
Abstract
Background
In retinal degenerative disease, progressive and debilitating conditions result in deterioration of retinal cells and visual loss. In human, retina lacks the inherent capacity for regeneration. Therefore, regeneration of retinal layer from human retinal progenitor cells (hRPCs) is a challenging task and restricted in vitro maintenance of hRPCs remains as the main hurdle. Retina and anterior neural fold homeobox gene (RAX) play critical roles in developing retina and maintenance of hRPCs. In this study, for the first time regulatory regions of human RAX gene with potential promoter activity were experimentally investigated.
Results
For this purpose, after in silico analysis of regulatory regions of human RAX gene, the expression of EGFP reporter derived by putative promoter sequences was first evaluated in 293 T cells and then in hRPCS derived from human embryonic stem cells. The candidate region (RAX-3258 bp) showed the highest EGFP expression in hRPCs. This reporter construct can be used for in vitro monitoring of hRPC identity and verification of an efficient culture medium for maintenance of these cells.
Conclusions
Furthermore, our findings provide a platform for better insight into regulatory regions of human RAX gene and molecular mechanisms underlying its vital functions in retina development.
Similar content being viewed by others


Background
Retinitis pigmentosa (RP) and age-related macular degeneration (AMD) are the most common types of retinal degeneration disease (RDD) [1]. In RDD, retinal cells are damaged and thereby visual ability is impaired [2]. The intrinsic regenerative capacity of the human retina is extremely restricted and there is a growing focus on using human retinal progenitor cells (hRPCs) as a potential therapeutic approach for restoring retinal function and visual ability [3]. In this regard, RPCs can reduce disease progression rate by secretion of growth factors and upon integration and differentiate into new rod and cone photoreceptors in retinal layer, can facilitate the process of visual rehabilitation [4,5,6]. In addition, transplanted RPCs exhibit low tumorigenic potential and have tendency to differentiate into retinal cell layers [7]. However, their restricted in vitro maintenance and proliferation capacity have hampered their application in the field of regenerative medicine. It has been shown that they lost their proliferative ability after maximum of seven passages, with reduced capacity for retinal cells formation [3]. One indirect approach to assess hRPCs preservation while being maintained in vitro, is to target their cell morphology and molecular based tracking such as immunostaining. However, a more direct alternative is to use the advantage of a fluorescent reporter under control of a specific retinal promoter [8]. Retina and anterior neural fold homeobox gene (RAX) is one of the initial genes expressed in prospective retina derived from the anterior neural plate [9]. RAX transcription factor consists of two conserved domains of homeodomain proteins. The first one is an octapeptide motif in the N-terminal and the second one is a C-terminal OAR (otp, aristaless, and rax) domain, which play prominent roles in the eye development [10]. RAX is essential for the eye field specification and normal development of retina. It is generally down-regulated during differentiation towards retinal cells [11]. Previous researches showed positive correlation between RAX expression and RPCs proliferation in mouse and xenapous [12, 13]. Human RAX promoter region has not been extensively examined yet [14]. In this study, after prediction of putative human RAX promoter regions, the expression of EGFP reporter derived by these regulatory regions was first evaluated in 293T cells and then in human embryonic stem cell (hESC) derived hRPCs. Distal region of human RAX gene containing − 3097 to + 161 resulted in the highest EGFP expression in hRPCs. Altogether, our findings provide a better understanding of regulatory regions of human RAX gene, and can be extended to study the mechanism behind RAX function in multipotent retinal progenitors. The identification of human RAX promoter sequence might be a valuable supplementary tool for assessment of molecular pathways involved in retinal proliferation and differentiation.
Results
Verification of hESCs differentiation into hRPCs
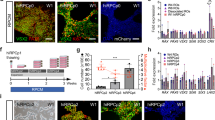
After differentiation of hESCs to hRPCs by RDM, the identity of hRPCs was confirmed by immunocytochemical analysis of eye filed transcription factors (EFTF) panel including LHX2, RAX, PAX6, SIX3 and also stemness markers like OCT4 and NANOG (Fig. 1F, G). According to Fig. 1, differentiation of hESCs towards retinal progenitors was verified by significant reduction of OCT4 and NANOG, whereas EFTFs had successfully higher protein expression in derived hRPCs. For further confirmation of proper retinal development, the relative expression of EFTFs were analyzed at mRNA level by quantitative RT-PCR. Based on our results, the expression of anterior neural and eye field genes including SIX3, RAX and PAX6 were increased when cells cultured in RDM compared to hESCs. Moreover, the expression of stemness markers including OCT4 and NANOG, were significantly decreased after differentiation of ESCs into hRPCs (Fig. 1G). These results successfully confirmed the potential of selected medium for differentiation of human stem cells towards retinal progenitor cells.

