natural and disturbed forest tree communities. New Phytol. 2011;193:779–86.
Tang CQ, Ohsawa M. Tertiary relic deciduous forests on a humid subtropical mountain, Mt. Emei, Sichuan, China. Folia Geobot. 2002;37:93–106.
Ge J, Dai Y, Zhang Z. Major changes in East Asian climate in the mid-Pliocene: triggered by the uplift of the Tibetan Plateau or global cooling? J Asian Earth Sci. 2013;69:48–59.
Maslin MA, Brierley CM, Millner AM, Shultz, Trauth MH, Wilson KE. East African pulses and early human evolution. Quaternary Sci Rev. 2014;101:1–17.
Tian S, Lei SQ, Hu W, Deng LL, Li B, Meng QL. Repeated range expansions and inter-/postglacial recolonization routes of Sargentodoxa cuneata (Oliv.) Rehd. et Wils. (Lardizabalaceae) in subtropical China revealed by chloroplast phylogeography. Mol Phylogenet Evol. 2015;85:238–46.
Hsü J. Late Cretaceous and Cenozoic vegetation in China, emphasizing their connections with North America. Ann Mo Bot Gard. 1983;70:490–508.
Liu H, Xing Q, Ji Z, Xu L, Tian Y. An outline of Quaternary development of Fagus forest in China: palynological and ecological perspectives. Flora. 2003;198:249–59.
McCormack JE, Peterson AT, Bonaccorso E, Smith TB. Speciation in thehighlands of Mexico: genetic and phenotypic divergence in the Mexican jay (Aphelocoma ultramarina). Mol Ecol. 2008;17:2505–21.
Galbreath KE, Hafner DJ, Zamudio KR. When cold is better: climate-driven elevation shifts yield complex patterns of diversification and demography in an alpine specialist (American Pika, Ochotona princeps). Evolution. 2009;803:2848–63.
Wood DA, Vandergast AG, Lemos Espinal JA, Fisher RN, Holycross AT. Refugial isolation and divergence in the Narrowheaded Gartersnake species complex (Thamnophis rufipunctatus) as revealed by multilocus DNA sequence data. Mol Ecol. 2011;20:3856–78.
Bai WN, Wang WT, Zhang DY. Contrasts between the phylogeographic patterns of chloroplast and nuclear DNA highlight a role for pollen-mediated gene flow in preventing population divergence in an East Asian temperate tree. Mol Phylogenet Evol. 2014;81:37–48.
Gugger PF, González-Rodŕguez A, Rodŕguez-Correa H, Sugita S, Cavender-Bares J. Southward Pleistocene migration of Douglas-fir into Mexico: phylogeography, ecological niche modeling, and conservation of ‘rear edge’ populations. New Phytol. 2011;189:1185–99.
Wu ZY, Wu SG. A proposal for a new floristic kingdom (realm) – the E. Asiatic kingdom, its delimitation and characteristics. In: Zhang AL, Wu SG, editors. Proceedings of the First International Symposium on Floristic Characteristics and Diversity of East Asian Plants. Beijing, China/Berlin, Heidelberg, Germany: China Higher Education Press/Springer; 1996. p. 3–42.
Gao LM, Möller M, Zhang XM, Hollingsworth ML, Liu J, Mill RR, Gibby M, Li DZ. High variation and strong phylogeographic pattern among cpDNA haplotypes in Taxus wallichiana (Taxaceae) in China and North Vietnam. Mol Ecol. 2007;16:4684–98.
Fan DM, Yue JP, Nie ZL, Li ZM, Comes HP, Sun H. Phylogeography of Sophora davidii (Leguminosae) across the ‘Tanaka-Kaiyong Line’, an important phytogeographic boundary in Southwest China. Mol Ecol. 2013;22:4270–88.
Momohara A. Floral and paleoenvironmental history from the late Pliocene to middle Pleistocene in and around central Japan. Palaeogeogr Palaeocl. 1994;108:281–93.
Takeuchi A. Basement-involved tectonics in North Fossa Magna, central Japan: The significance of the northern Itoigawa-Shizuoka Tectonic Line. EPS. 2004;56:1261–9.
Aoki K, Kato M, Murakami N. Phylogeographical patterns of a generalist acorn weevil: insight into the biogeographical history of broadleaved deciduous and evergreen forests. BMC Evol Biol. 2009;9:103.
Kikuchi R, Jae-Hong P, Takahashi H, Maki M. Disjunct distribution of chloroplast DNA haplotypes in the understory perennial Veratrum album ssp. oxysepalum (Melanthiaceae) in Japan as a result of ancient introgression. New Phytol. 2010;188:879–91.
Yi S, Kim SJ. Vegetation changes in western central region of Korean Peninsula during the last glacial (ca. 21.1–26.1 cal kyr BP). Geosci J. 2010;14:1–10.
