
Abstract
Background
Viral genomics and epidemiology have been increasingly important tools for analysing the spread of key pathogens affecting daily lives of individuals worldwide. With the rapidly expanding scale of pathogen genome sequencing efforts for epidemics and outbreaks efficient workflows in extracting genomic information are becoming increasingly important for answering key research questions.
Results
Here we present Genofunc, a toolkit offering a range of command line orientated functions for processing of raw virus genome sequences into aligned and annotated data ready for analysis. The tool contains functions such as genome annotation, feature extraction etc. for processing of large genomic datasets both manual or as part of pipeline such as Snakemake or Nextflow ready for down-stream phylogenetic analysis. Originally designed for a large-scale HIV sequencing project, Genofunc has been benchmarked against annotated sequence gene coordinates from the Los Alamos HIV database as validation with downstream phylogenetic analysis result comparable to past literature as case study.
Conclusion
Genofunc is implemented fully in Python and licensed under the MIT license. Source code and documentation is available at: https://github.com/xiaoyu518/genofunc.
Similar content being viewed by others

Introduction
In the past decades, next generation sequencing and the evolution of computational biology have brought forth traction in the field of large scale genomics with public health implications [1, 2] alongside epidemiological surveillance and control [3, 4]. Viral phylodynamics uses genomic and epidemiological data in combination with mathematical modelling allowing estimation of phylogenies inferring valuable information of viruses through time and geographical locations based on metadata [5]. Currently, with the Covid-19 pandemic, the field of viral phylodynamics and automated bioinformatic pipelines have accelerated with a plethora of toolkits created to achieve this in real-time as a way of pandemic response [6,7,8].
Human immunodeficiency virus (HIV) is a rapidly evolving RNA virus being the largest viral pandemic pre-Covid with more than 37 million infected individuals in 2020 based on UNAIDS [9]. Initiatives such as PANGEA (Phylogenetics And Networks for Generalised Epidemics in Africa) have laid the groundwork in an attempt to both increase knowledge through large-scale sequencing to tackle HIV through prevention and treatment measures in key populations in an attempt to slow down the pandemic [10, 11]. Los Alamos HIV database also provides a platform with diversified whole genome HIV sequences from around the world with sample times over decades [12].
Here, we present Genofunc, a tool mainly for the annotating of raw sequences with protein coding features by aligning them in-frame based on reference sequence(s). In combination with several other command line functions such as feature extraction and filtering, key genomic features can be extracted for downstream phylogenetic analysis through tools such as Nextstrain augur [13] or BEAST [14].
Genofunc is an easily utilizable single command line tool fully scripted using python3 and can be installed through Python Package Index (PyPI) through pip install Genofunc. We test the Genofunc pipeline using whole genome HIV sequences downloaded from the Los Alamos HIV database as well as benchmark the estimated annotations from Genofunc against the Los Alamos annotated information. We validate the pol dataset extracted and infer the HIV root date and location with estimated clock rate using BEAST and compare results to past literature [15, 16].
Implementation
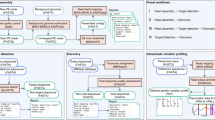
To allow easy utility and installation of Genofunc, the toolkit can be installed through GitHub or pip. This toolkit is fully written in python3.9 with functions constructed in a sequential fashion for easy pipelining mechanics of manipulating large raw HIV whole genome sequence datasets. Genofunc includes single command line functions consisting of two main components, FASTA/metadata file manipulation and HIV sequence processing (Fig. 1). For the prior component, single-task functions are scripted for easy manipulation of FASTA files and metadata files in CSV or TSV formats. The general functions include FASTA concatenation and filtering, metadata extraction and merging, sequence ID encoding and renaming etc. The latter functions are specific for HIV sequences including matching raw sequence to an existing reference file created by the user using minimap2 (reference_matcher) [17]; annotating raw sequence to the closest reference genome through local pairwise alignment using parasail (genome_annotator) [18] and finally extracting gene regions of interest based on annotated information (feature_extractor). Documentation and example command line references can be found on the Genofunc GitHub page.