Overview of retinal differentiation and characterization of hPSCs-derived RPCs. A Stepwise process of hPSCs differentiation into RPCs. B Phase contrast images of hPSCs cultured on Matrigel coated dish, C hPSC derived EBs cultured in low adherent dishes, D Expansion of RPCs from EBs on Matrigel coated dishes and (E) Expansion of hRPCs 21 days after seeding. F Immunocytochemistry of eye field-associated transcription factors: RAX, PAX6, LHX2 (red) and NESTIN (green). Scale bars: 100 μm and 50 μm (G) qPCR analysis of stemness and EFTF biomarkers in hPSCs and RPCs (*p < 0.05 vs control, n = 3)
In silico analysis of RAX promoter
In this study UCSC genome browser was applied for analysis of chromatin structure (DNase hypersensitivity), and chromatin state of human RAX gene upstream region.
DNase I hypersensitivity marks different classes of cis regulatory elements within genome, such as promoters and enhancers [15]. UCSC analysis showed several subset of DNase I clusters within ~ 3 kb region upstream of human RAX gene (chr18: 59,273,233–59,276,490). Also, Fig. 2A demonstrated the distribution of H3K4me1, H3K4me3 and H3K27ac in 5′ upstream region of RAX gene, which are marks of active promoter and enhancer regions.

Analysis of putative regulatory regions of human RAX gene to derive EGFP reporter plasmids. A Screenshot from UCSC genome browser indicating human RAX gene upstream region, H3K4me1, H3K4me3, H3K27ac and Pol2 patterns, CpG islands and DNase clusters. B Schematic representation of pEGFP-C1 plasmid in which CMV promoter was substituted with putative promoter regions of human RAX gene. C The final expression plasmids were confirmed by restriction digestion analysis
Based on ChIP-seq analysis in UCSC database, there are three fragments upstream of transcription start site of human RAX gene enriched of RNA polymerase II which can be indicator of promoter activity of this region [16].
CpG islands are typically located near TSS and might be associated with promoter regions [17]. In silico analysis of upstream region of RAX gene by UCSC genome browser confirmed the presence of a CpG island of around 1 kb length. Moreover, these results showed high evolutionarily conservation of this sequence in primates which reflects the significance of elements contained in this genomic regulatory region. Collectively based on these results 3258 bp upstream of human RAX gene was selected as a putative region with promoter activity.
In order to have a better view of potential regulation of RAX gene at transcriptional level, distal region was also analyzed for putative binding sites of transcription factors involved in retinal progenitor cell proliferation or development. For this purpose, GTRD which is a collection of ChiP-seq database for identification of TFBS in human and mouse, and also JASPAR data base were analyzed.
This sequence analysis revealed the presence of several presumptive binding sites for SOX2 and OTX2 in distal region of human RAX gene in accordance with previous studies in Xenopus which was introduced as a conserved noncoding sequence (CNS1) by Danno et.al (Fig. 3A) [18]. UCSC analysis revealed that CNS1 is highly conserved in primates and rodents (Fig. 2A). Also a number of putative binding sites were predicted for SMAD2/3 within human RAX gene. SMAD2/3 has been introduced as a key mediator for in vitro differentiation of mouse ESCs into retinal cells by direct binding to the regulatory elements of vital genes like Rax, Pax6 and Otx2 [19].