Han WX, Fang XM, Berger A. Tibet forcing of mid-Pleistocene synchronous enhancement of East Asian winter and summer monsoons revealed by Chinese loess record. Quaternary Res. 2003;78:174–84.
Gotanda K, Yasuda Y. Spatial biome changes in southwestern Japan since the Last Glacial Maximum. Quatern Int. 2008;184:84–93.
Hewitt GM. Post-glacial re-colonization of European Biota. Biol J Linn Soc. 1999;68:87–112.
Stewart JR, Lister AM, Barnes I, Dalen L. Refugia revisited: individualistic responses of species in space and time. P Roy Soc Lond B Bio. 2010;277:661–71.
Ashcroft MB. Identifying refugia from climate change. J Biogeogr. 2010;37:1407–13.
Park YC, Kitade O, Schwarz M, Kim JP, Kim W. Intraspecific molecular phylogeny, genetic variation and phylogeography of Reticulitermes speratus (Isoptera: Rhinotermitidae). Mol Cell. 2006;21:89–103.
Acknowledgments
The authors thank Y. H. Zhang and Y. R. Mao for their great help in collecting plant materials in China and two anonymous reviewers for insightful comments on an earlier version of this manuscript. This research was supported by the National Science Foundation of China (Grant No. 31570214) and the International Cooperation and Exchange of the National Natural Science Foundation of China (Grant Nos. 31511140095, 31561143015).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YXQ conceived the ideas; YNC, LYC and SS contributed to the sampling; YNC collected and analysed the data. The manuscript was written by YXQ, YNC and HPC. All authors read and approved the final manuscript.
Additional files
Additional file 1: Figure S1.
(a) Map showing the location of places quoted in the text including 26 sampled populations of E. pleiosperma (red dots) and 10 sampled populations of E. polyandra (blue dots). More information about the sampled populations is available in Additional file 2: Table S1. (b) Fossil records of extinct and potentially extant (‘Euptelea-like’) species of Eupteleaceae in North America and East Asia [2]. (PDF 1037 kb)
Additional file 2: Table S1.
Geographic and genetic characteristics of 26 E. pleiosperma and 10 E. polyandra populations used in this study. (DOC 130 kb)
Additional file 3: Table S2.
GenBank accession numbers of newly obtained cpDNA and ITS sequences (prefix ‘KR’), and some downloaded sequences from GenBank used in the present study. (DOC 158 kb)
Additional file 4: Table S3.
Chloroplast DNA sequence polymorphisms detected in Euptelea at two IGS and one intron regions, identifying 35 chlorotypes (H1–35). (DOC 251 kb)
Additional file 5: Table S4.
ITS sequence polymorphisms detected in Euptelea at ITS1 and ITS2 (the 5.8S excluded) regions and identifying 10 ribotypes (R1–10). (DOC 63 kb)
Additional file 6: Table S5.
Mean estimates of genetic diversity and differentiation at eight nuclear microsatellite loci surveyed across 33 Euptelea populations (marked with an asterisk in Additional file 2: Table S1). (DOC 30 kb)
Additional file 7: Figure S2.
structure results for identifying the optimal number of clusters (K) among 440 individuals (36 populations) of Euptelea based on nSSR data. The number of K was varied from 1 to 36 in 10 independent runs. The dot plot indicates the mean posterior probability [lnP(D)] for each value of K [64], and the superimposed line diagram represents the corresponding ΔK statistics calculated according to Evanno et al. [65]. (PDF 337 kb)
Additional file 8: Figure S3.
Bayesian Skyline Plots (BSPs) inferred from cpDNA (psbA–trnH, rpoB–trnC, rpL16, petN–trnC, matK, rbcL) sequences, and depicting changes of effective population size (N e; y–axis) through time [in million of years ago (Ma); x–axis] in: (a) E. pleiosperma; (b) E. polyandra; and (c) the J1 and (d) the J2 phylogroups of the latter species. The black solid line is the median estimate, and the gray dashed line marks the 95 % highest probability density (HPD) intervals. (PDF 475 kb)
Additional file 9: Figure S4.
Box-plots of 19 BIOCLIM variables and results of t-tests for 26 populations of E. pleiosperma in China and 10 populations of E. polyandra in Japan. Each box plot shows the maximum, 75th percentile, 25th percentile, and minimum value around the median value for each species. (PDF 443 kb)
Additional file 10: Figure S5.
Box-plots of 19 BIOCLIM variables and results of t-tests for two cpDNA lineages of E. pleiosperma in China (W: western populations; CE: central-eastern populations). Each box plot shows the maximum, 75th percentile, 25th percentile, and minimum value around the median value for each population group (‘lineage’). (PDF 392 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cao, YN., Comes, H.P., Sakaguchi, S. et al. Evolution of East Asia’s Arcto-Tertiary relict Euptelea (Eupteleaceae) shaped by Late Neogene vicariance and Quaternary climate change. BMC Evol Biol 16, 66 (2016). https://doi.org/10.1186/s12862-016-0636-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-016-0636-x