Function workflow in processing raw HIV sequences to aligned sequences through Genofunc scripts ready for phylogenetic analysis using tools such as augur or BEAST. Light orange boxes contain the main workflow for processing raw sequence file to phylogeneticly ready alignment with input/output file type at each step declared within the brackets split by a hyphen respectively. The main functions are, Reference_matcher matching raw HIV sequences to the closest annotated sequence within a pre-constructed reference file. Genome_annotator map the raw sequence to the best reference from the output from reference_matcher and annotate the mapped sequence with the information on the best reference. Feature_extractor allows users to extract gene regions based on the annotated information from genome_annotator. Sequence alignment can be done through group_align which aligns multiple groups of sequences against a consensus sequence and then concatenated together for more efficient alignment or use any other alignment tools such as MAFFT [19]. The aligned file can be feed to BEAST [14] or augur [13] for phylogenetic post-analysis. Other task orientated functions (green box) can be added to the main pipeline at any phase of the workflow if the input (blue) and output (orange) files fit the up and downstream functions
Results
To identify the versatility and accuracy of the annotation and extraction of gene features using Genofunc, we downloaded and tested on three viral datasets, the West Nile virus, the Zaire ebolavirus and the Monkeypox virus, from GenBank and one HIV-1 dataset from Los Alamos. All complete sequences with fully annotated gene features were used as the test dataset summing up to 1211, 512, 914 and 6030 sequences respectively. Accession ID HQ596519, KJ660348 and NC_003310 were used as reference sequences for West Nile virus, Zaire ebolavirus and Monkeypox virus according to past literature [19,20,21] while a reference list of 237 sequences was selected consisting of the newest sequence from each subtype for HIV-1. The pipeline shown in Fig. 1 was performed without the final alignment step with all gene features extracted for all four datasets. Due to the large number of ORFs contained within the Monkeypox virus which many does not have well defined functions, we selected the first 6 genes which are associated with virulence and immune evasion within literature as a subset to represent the feasibility of Genofunc feature identification function for larger DNA genomes [22]. Similarly, complete genome sequences were imported, annotated based on the same reference and extracted using Geneious Prime using default settings [23]. Local alignments were performed for each viral sequence gene feature extracted by Genofunc and Geneious Prime to calculate a match ratio (sum of matching nucleotides/alignment length) against the corresponding annotated gene regions on GenBank. A summary comparing the performance of Genofunc against Geneious for the four viral datasets in terms of accuracy and run time are shown in Table 1.
Next, we test the efficiency of the Genofunc pipeline using 16,832 whole genome sequences from the Los Alamos HIV database [12]. Through filtering of missing metadata and removing duplicated sequences, we retained a raw dataset of 10,358 sequences ranging over a variety of subtypes and countries in an attempt to estimate the root of HIV-1. Following a similar pipeline to the benchmarking test, the pol gene region were extracted from the raw Los Alamos HIV-1 dataset using Snakemake [24] combining the Genofunc functions reference_matcher, genome_annotator, feature_extractor and filter_fasta resulting in a dataset of 10,293 pol sequence. 65 sequences were filtered out with coverage of less than 95% un-ambiguous sites (non-N) and/or length span of under 2500 nucleotide bases. Alignment was done using MAFFT [25] under default settings and regions with over 50% gaps were masked to create the final aligned dataset.
Phylogenetic analysis was done in another pipeline using augur [13] starting with a phylogenetic tree inferred using IQTREE with substitution model GTR + R6 [26, 27]. Timed phylogeny was inferred using TREETIME [28] with a clock rate of 0.001075 pre-estimated using BEAST on the 239 reference sequences [14]. A clock filter of 4.5 inter-quartile range was set to remove any sequence outliers which does not fit the clock regression model. Ancestral traits reconstruction was done using TREETIME for the estimation of root location. The inferred root date for HIV-1 is 1905 based on 9868 pol sequences resulting from sequences being removed as outliers not fitting to the molecular clock given. The ancestral location inferred is Africa between Cameroon and the Democratic Republic of Congo reflecting a similar region in past literature [15, 16] (Additional file 1: Fig. S2).