Deletion analysis of human RAX promoter regions. A A series of 5′ deletions from − 3258 to + 161 bp of human RAX gene. B, C Intensity of EGFP reporter in promoter deletion constructs was determined by flow cytometry in 293 T cells (B) and RPCs (C). Data are expressed as mean ± SD (*p < 0.05, ** p < 0.005, ***p < 0.0005, n = 3). D, E Comparison of EGFP expression derived by different RAX regulatory regions in 293 T cells (D) and RPCs (E) using flow cytometry and fluorescence microscopic analysis. The scale bar is 100 μm
Deletion analysis of human RAX promoter
To experimentally investigate the promoter activity of 5′ flanking region of human RAX gene, we performed EGFP reporter assay using deletion constructs of regulatory regions. For this purpose, different candidate regulatory fragments were cloned upstream of EGFP reporter (Fig. 3A) and transiently transfected in 293T cells (Fig. 3B, D) and hRPCs (Fig. 3C, E). Based on protein atlas database 293 T cells exhibited a low level of RAX mRNA expression. RT-PCR results also confirmed the amplification of RAX gene (NM_013435) with 107 bp size with synthesized cDNA from 293T total RNA (Fig. 4A, B). So this cell line with high transfection efficiency was selected for analysis of RAX promoter activity. Before transfecting putative promoter region into target cells, the integrity of expression constructs was examined by restriction digestions (Fig. 2B, C) and sequencing analysis (data not shown). Based on flow cytometry analysis 48 h post transfection, minimal upstream sequences of RAX gene with 267 and 525 bp length resulted in low rate of EGFP expression in 293T cells. After elimination of these two minimal regions, 3007 and 2761 bp fragments indicated lower EGFP expression compared to the minimal regions. On the other hand, 1665 and 3258 bp regions could highly derive the EGFP reporter and with a similar expression rate (Fig. 3B). Surprisingly, 3258 bp distal region indicated the highest RAX promoter activity in retinal progenitor cells and contributed to 2-fold EGFP enhancement in compared to 1665 proximal promoter sequence (Fig. 3C). These results suggest that there might be enhancer elements in distal region of human RAX gene which might cooperate with minimal regions and thereby modulating transcription of the associated RAX gene.

RAX expression result was confirmed by PCR amplification. A Phase contrast image of 293T cell line. B PCR amplification of RAX gene (107 bp) with synthesized cDNA from 293T isolated total RNA, 50 bp DNA ladder
Discussion
In retina, proliferation, differentiation and cell fate decision are among cellular events being controlled by complex extrinsic and intrinsic signals [20, 21]. Transcription factors such as PAX6, SIX3, OTX2, SOX2 and RAX are intrinsic regulators of maintenance and development of RPCs [22, 23].
In vertebrates, RAX transcription factor plays indispensable roles in early development of retina and has been implicated in RPCs maintenance [12]. Deletion studies of Rax in mice led to loss of optic vesicle development [24]. Moreover, studies have been showed that conditional knockout of Rax resulted in failure of laminar structure formation in retina, reduction of retinal progenitor cells, and retinal cell fate changes in conditional knockout mouse model [25]. However, there are still ambiguous aspects of RAX transcription factor roles in mammalian eye formation.
Although, most of the retinal disorders are resulted from destruction of different retinal cell types [26], providing a sufficient pool of RPCs for cell therapy as clinical approach is presently unachievable. Since in vitro culture of ESC derived RPCs, leads to down regulation of pivotal EFTFs in these progenitor cells and loss of their identity over passages [27,28,29], this limitation results in failure of providing adequate pool of RPCs for downstream studies. Therefore, to monitor in vitro maintenance of ESCs-derived hRPCs, an expression vector driving EGFP reporter by human RAX promoter was designed. For this purpose, we experimentally investigated the regulatory regions upstream of human RAX gene with potential promoter activity. This approach can facilitate further researches in optimization of in vitro culture condition, like analysis of different growth factors and small molecules, required for maintenance of RPCs with high capability of proliferation and multipotency. With providing adequate number of RPCs, animal studies and clinical trials can be more successfully performed in hope of achieving several cell-based therapy approaches for diseases such as AMD and RP, as most common retinal disease.
First we characterized hRPCs derived from hESCs by expression analysis of eye field markers [30]. In our previous studies, Noggin, IWR and IGF-1 were included to the culture medium to differentiation of hESCs into hRPCs [31]. In this protocol, several sequential induction steps are needed to achieve retinal progenitor cells. For development of forebrain derivatives, BMP and Wnt/ß-catenin pathways should be antagonized [32, 33]. Therefore, in order to direct ESCs to the anterior neural fate, EBs were treated with combination of noggin (a potent inhibitor of BMP pathway), IWR (an antagonist of Wnt/ß-catenin signaling pathway) and IGF-1as an inducer of retinal progenitors from ESCs under 3D culture conditions [34].
The prediction of putative promoter region was performed by using different bioinformatics tools. Human RAX promoter region has not been thoroughly studied so far. For this purpose, upstream flanking region of human RAX gene was analyzed by UCSC in terms of chromatin state (H3K4me1, H3K4me3 and H3K27ac), DNase hypersensitive sites, CpG islands, POL II enrichment and sequence conservation [35]. DNase clusters indicated that transcriptional machinery might be enriched at these particular sequences with open chromatin structure [36]. Furthermore, our in silico analysis demonstrated the distribution of H3K4me1, H3K4me3 and H3K27ac in 5′ upstream region of human RAX gene. H3K4me1 and H3K4me3 epigenetic modifications are normally signature of active promoter and enhancer regions. Enrichment of H3K27ac is also an active chromatin mark [37, 38]. Basically, RNA polymerase II binds to promoter region of genes for transcription initiation with aid of transcription factors. Most mammalian RNA polymerase II initiate transcription at CpG islands, which are devoid of DNA methylation [16]. UCSC analysis confirmed the presence of sites enriched by RNAP II and a CpG island in regulatory region of RAX gene. Collectively, ~ 3.2 kb upstream of RAX gene was considered as the putative promoter region. The promoter activity of this region was experimentally investigated by using deletion constructs deriving EGFP reporter. Deletion analysis of ~ 3.2 kb human RAX promoter regions (− 3097 to the + 167) was analyzed in 293T and ESCs-derived hRPCs. The results of deletion construct analysis showed that region from − 3097 to − 336 (RAX-2761) and − 3097 to − 90 (RAX-3007) of human RAX gene could not independently drive downstream EGFP expression after elimination of minimal regions which basically include general regulatory binding sites required to trigger transcription. These findings indicated that the minimal RAX regulatory promoter regions, from − 106 to + 161 (RAX-267) and − 364 to + 161 (RAX-525), consist of critical elements to derive RAX gene expression. Candidate distal region − 3097/+ 161 (RAX-3258) and proximal region 1504/+ 161 (RAX-1665) showed a significantly higher EGFP expression in both 293T cells and hRPCs. Interestingly, distal region (RAX-3258 bp) demonstrated a remarkable more EGFP expression than RAX-1665 bp in hRPCs. Based on our results, we speculated that potential elements which are located in distal region, mediate the transcriptional stimulation and contributes to the higher promoter activity of RAX-3258 compared to RAX-1665.
Previous studies confirmed the necessity of conserved noncoding sequence 1 (CNS1) which is located ~ 2 kb upstream of Rax promoter as a regulatory region for expression of Rax in mice. CNS1 contains a highly conserved binding sites for Sox2 and Otx2 transcription factors across vertebrates, which are required for Rax transcription in mice [18]. These studies identified that Sox2 and Otx2 are potent modulators of Rax expression by direct binding to the promoter region and synergistically activate its transcription. Interactions between Sox2 and Otx2 proteins, regulate the expression of Rax during eye development [18].
Our bioinformatic analysis using JASPAR, GTRD and UCSC was also predicted binding sites for SOX2 and OTX2 in distal regulatory region of RAX gene. This key conserved region was included in RAX-3258 bp distal promoter which exhibited the most promoter activity in hRPCs with high expression of SOX2 and OTX2. Interestingly, this candidate region was resulted in substantial reduced expression of EGFP in 293T cells which do not express SOX2 or OTX2 endogenously [18]. Albeit, the roles of other transcription factors in the regulation of human RAX expression should not been ignored.
Our in silico analysis also revealed several binding sites for SMAD2/3 in distal region of human RAX gene. Previous studies of in vitro differentiation of mouse ESCs into retinal cells, identified SMAD2/3 as a key regulator of several retinal genes like Rax [39, 40]. Moreover, this study showed that SMAD2/3 was able to directly bind to regulatory elements of retinal and photoreceptor precursor genes. In fact, SMAD2/3 binds to Smad binding elements (SBEs) which are located in distal promoter regions of target genes such as RAX and activates their expression (Fig. 5A, B).

TFBS analysis of human RAX distal promoter region (− 1665 to − 3078) (A) Putative binding sites of SOX2, OTX2 and SMAD2/3 within this region were analyzed using the EPD database. (B) Profile summery of transcription factor binding sites using JASPAR data base
For further studies-regarding these molecular mechanisms in human, the effect of corresponding transcription factor overexpression on RAX promoter activity with mutated TFBSs can be investigated.
Conclusion
In summary, the present study introduced the regulatory region of RAX gene with high promoter activity. When, this region is included in an expression vector expressing EGFP, it may provide a molecular tool for monitoring retinal progenitor cell maintenance during in vitro culture. Furthermore, our study can be extended towards future investigations regarding the molecular mechanisms by which RAX play key roles in proliferation and development of retinal progenitor cells and reveal more aspects of hRPCs regulation.
Methods
Bioinformatics analysis
The sequence of human RAX gene was obtained from National Center for Biotechnology Information (NCBI). Different bioinformatic tools were used to predict the potential promoter regions of human RAX gene. These software are as follows; UCSC (http://genome.ucsc.edu/), and Genomatix (https://www.genomatix.de/). Also, the putative transcription factor binding sites (TFBS) within the RAX promoter region were analyzed using the gene transcription regulation database GTRD (http://gtrd.biouml.org/) and JASPAR (http:// http://jaspar.genereg.net/).