Discussion
Although Genofunc was written originally written for processing HIV genome sequences, the package can be easily utilised for other viral sequences with a given reference genome. This is shown through the benchmarking test of different viral datasets against another annotation tool Geneious and comparable to the annotation information on GenBank/Los Alamos. The results are robust not only for a diverse viral dataset such as HIV-1 using a short list of reference sequences but also proving true for viruses that are annotated using only a single reference for estimating gene feature coordinates with high accuracy over all genes analysed. Genofunc also remains robust for annotating larger DNA genomes but at a slightly lower accuracy compared to shorter RNA viruses for extracting specific gene features (Additional file 1: Fig. S1). However, it is worth taking note that the reference(s) chosen should be well annotated and represent the input raw dataset for better accuracy on estimated gene features.
We believe that based on our case study, Genofunc could be easily used on all viruses for the annotation and identification of gene features especially suitable for processing large raw sequence datasets. The setup of the pipeline using Genofunc in this case study also showed efficiency and simplicity in annotating and extracting key genomic regions from raw sequences which can be used in all forms of applications. This is shown through the runtime comparison to Geneious whereby Genofunc scale better with increase in dataset size but poorly for long viral genome sequences. Therefore, the current version of Genofunc is highly recommended for large scale short genome analysis (< 50 Kb). Future development of Genofunc lies in improving algorithm for reducing runtime in analysing longer genomes, improving accuracy in annotating gene features and correcting artefactual frameshifts that may be due to errors in sequencing or consensus genome calling pipelines.
In summary, Genofunc is a single command line toolkit suitable for constructing an automated pipeline to process large raw virus sequence datasets efficiently and readily for large scaled phylogenetics. Genofunc is open source and available with documentation at https://github.com/xiaoyu518/genofunc.
Availability of data and materials
All data used in this research can be found on the Los Alamos HIV database found at https://www.hiv.lanl.gov/content/index and GenBank on https://www.ncbi.nlm.nih.gov/. Project name: Genofunc. Project home page: https://github.com/xiaoyu518/genofunc. Operating system(s): iOS/Linux. Programming language: Python. Other requirements: None. License: MIT. Any restrictions to use by non-academics: None.
References
Chato C, Feng Y, Ruan Y, Xing H, Herbeck J, Kalish M, Poon AFY: Optimized phylogenetic clustering of HIV-1 sequence data for public health applications. bioRxiv 2022:2022.2001.2014.476062.
Ratmann O, Kagaayi J, Hall M, Golubchick T, Kigozi G, Xi X, Wymant C, Nakigozi G, Abeler-Dörner L, Bonsall D, et al. Quantifying HIV transmission flow between high-prevalence hotspots and surrounding communities: a population-based study in Rakai, Uganda. Lancet HIV. 2020;7(3):e173–83.
Souto B, Triunfante V, Santos-Pereira A, Martins J, Araújo PMM, Osório NS. Evolutionary dynamics of HIV-1 subtype C in Brazil. Sci Rep. 2021;11(1):23060.
Ratmann O, Wymant C, Colijn C, Danaviah S, Essex M, Frost SDW, Gall A, Gaiseitsiwe S, Grabowski M, Gray R, et al. HIV-1 full-genome phylogenetics of generalized epidemics in sub-Saharan Africa: impact of missing nucleotide characters in next-generation sequences. AIDS Res Hum Retroviruses. 2017;33(11):1083–98.
Volz EM, Koelle K, Bedford T. Viral phylodynamics. PLoS Comput Biol. 2013;9(3): e1002947.
Turakhia Y, Thornlow B, Hinrichs AS, De Maio N, Gozashti L, Lanfear R, Haussler D, Corbett-Detig R. Ultrafast sample placement on existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat Genet. 2021;53(6):809–16.
Gill MS, Lemey P, Suchard MA, Rambaut A, Baele G. Online Bayesian phylodynamic inference in BEAST with application to epidemic reconstruction. Mol Biol Evol. 2020;37(6):1832–42.
Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, Sagulenko P, Bedford T, Neher RA. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34(23):4121–3.
UNAIDS: AIDSinfo global data on HIV epidemiology and response. 2022.
Pillay D, Herbeck J, Cohen MS, de Oliveira T, Fraser C, Ratmann O, Brown AL, Kellam P, Consortium P-H. PANGEA-HIV: phylogenetics for generalised epidemics in Africa. Lancet Infect Dis. 2015;15(3):259–61.
Abeler-Dörner L, Grabowski MK, Rambaut A, Pillay D, Fraser C. PANGEA-HIV 2: phylogenetics and networks for generalised epidemics in Africa. Curr Opin HIV AIDS. 2019;14(3):173–80.
LosAlamos: HIV sequence database. 2022.
Huddleston J, Hadfield J, Sibley TR, Lee J, Fay K, Ilcisin M, Harkins E, Bedford T, Neher RA, Hodcroft EB. Augur: a bioinformatics toolkit for phylogenetic analyses of human pathogens. J Open Source Softw. 2021;6(57):2906.
Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214.
Faria NR, Rambaut A, Suchard MA, Baele G, Bedford T, Ward MJ, Tatem AJ, Sousa JD, Arinaminpathy N, Pépin J, et al. The early spread and epidemic ignition of HIV-1 in human populations. Science. 2014;346(6205):56–61.
Worobey M, Gemmel M, Teuwen DE, Haselkorn T, Kunstman K, Bunce M, Muyembe J-J, Kabongo J-MM, Kalengayi RM, Van Marck E, et al. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature. 2008;455(7213):661–4.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100.
Daily J. Parasail: SIMD C library for global, semi-global, and local pairwise sequence alignments. BMC Bioinform. 2016;17(1):81.
Petersen LR. Global epidemiology of West Nile virus. In: West Nile encephalitis virus infection: viral pathogenesis and the host immune response. New York, NY: Springer New York; 2009, p. 1–23.
Gire SK, Goba A, Andersen KG, Sealfon RS, Park DJ, Kanneh L, Jalloh S, Momoh M, Fullah M, Dudas G, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345(6202):1369–72.
Shchelkunov SN, Totmenin AV, Babkin IV, Safronov PF, Ryazankina OI, Petrov NA, Gutorov VV, Uvarova EA, Mikheev MV, Sisler JR, et al. Human monkeypox and smallpox viruses: genomic comparison. FEBS Lett. 2001;509(1):66–70.
Lum FM, Torres-Ruesta A, Tay MZ, Lin RTP, Lye DC, Rénia L, Ng LFP. Monkeypox: disease epidemiology, host immunity and clinical interventions. Nat Rev Immunol. 2022;22(10):597–613.
Prime G. Geneious. In, 2023.0.1 edn; 2023.
Mölder F, Jablonski KP, Letcher B, Hall MB, Tomkins-Tinch CH, Sochat V, Forster J, Lee S, Twardziok SO, Kanitz A, et al. Sustainable data analysis with snakemake. FRes. 2021;10:33.
Katoh K, Misawa K, Kuma K, Miyata T: MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2014;32(1):268–74.
Soubrier J, Steel M, Lee MS, Der Sarkissian C, Guindon S, Ho SY, Cooper A. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol Biol Evol. 2012;29(11):3345–58.
Sagulenko P, Puller V, Neher RA. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018;4(1):vex042.
Acknowledgements
Thanks to Andrew Rambaut for the discussions and concepts that are implemented in Genofunc and to all of the data producers that have contributed to the GenBank and Los Alamos HIV database used for the benchmarking test.
Funding
This work was supported by the PANGEA HIV-1 project funded by Bill and Melinda Gates Foundation Grant [OPP1175094].
Author information
Authors and Affiliations
Contributions
XY was responsible for the research, wrote the manuscript and the software. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary Figures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yu, X. Genofunc: genome annotation and identification of genome features for automated pipelining analysis of virus whole genome sequences. BMC Bioinformatics 24, 218 (2023). https://doi.org/10.1186/s12859-023-05356-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-023-05356-3