Primers design for amplification of putative human RAX promoter regions
Primers used to amplify the potential promoter regions of the RAX gene were designed from National Centre for Biotechnology Information (NCBI) database and confirmed by Primer-BLAST and Oligo7. The primers were reconstituted in nuclease-free water to a concentration of 10 pM/μl. Then, 25 μl PCR reactions containing 2 μl of human genomic DNA (50 ng) as template, 1 μl of each forward and reverse primers (10 pM/μl), 1X Multiplex PCR Master Mix (Yekta Tajhiz Azma Co., Tehran, Iran), and 6 μl of nuclease-free water were prepared, Amplification was carried out in thermocycler (Thermoscientific) with amplification conditions shown in Table 1.
Cloning of putative human RAX promoter regions into pEGFP-C1 vector
Using the primers listed in Table 1, the candidate promoter region − 3097/+ 161 of the human RAX gene, and a series of control fragments (− 106/+ 161, − 364/+ 161, − 1504/+ 161, − 3097/− 336, − 3097/− 90) were amplified by PCR from human genomic DNA and inserted into SalI/BglII site of the pEGFP-C1 vector upstream of EGFP reporter. The final expression vector was transformed into E. coli DH5-α cells. The integrity of all target sequences was verified by sequencing before evaluation of promoter activity in target cells.
In vitro cell culture of human embryonic stem cells
RH6 human embryonic stem cells (hESCs, Royan institute) were seeded on 0.3 mg/ml Matrigel (Sigma-Aldrich, St. Louis, MO)-coated tissue culture dishes containing DMEM/F12 medium supplemented with 20% knockout serum replacement (KSR), 0.1 mM nonessential amino acids, 2 mM L-glutamine, 1% ITS and 100 ng/ml bFGF with daily medium exchange (Fig. 1B).
Differentiation of hESCs into hRPCs
The process of retinal differentiation was briefly demonstrated in Fig. 1A. For neural retinal differentiation, the over confluent feeder-free hESCs were dissociated and then a mechanical approach was used to initiate embryoid body formation (EBs) (Fig. 1C). So hESCs were transferred to low adherent dishes in order to form EBs in neural induction medium (NIM) containing 1 ng/ml noggin (R&D, 1976-NG), 3 μM IWR (R&D, 5439-DK/CF), and 5 ng/mL human recombinant insulin-like growth factor-1 (IGF-1) (R&D, 291-GI) in DMEM/F12 medium supplemented with 10% KSR, 0.1 mM nonessential amino acids, 2 mM L-glutamine, and 1% B27 (Gibco, 17,504–044) for 3 days. In the next step, the EBs were dissociated by Accutase (Millipore, SCR005) and were replated on 1 mg/ml laminin and 15 mg/ml Poly-L-ornithine (both from Sigma-Aldrich) coated 6-well tissue culture plates. The culture medium was replaced with retinal determination medium (RDM) containing DMEM/F12 supplemented with 1% B27, 2% N2 (Gibco, 17,502–048), 10 ng/ml noggin, 3 μM IWR, 10 ng/ml IGF-1, and 10 ng/ml bFGF as previously explained. This medium was exchanged every 2 days up to 21 days (Fig. 1D, E).
Transient transfection
For investigation the promoter activity of expression vectors harboring the potential promoter regions of RAX gene by transfection into 293T cell line, these cells were maintained in DMEM supplemented with 1% L-glutamine, 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin and incubated at 37 °C in 5% CO2. Next equal moles of expression vectors were transfected into 293T cells [41] by Lipofectamine LTX Transfection Reagent according to the manufacturer’s instructions (Invitrogen, Germany) in a 24-well cell culture plate. Also control groups were transfected by pEGFP-C1 and promoter-less vector (pEGFP-np) for analysis the efficiency of transfection and confirmation of target promoter’s activity respectively. The expression of EGFP reporter resulting from promoter activity of RAX regulatory regions were assayed 48 h post transfection. To explore the potential promoter activity of these regions in retinal progenitors, they were also transfected into hESCs- derived hRPCs, as described above.
Immunocytochemical analysis
To evaluate the expression of stem cell and retinal progenitor markers, immunocytochemistry staining was carried out using the primary antibodies: PAX6 (SC-11357, 1/50), RAX (LS-C53650, 1/200), NESTIN (ab-22,035, 1/100), LHX2 (SC-81311, 1/100) and secondary antibodies Anti-Mouse IgG-FITC (Sigma, AP124F) and Anti-Rabbit IgG-TRITC (Sigma, T6778). Briefly, 5 × 104 cells/well coverslips coated with Matrigel were plated in 24-well plates. One day later, samples were fixed with 4% paraformaldehyde for 30 min at room temperature. Subsequently, these cells were permeabilized by 0.4% Triton X-100 for 30 min and were stained with blocking solution-diluted primary antibodies (BSA, 10 mg/ml) and kept at 4 °C overnight. Then, they were treated with secondary antibodies at 37 °C for 1 h. Furthermore, cell nuclei were stained with DAPI (3 ng/ml, Invitrogen). The images were taken by fluorescent microscope (Olympus, Center Valley, PA, USA) equipped with an Olympus DP70 camera.
Quantitative PCR (qPCR)
The total RNA of hRPCs and RH6 cells (as negative control) was extracted using RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). Then, cDNA was synthesized using Takara cDNA Synthesis kit using random hexamer primers. All qRT-PCR reactions were performed in triplicate, and data were normalized to human GAPDH mRNA. Relative fold changes in target gene expression was calculated using 2−ΔΔCt method. Table 1, represented primer sequences used in quantitative PCR. Moreover, to investigate the stemness state and multipotency capacity of hESCs-derived hRPCs, the relative expression of several markers including OCT4, NANOG and eye field markers like NESTIN, SIX3, PAX6 and RAX were evaluated [11]. Each experiment, had a negative template control (NTC) for primer specificity analysis and lack of DNA contamination. SYBR Green I Master reaction mix (Thermo Fisher Scientific) was used for qPCR analysis of gene expression, and amplification was detected with Light Cycler 480 ABi System.
Flow cytometry analysis
Flow cytometry assessment was performed to evaluate the quantification of EGFP reporter derived by promoter regions of RAX gene. For this purpose, 2 days after transfection of target cells with expression vectors, cells were detached and re-suspended in cold PBS− and analyzed by FACS Vantage flow cytometry (Becton Dickinson). Data were analyzed using BD Cell Quest Pro and WinMDI 2.9 software.
Statistical analysis
All data were analyzed using one-way ANOVA, Tukey’s post-hoc analysis and Student’s t-test and are shown as the mean ± SD. In each experiment, at least three biological replicates were examined. In this study, P ≤ 0.05 was considered as statistically significant.
Availability of data and materials
The datasets analysed during the current study are available in the [GenBank] repository, [accession number MZ427311].
Abbreviations
- hRPCs:
-
Human retinal progenitor cells
- RAX:
-
Retina and anterior neural fold homeobox
- RP:
-
Retinitis pigmentosa
- AMD:
-
Age-related macular degeneration
- RDD:
-
Retinal degeneration disease
- EFTF:
-
Eye field transcription factor
- CNS:
-
Conserved noncoding sequence
- hESC:
-
Human embryonic stem cell
- TFBS:
-
Transcription factor binding site
References
Gagliardi G, M'Barek KB, Goureau O. Photoreceptor cell replacement in macular degeneration and retinitis pigmentosa: a pluripotent stem cell-based approach. Prog Retin Eye Res. 2019;71:1–25. https://doi.org/10.1016/j.preteyeres.2019.03.001.
Sottile F, Pesaresi M, Simonte G, Cosma MP. Cell therapy for degenerative retinal disease: special focus on cell fusion-mediated regeneration. In: Cell-Based Therapy for Degenerative Retinal Disease: Springer; 2019. p. 217–44.
Young MJ, Tucker BA, Baranov PY. Low oxygen culture conditions for maintaining retinal progenitor cell multipotency. Tissue Eng Part A. 2017;20(9-10):1465.
Zhou J, Benito-Martin A, Mighty J, Chang L, Ghoroghi S, Wu H, et al. Retinal progenitor cells release extracellular vesicles containing developmental transcription factors, microRNA and membrane proteins. Sci Rep. 2018;8(1):1–15.
Liu Y, Chen SJ, Li SY, Qu LH, Meng XH, Wang Y, et al. Long-term safety of human retinal progenitor cell transplantation in retinitis pigmentosa patients. Stem Cell Res Ther. 2017;8(1):209. https://doi.org/10.1186/s13287-017-0661-8.
Wang S-T, Chen L-l, Zhang P, Wang X-B, Sun Y, Ma L-X, et al. Transplantation of retinal progenitor cells from optic cup-like structures differentiated from human embryonic stem cells in vitro and in vivo generation of retinal ganglion-like cells. Stem Cells Dev. 2019;28(4):258–67. https://doi.org/10.1089/scd.2018.0076.
Singh MS, Park SS, Albini TA, Canto-Soler MV, Klassen H, MacLaren RE, et al. Retinal stem cell transplantation: balancing safety and potential. Prog Retin Eye Res. 2020;75:100779. https://doi.org/10.1016/j.preteyeres.2019.100779.
Bervoets I, Charlier D. A novel and versatile dual fluorescent reporter tool for the study of gene expression and regulation in multi-and single copy number. Gene. 2018;642:474–82. https://doi.org/10.1016/j.gene.2017.11.061.
Kon T, Furukawa T. Origin and evolution of the Rax homeobox gene by comprehensive evolutionary analysis. FEBS Open Bio. 2020;10(4):657–73. https://doi.org/10.1002/2211-5463.12832.
Furukawa T, Kozak CA, Cepko CL. Rax, a novel paired-type homeobox gene, shows expression in the anterior neural fold and developing retina. Proc Natl Acad Sci. 1997;94(7):3088–93. https://doi.org/10.1073/pnas.94.7.3088.
Zuber ME, Gestri G, Viczian AS, Barsacchi G, Harris WA. Specification of the vertebrate eye by a network of eye field transcription factors. Development. 2003;130(21):5155–67. https://doi.org/10.1242/dev.00723.
Muranishi Y, Terada K, Furukawa T. An essential role for Rax in retina and neuroendocrine system development. Develop Growth Differ. 2012;54(3):341–8. https://doi.org/10.1111/j.1440-169X.2012.01337.x.
Giudetti G, Giannaccini M, Biasci D, Mariotti S, Degl'Innocenti A, Perrotta M, et al. Characterization of the Rx1-dependent transcriptome during early retinal development. Dev Dyn. 2014;243(10):1352–61. https://doi.org/10.1002/dvdy.24145.
Harding P, Moosajee M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J Dev Biol. 2019;7(3):16.
Wilken MS, Brzezinski JA, La Torre A, Siebenthall K, Thurman R, Sabo P, et al. DNase I hypersensitivity analysis of the mouse brain and retina identifies region-specific regulatory elements. Epigenetics Chromatin. 2015;8(1):8.
Hampsey M. Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiol Mol Biol Rev. 1998;62(2):465–503.
Ioshikhes IP, Zhang MQ. Large-scale human promoter mapping using CpG islands. Nat Genet. 2000;26(1):61–3.
Danno H, Michiue T, Hitachi K, Yukita A, Ishiura S, Asashima M. Molecular links among the causative genes for ocular malformation: Otx2 and Sox2 coregulate Rax expression. Proc Natl Acad Sci. 2008;105(14):5408–13. https://doi.org/10.1073/pnas.0710954105.
Lu AQ, Barnstable CJ. Pluripotent stem cells as models of retina development. Mol Neurobiol. 2019;56(9):6056–70. https://doi.org/10.1007/s12035-019-1504-7.
Dyer MA, Cepko CL. Regulating proliferation during retinal development. Nat Rev Neurosci. 2001;2(5):333–42. https://doi.org/10.1038/35072555.
Zhang SS-M, Fu X-Y, Barnstable CJ. Tissue culture studies of retinal development. Methods. 2002;28(4):439–47. https://doi.org/10.1016/S1046-2023(02)00263-3.
Heavner W, Pevny L. Eye development and retinogenesis. Cold Spring Harb Perspect Biol. 2012;4(12):a008391.
Levine EM, Green ES. Cell-intrinsic regulators of proliferation in vertebrate retinal progenitors. Semin Cell Dev Biol. 2004;15(1):63–74.
Medina-Martinez O, Amaya-Manzanares F, Liu C, Mendoza M, Shah R, Zhang L, et al. Cell-autonomous requirement for rx function in the mammalian retina and posterior pituitary. PLoS One. 2009;4(2):e4513.
Mathers P, Grinberg A, Mahon K, Jamrich M. The Rx homeobox gene is essential for vertebrate eye development. Nature. 1997;387(6633):603–7.
Ramsden CM, Powner MB, Carr A-JF, Smart MJ, da Cruz L, Coffey PJ. Stem cells in retinal regeneration: past, present and future. Development. 2013;140(12):2576–85.
Klassen HJ, Ng TF, Kurimoto Y, Kirov I, Shatos M, Coffey P, et al. Multipotent retinal progenitors express developmental markers, differentiate into retinal neurons, and preserve light-mediated behavior. Invest Ophthalmol Vis Sci. 2004;45(11):4167–73.
Yun C, Oh J, Lee B, Lee J-M, Ariunaa T, Huh K. Generation of Retinal Progenitor Cells from Human Induced Pluripotent Stem Cell-Derived Spherical Neural Mass. Tissue Eng Regen Med. 2017;14(1):39–47.
Qu L, Gao L, Xu H, Duan P, Zeng Y, Liu Y, et al. Combined transplantation of human mesenchymal stem cells and human retinal progenitor cells into the subretinal space of RCS rats. Sci Rep. 2017;7(1):1–14.
Amirpour N, Karamali F, Rabiee F, Rezaei L, Esfandiari E, Razavi S, et al. Differentiation of human embryonic stem cell–derived retinal progenitors into retinal cells by sonic hedgehog and/or retinal pigmented epithelium and transplantation into the subretinal space of sodium iodate–injected rabbits. Stem Cells Dev. 2012;21(1):42–53.
Cifuentes H. Induction of human embryonic stem cell derived retinal stem cells in vitro using transient overexpression of messenger RNA for BLIMP, ONECUT1 and OTX2; 2016.
Bertacchi M, Pandolfini L, D'Onofrio M, Brandi R, Cremisi F. The double inhibition of endogenously produced BMP and W nt factors synergistically triggers dorsal telencephalic differentiation of mouse ES cells. Dev Neurobiol. 2015;75(1):66–79.
Bae D, Mondragon-Teran P, Hernandez D, Ruban L, Mason C, Bhattacharya SS, et al. Hypoxia enhances the generation of retinal progenitor cells from human induced pluripotent and embryonic stem cells. Stem Cells Dev. 2012;21(8):1344–55.
Lamba DA, Karl MO, Ware CB, Reh TA. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci U S A. 2006;103(34):12769–74.
Laverrière J-N, L’Hôte D, Tabouy L, Schang A-L, Quérat B, Cohen-Tannoudji J. Epigenetic regulation of alternative promoters and enhancers in progenitor, immature, and mature gonadotrope cell lines. Mol Cell Endocrinol. 2016;434:250–65.
Mercer TR, Edwards SL, Clark MB, Neph SJ, Wang H, Stergachis AB, et al. DNase I–hypersensitive exons colocalize with promoters and distal regulatory elements. Nat Genet. 2013;45(8):852.
Pradeepa MM, Grimes GR, Kumar Y, Olley G, Taylor GC, Schneider R, et al. Histone H3 globular domain acetylation identifies a new class of enhancers. Nat Genet. 2016;48(6):681.
Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107(50):21931–6.
Lu AQ, Popova EY, Barnstable CJ. Activin signals through SMAD2/3 to increase photoreceptor precursor yield during embryonic stem cell differentiation. Stem Cell Reports. 2017;9(3):838–52.
Sakaki-Yumoto M, Liu J, Ramalho-Santos M, Yoshida N, Derynck R. Smad2 is essential for maintenance of the human and mouse primed pluripotent stem cell state. J Biol Chem. 2013;288(25):18546–60.
Hornstein BD, Roman D, Arévalo-Soliz LM, Engevik MA, Zechiedrich L. Effects of circular DNA length on transfection efficiency by electroporation into HeLa cells. PLoS One. 2016;11(12):e0167537. https://doi.org/10.1371/journal.pone.0167537.
Acknowledgements
We are so thankful for our colleagues, Dr. Kianoush Dormianiand Fatemeh Haghayegh for their assistances that greatly improved the manuscript.
Funding
The materials supported for this study were mainly supported from national institute for medical research development (NIMAD; Grant No. 971117). But this grant had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the analysis were done by MHNE & FK, Collected the data was done by AA, PSH, Performed the analysis was done by AA, PSH & FK, Wrote the manuscript was done by PSH, AA, Interpretation of the obtained information was done by MHNE & SHI. All authors read and approved the final manuscripts.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Ethical Committee of Royan institute (IR.ACECR.ROYAN.REC.1396.41).
Consent for publication
Not applicable.
Competing interests
There are no competing interests to report.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Atefi, A., Kojouri, P.S., Karamali, F. et al. Construction and characterization of EGFP reporter plasmid harboring putative human RAX promoter for in vitro monitoring of retinal progenitor cells identity. BMC Mol and Cell Biol 22, 40 (2021). https://doi.org/10.1186/s12860-021-00378-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12860-021-00378-2